
The Determination of Cannabinoids in Urine Samples Using Microextraction by Packed Sorbent and Gas Chromatography-Mass Spectrometry
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
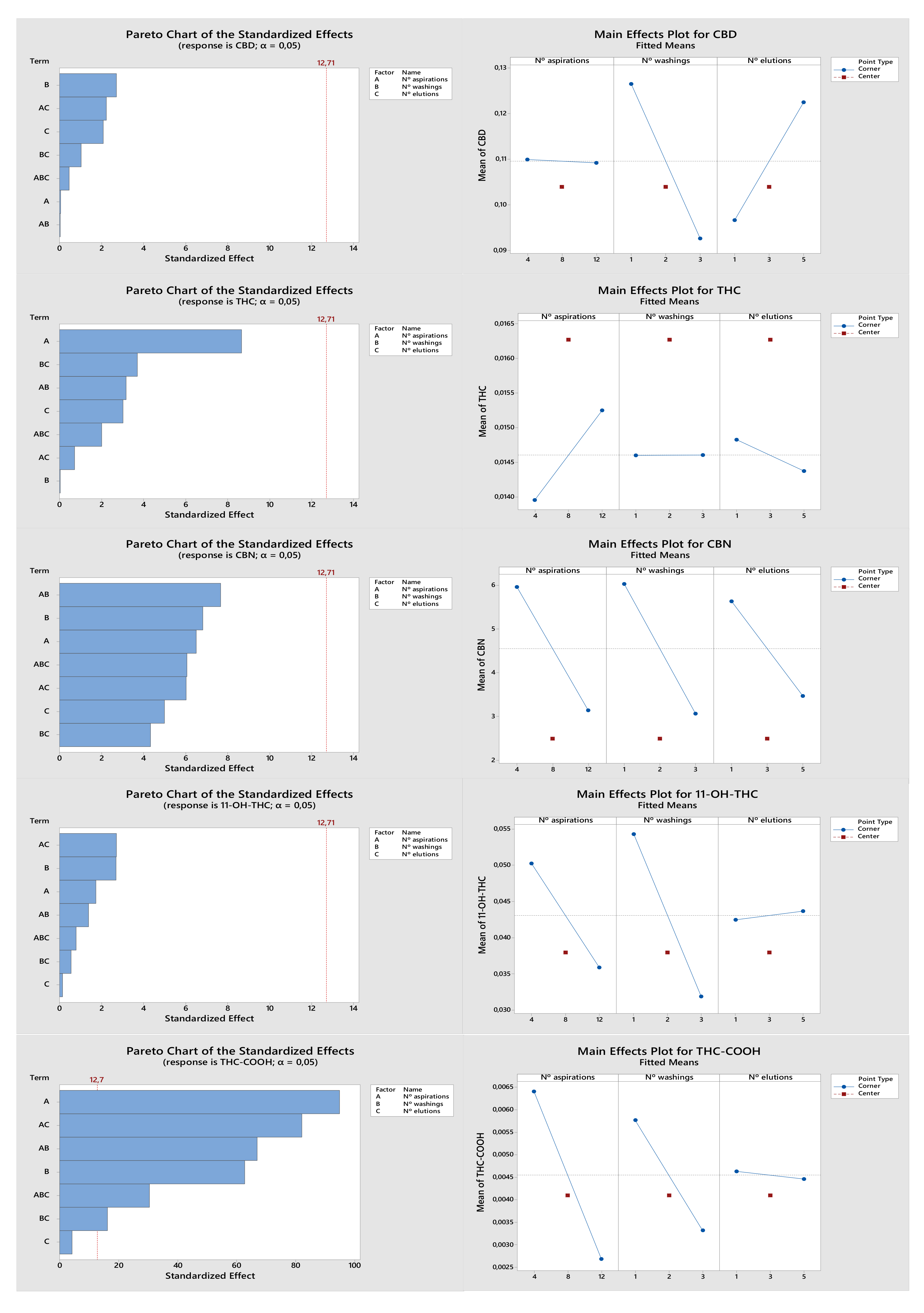
2.1. Optimization of the Extraction Procedure
2.1.1. Extraction Procedure Selection
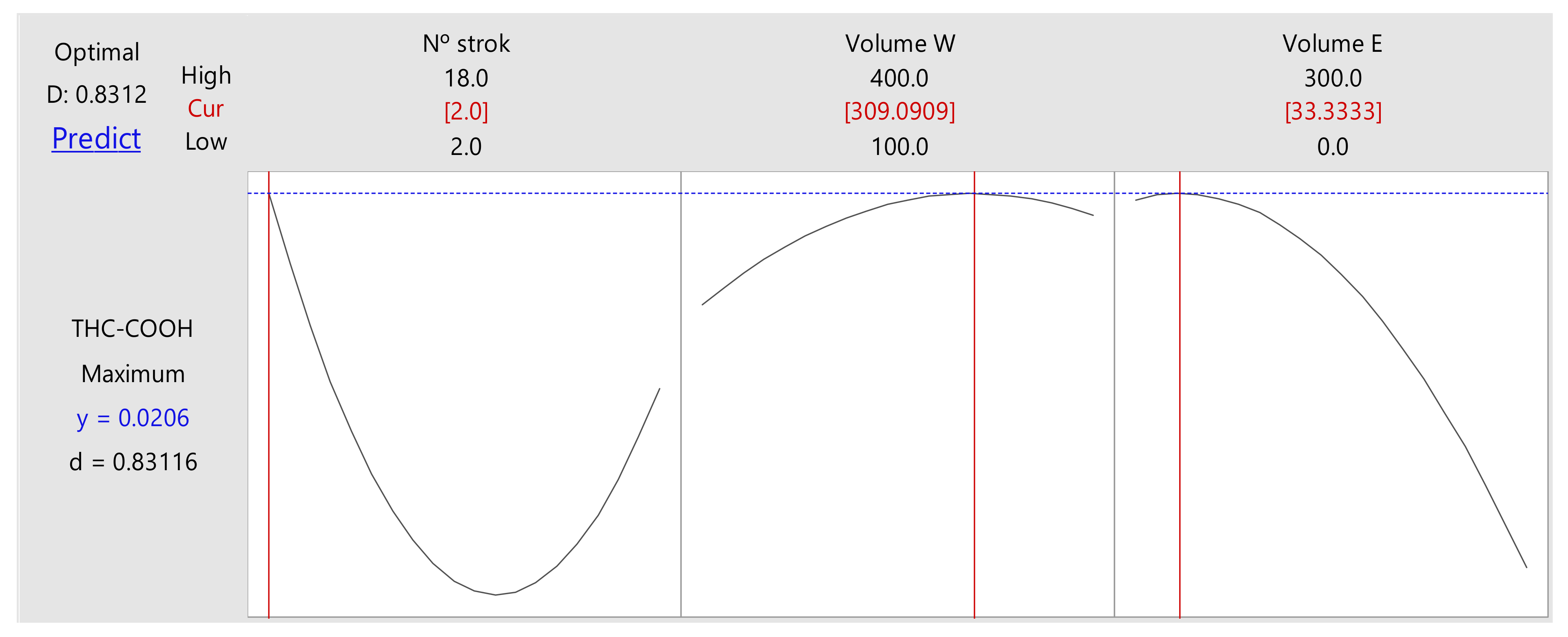
2.1.2. Optimization of the Experimental Design
2.2. Method Validation Parameters
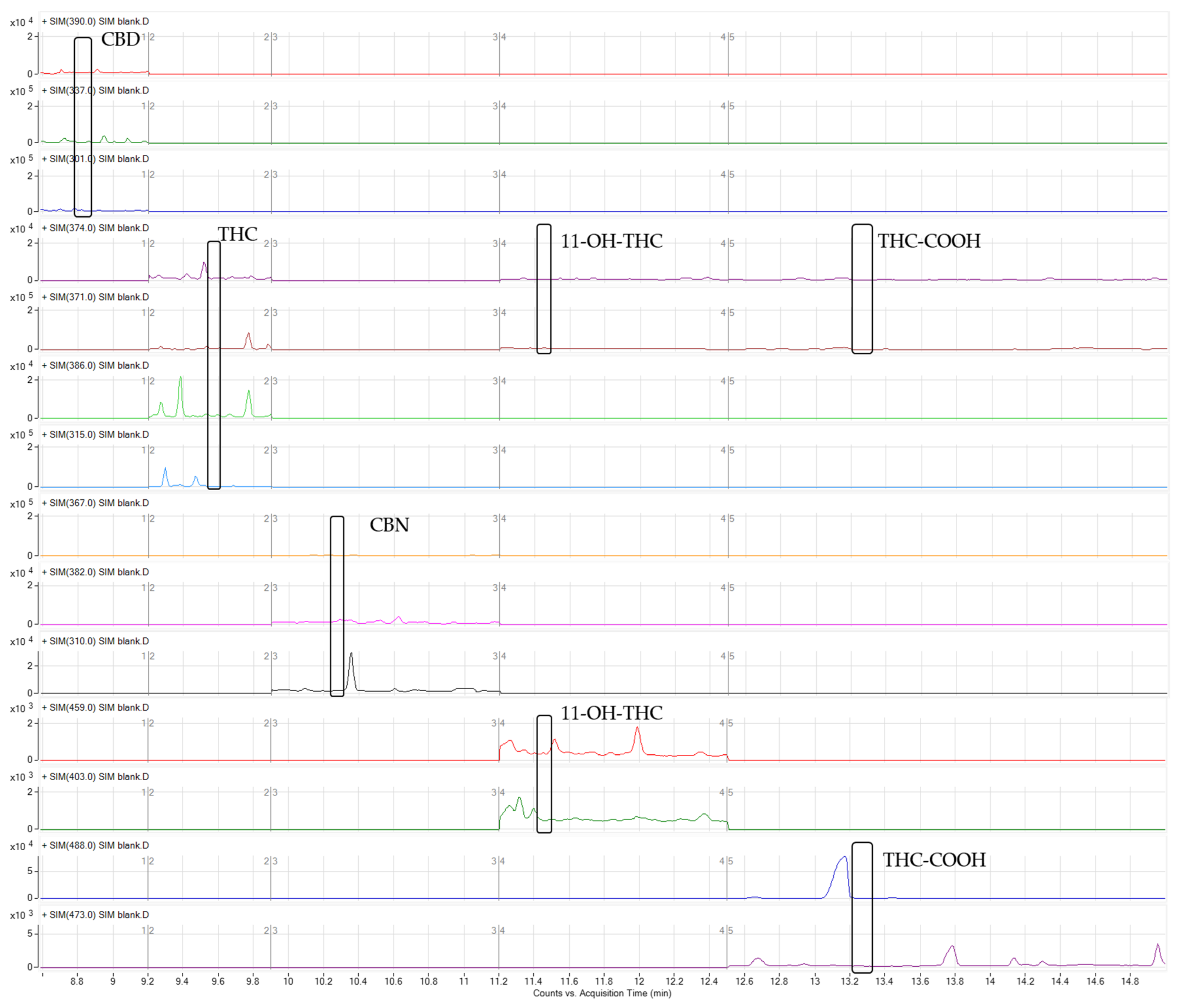
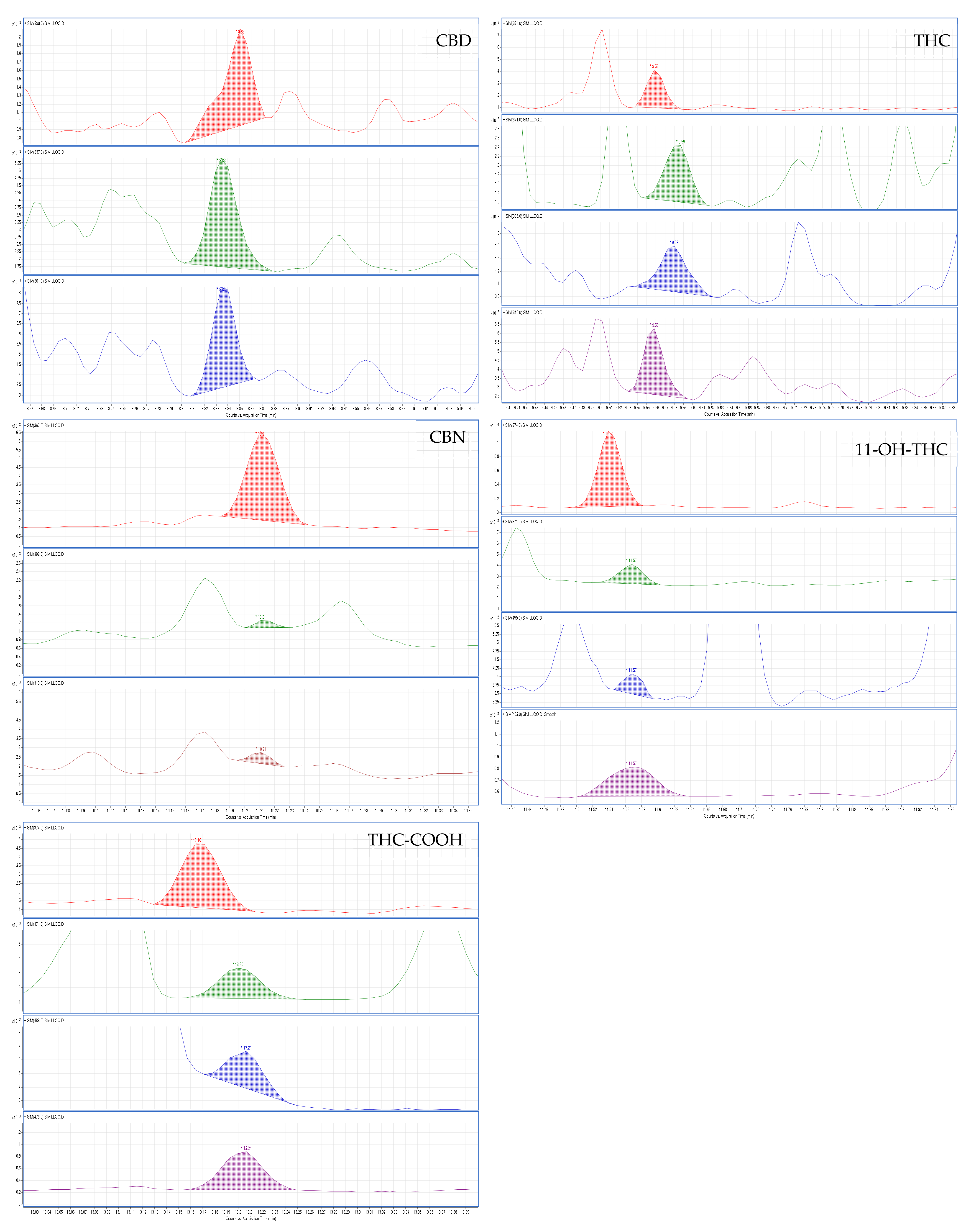
2.2.1. Selectivity
2.2.2. Linearity and Calibration Model
2.2.3. Limits of Detection and Quantification
2.2.4. Intra-Day, Inter-Day, and Intermediate Precision and Accuracy
2.2.5. Extraction Efficiency
2.2.6. Stability
2.2.7. Carryover
2.2.8. Method Applicability
3. Materials and Methods
3.1. Reagents and Standards
3.2. Biological Specimens/Urine Samples
3.3. Gas Chromatographic and Mass Spectrometric Conditions
3.4. Sample Preparation
3.5. MEPS Procedure Optimization
3.6. Validation Procedure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cannabis: Health and Social Responses. Available online: https://www.emcdda.europa.eu/publications/mini-guides/cannabis-health-and-social-responses_en (accessed on 9 July 2022).
- Gonçalves, J.; Rosado, T.; Soares, S.; Simão, A.; Caramelo, D.; Luís, Â.; Fernández, N.; Barroso, M.; Gallardo, E.; Duarte, A. Cannabis and Its Secondary Metabolites: Their Use as Therapeutic Drugs, Toxicological Aspects, and Analytical Determination. Medicines 2019, 6, 31. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime (ONUDC) Global Overview: Drug Demand; ONUDC: Vienna, Austria, 2021; ISBN 9789211483611.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). Cannabis Legislation in EUROPE; European Union: Lisbon, Portugal, 2018; ISBN 03029743. [Google Scholar]
- Preuss, U.W.; Huestis, M.A.; Schneider, M.; Hermann, D.; Lutz, B.; Hasan, A.; Kambeitz, J.; Wong, J.W.M.; Hoch, E. Cannabis Use and Car Crashes: A Review. Front. Psychiatry 2021, 12, 643315. [Google Scholar] [CrossRef]
- Molinaro, S.; Vicente, J.; Benedetti, E.; Cerrai, S.; Colasante, E.; Arpa, S.; Chomynová, P.; Kraus, L.; Monshouwer, K. ESPAD Report; ESPAD: Lisbon, Portugal, 2019. [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report: Trends and Developments; 2021. Available online: https://www.emcdda.europa.eu/publications/edr/trends-developments/2021_en (accessed on 27 October 2021).
- Matheson, J.; Mann, R.E.; Sproule, B.; Huestis, M.A.; Wickens, C.M.; Stoduto, G.; George, T.P.; Rehm, J.; Le Foll, B.; Brands, B. Acute and residual mood and cognitive performance of young adults following smoked cannabis. Pharmacol. Biochem. Behav. 2020, 194, 172937. [Google Scholar] [CrossRef]
- Wickens, C.M.; Wright, M.; Mann, R.E.; Brands, B.; Di Ciano, P.; Stoduto, G.; Fares, A.; Matheson, J.; George, T.P.; Rehm, J.; et al. Separate and combined effects of alcohol and cannabis on mood, subjective experience, cognition and psychomotor performance: A randomized trial. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 118, 110570. [Google Scholar] [CrossRef]
- Kancherla, N.; Jeyanthi, K.; Abbas, R.; Sathi, T.; Upadhyay, A.; Garlapati, S. Cannabis Associated Mental Health Effects: A Review. J. Pharm. Bioallied Sci. 2021, 13, S943–S946. [Google Scholar] [CrossRef]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef]
- Legare, C.A.; Raup-Konsavage, W.M.; Vrana, K.E. Therapeutic Potential of Cannabis, Cannabidiol, and Cannabinoid-Based Pharmaceuticals. Pharmacology 2022, 107, 131–149. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Progress in the Chemistry of Organic Natural Products; Springer: Berlin/Heidelberg, Germany, 2017; Volume 103, pp. 1–36. ISBN 9783319455419. [Google Scholar]
- Filipiuc, L.E.; Ababei, D.C.; Alexa-Stratulat, T.; Pricope, C.V.; Bild, V.; Stefanescu, R.; Stanciu, G.D.; Tamba, B.I. Major phytocannabinoids and their related compounds: Should we only search for drugs that act on cannabinoid receptors? Pharmaceutics 2021, 13, 1823. [Google Scholar] [CrossRef]
- Lucas, C.J.; Galettis, P.; Schneider, J. The pharmacokinetics and the pharmacodynamics of cannabinoids. Br. J. Clin. Pharmacol. 2018, 84, 2477–2482. [Google Scholar] [CrossRef]
- Karschner, E.L.; Swortwood-Gates, M.J.; Huestis, M.A. Identifying and Quantifying Cannabinoids in Biological Matrices in the Medical and Legal Cannabis Era. Clin. Chem. 2020, 66, 888–914. [Google Scholar] [CrossRef]
- D’Orazio, A.L.; Mohr, A.L.A.; Chan-Hosokawa, A.; Harper, C.; Huestis, M.A.; Limoges, J.F.; Miles, A.K.; Scarneo, C.E.; Kerrigan, S.; Liddicoat, L.J.; et al. Recommendations for Toxicological Investigation of Drug-Impaired Driving and Motor Vehicle Fatalities-2021 Update. J. Anal. Toxicol. 2021, 45, 529–536. [Google Scholar] [CrossRef]
- Abdel-Rehim, M. Microextraction by packed sorbent (MEPS): A tutorial. Anal. Chim. Acta 2011, 701, 119–128. [Google Scholar] [CrossRef]
- Moein, M.M.; Abdel-Rehim, A.; Abdel-Rehim, M. Microextraction by packed sorbent (MEPS). TrAC Trends Anal. Chem. 2015, 67, 34–44. [Google Scholar] [CrossRef]
- Rosado, T.; Gallardo, E.; Vieira, D.N.; Barroso, M. Microextraction by Packed Sorbent. In Microextraction Techniques in Analytical Toxicology; CRC Press: Boca Raton, FL, USA, 2021; pp. 71–115. ISBN 9781003128298. [Google Scholar]
- Abdel-Rehim, M. Recent advances in microextraction by packed sorbent for bioanalysis. J. Chromatogr. A 2010, 1217, 2569–2580. [Google Scholar] [CrossRef]
- Santos, C.; Oppolzer, D.; Gonçalves, A.; Barroso, M.; Gallardo, E. Determination of Organophosphorous Pesticides in Blood Using Microextraction in Packed Sorbent and Gas Chromatography-Tandem Mass Spectrometry. J. Anal. Toxicol. 2018, 42, 321–329. [Google Scholar] [CrossRef]
- Rosado, T.; Gallardo, E.; Vieira, D.N.; Barroso, M. Microextraction by Packed Sorbent as a Novel Strategy for Sample Clean-Up in the Determination of Methadone and EDDP in Hair. J. Anal. Toxicol. 2020, 44, 840–850. [Google Scholar] [CrossRef]
- Rosado, T.; Gonçalves, A.; Margalho, C.; Barroso, M.; Gallardo, E. Rapid analysis of cocaine and metabolites in urine using microextraction in packed sorbent and GC/MS. Anal. Bioanal. Chem. 2017, 409, 2051–2063. [Google Scholar] [CrossRef]
- Malaca, S.; Rosado, T.; Restolho, J.; Rodilla, J.M.; Rocha, P.M.M.; Silva, L.; Margalho, C.; Barroso, M.; Gallardo, E. Determination of amphetamine-type stimulants in urine samples using microextraction by packed sorbent and gas chromatography-mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1120, 41–50. [Google Scholar] [CrossRef]
- Simão, A.Y.; Monteiro, C.; Marques, H.; Rosado, T.; Margalho, C.; Barroso, M.; Andraus, M.; Gallardo, E. Analysis of opiates in urine using microextraction by packed sorbent and gas Chromatography-Tandem mass spectrometry. J. Chromatogr. B 2022, 1207, 123361. [Google Scholar] [CrossRef]
- Sorribes-Soriano, A.; Verdeguer, J.; Pastor, A.; Armenta, S.; Esteve-Turrillas, F.A. Determination of Third-Generation Synthetic Cannabinoids in Oral Fluids. J. Anal. Toxicol. 2021, 45, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Pautova, A.K.; Khesina, Z.B.; Litvinova, T.N.; Revelsky, A.I.; Beloborodova, N.V. Metabolic profiling of aromatic compounds in cerebrospinal fluid of neurosurgical patients using microextraction by packed sorbent and liquid–liquid extraction with gas chromatography–mass spectrometry analysis. Biomed. Chromatogr. 2021, 35, 4969. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rehim, M. New trend in sample preparation: On-line microextraction in packed syringe for liquid and gas chromatography applications. J. Chromatogr. B 2004, 801, 317–321. [Google Scholar] [CrossRef]
- Silva, C.; Cavaco, C.; Perestrelo, R.; Pereira, J.; Câmara, J. Microextraction by Packed Sorbent (MEPS) and Solid-Phase Microextraction (SPME) as Sample Preparation Procedures for the Metabolomic Profiling of Urine. Metabolites 2014, 4, 71–97. [Google Scholar] [CrossRef] [PubMed]
- Sergi, M.; Montesano, C.; Odoardi, S.; Mainero Rocca, L.; Fabrizi, G.; Compagnone, D.; Curini, R. Micro extraction by packed sorbent coupled to liquid chromatography tandem mass spectrometry for the rapid and sensitive determination of cannabinoids in oral fluids. J. Chromatogr. A 2013, 1301, 139–146. [Google Scholar] [CrossRef]
- Rosado, T.; Fernandes, L.; Barroso, M.; Gallardo, E. Sensitive determination of THC and main metabolites in human plasma by means of microextraction in packed sorbent and gas chromatography–tandem mass spectrometry. J. Chromatogr. B 2017, 1043, 63–73. [Google Scholar] [CrossRef]
- Sartore, D.M.; Vargas Medina, D.A.; Costa, J.L.; Lanças, F.M.; Santos-Neto, Á.J. Automated microextraction by packed sorbent of cannabinoids from human urine using a lab-made device packed with molecularly imprinted polymer. Talanta 2020, 219, 121185. [Google Scholar] [CrossRef]
- Scientific Working Group for Forensic Toxicology (SWGTOX). Standard Practices for Method Validation in Forensic Toxicology. J. Anal. Toxicol. 2013, 37, 452–474. [Google Scholar] [CrossRef]
- Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 27 October 2021).
- Substance Abuse and Mental Health Services Administration (SAMHSA). Clinical Drug Testing in Primary Care. In Technical Assistance Publication Series; SAMHSA: Rockville, MD, USA, 2012. [Google Scholar]
- European Workplace Drug Testing Society. European Guidelines for Workplace Drug Testing in Urine 2015-11-01; European Workplace Drug Testing Society, 2015; pp. 1–40. [Google Scholar]
- Reber, J.D.; Karschner, E.L.; Seither, J.Z.; Knittel, J.L.; Walterscheid, J.P. Screening and confirmation methods for the qualitative identification of nine phytocannabinoids in urine by LC-MS/MS. Clin. Biochem. 2021, 98, 54–62. [Google Scholar] [CrossRef]
- Montesano, C.; Sergi, M.; Odoardi, S.; Simeoni, M.C.; Compagnone, D.; Curini, R. A μ-SPE procedure for the determination of cannabinoids and their metabolites in urine by LC–MS/MS. J. Pharm. Biomed. Anal. 2014, 91, 169–175. [Google Scholar] [CrossRef]
- Jagerdeo, E.; Montgomery, M.A.; Karas, R.P.; Sibum, M. A fast method for screening and/or quantitation of tetrahydrocannabinol and metabolites in urine by automated SPE/LC/MS/MS. Anal. Bioanal. Chem. 2010, 398, 329–338. [Google Scholar] [CrossRef]
- Korac, N.; Vidic, D.; Sutlović, D. Modified QuEChERS extraction and GC-MS analysis of selected cannabinoids from human urine. Glas. Hem. Tehnol. Bosne Hercegovine 2020, 54, 37–44. [Google Scholar] [CrossRef]
- Sempio, C.; Scheidweiler, K.B.; Barnes, A.J.; Huestis, M.A. Optimization of recombinant β-glucuronidase hydrolysis and quantification of eight urinary cannabinoids and metabolites by liquid chromatography tandem mass spectrometry. Drug Test. Anal. 2018, 10, 518–529. [Google Scholar] [CrossRef]
- Nestić, M.; Babić, S.; Pavlović, D.M.; Sutlović, D. Molecularly imprinted solid phase extraction for simultaneous determination of δ9-tetrahydrocannabinol and its main metabolites by gas chromatography-mass spectrometry in urine samples. Forensic Sci. Int. 2013, 231, 317–324. [Google Scholar] [CrossRef]
- Abraham, T.T.; Lowe, R.H.; Pirnay, S.O.; Darwin, W.D.; Huestis, M.A. Simultaneous GC-EI-MS determination of Δ9-tetrahydrocannabinol, 11-hydroxy-Δ9-tetrahydrocannabinol, and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol in human urine following tandem enzyme-alkaline hydrolysis. J. Anal. Toxicol. 2007, 31, 477–485. [Google Scholar] [CrossRef]
- Lendoiro, E.; De Castro, A.; Fernández-Vega, H.; Cela-Pérez, M.C.; López-Vilariño, J.M.; González-Rodríguez, M.V.; Cruz, A.; López-Rivadulla, M. Molecularly imprinted polymer for selective determination of Δ9-tetrahydrocannabinol and 11-nor-Δ9-tetrahydrocannabinol carboxylic acid using LC-MS/MS in urine and oral fluid Forensic Toxicology. Anal. Bioanal. Chem. 2014, 406, 3589–3597. [Google Scholar] [CrossRef]
- Rumpler, M.J. Quantitative analysis of 11-nor-9-carboxy-tetrahydrocannbinol (THC-COOH) in urine by LC-MS/MS following a simple filtration. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 957, 77–83. [Google Scholar] [CrossRef]
- Felli, M.; Martello, S.; Chiarotti, M. LC-MS-MS method for simultaneous determination of THCCOOH and THCCOOH-glucuronide in urine: Application to workplace confirmation tests. Forensic Sci. Int. 2011, 204, 67–73. [Google Scholar] [CrossRef]
- Morisue Sartore, D.; Costa, J.L.; Lanças, F.M.; Santos-Neto, Á.J. Packed in-tube SPME–LC–MS/MS for fast and straightforward analysis of cannabinoids and metabolites in human urine. Electrophoresis 2022, 43, 1555–1566. [Google Scholar] [CrossRef]
- Moretti, M.; Freni, F.; Carelli, C.; Previderé, C.; Grignani, P.; Vignali, C.; Cobo-Golpe, M.; Morini, L. Analysis of Cannabinoids and Metabolites in Dried Urine Spots (DUS). Molecules 2021, 26, 5334. [Google Scholar] [CrossRef]
- De M Prata, V.; Emídio, E.S.; Dorea, H.S. New catalytic ultrasound method for derivatization of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine, with analysis by GC-MS/MS. Anal. Bioanal. Chem. 2012, 403, 625–632. [Google Scholar] [CrossRef]
- Frei, P.; Frauchiger, S.; Scheurer, E.; Mercer-Chalmers-Bender, K. Quantitative determination of five cannabinoids in blood and urine by gas chromatography tandem mass spectrometry applying automated on-line solid phase extraction. Drug Test. Anal. 2022, 14, 1223–1233. [Google Scholar] [CrossRef]
- Meier, U.; Dussy, F.; Scheurer, E.; Mercer-Chalmers-Bender, K.; Hangartner, S. Cannabinoid concentrations in blood and urine after smoking cannabidiol joints. Forensic Sci. Int. 2018, 291, 62–67. [Google Scholar] [CrossRef]
- Rodrigues, L.C.; Kahl, J.M.; de Chinaglia, K.O.; de Campos, E.G.; Costa, J.L. Dispersive liquid–liquid microextraction of 11-nor-Δ9-tetrahydrocannabinol-carboxylic acid applied to urine testing. Bioanalysis 2022, 14, 87–100. [Google Scholar] [CrossRef]
- Saenz, S.R.; Lewis, R.J.; Angier, M.K.; Wagner, J.R. Postmortem Fluid and Tissue Concentrations of THC, 11-OH-THC and THC-COOH†. J. Anal. Toxicol. 2017, 41, 508–516. [Google Scholar] [CrossRef]
- Andersson, M.; Scheidweiler, K.B.; Sempio, C.; Barnes, A.J.; Huestis, M.A. Simultaneous quantification of 11 cannabinoids and metabolites in human urine by liquid chromatography tandem mass spectrometry using WAX-S tips. Anal. Bioanal. Chem. 2016, 408, 6461–6471. [Google Scholar] [CrossRef]
- Stephanson, N.; Josefsson, M.; Kronstrand, R.; Beck, O. Accurate identification and quantification of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine drug testing: Evaluation of a direct high efficiency liquid chromatographic-mass spectrometric method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 101–108. [Google Scholar] [CrossRef]
- Chebbah, C.; Pozo, O.J.; Deventer, K.; Van Eenoo, P.; Delbeke, F.T. Direct quantification of 11-nor-Δ 9 -tetrahydrocannabinol-9-carboxylic acid in urine by liquid chromatography/tandem mass spectrometry in relation to doping control analysis. Rapid Commun. Mass Spectrom. 2010, 24, 1133–1141. [Google Scholar] [CrossRef]
- Sánchez-González, J.; Salgueiro-Fernández, R.; Cabarcos, P.; Bermejo, A.M.; Bermejo-Barrera, P.; Moreda-Piñeiro, A. Cannabinoids assessment in plasma and urine by high performance liquid chromatography–tandem mass spectrometry after molecularly imprinted polymer microsolid-phase extraction. Anal. Bioanal. Chem. 2017, 409, 1207–1220. [Google Scholar] [CrossRef]
- Zanchetti, G.; Floris, I.; Piccinotti, A.; Tameni, S.; Polettini, A. Rapid and robust confirmation and quantification of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid (THC-COOH) in urine by column switching LC-MS-MS analysis. J. Mass Spectrom. 2012, 47, 124–130. [Google Scholar] [CrossRef]
- Moradi, M.; Yamini, Y.; Baheri, T. Analysis of abuse drugs in urine using surfactant-assisted dispersive liquid-liquid microextraction. J. Sep. Sci. 2011, 34, 1722–1729. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Wojciech Wojnowski—Science Profile—MOST Wiedzy. Available online: https://mostwiedzy.pl/en/wojciech-wojnowski,174235-1/AGREE? (accessed on 20 August 2022).
- Schroeder, J.L.; Marinetti, L.J.; Smith, R.K.; Brewer, W.E.; Clelland, B.L.; Morgan, S.L. The analysis of Δ9-tetrahydrocannabinol and metabolite in whole blood and 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine using disposable pipette extraction with confirmation and quantification by gas chromatography-mass spectrometry. J. Anal. Toxicol. 2008, 32, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.E.; Andrews, A.R.J. Screening method for 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine using hollow fiber membrane solvent microextraction with in-tube derivatization. J. Chromatogr. B Biomed. Sci. Appl. 2001, 760, 27–36. [Google Scholar] [CrossRef]
- At a Glance—Estimates of Drug Use in The European Union (Updated June 2021). Available online: https://www.emcdda.europa.eu/media-library/glance-—-estimates-drug-use-european-union-updated-june-2021_en (accessed on 27 October 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Weight | Linear Range (ng/mL) | Linearity | R2 ª | LOD (ng/mL) | LLOQ (ng/mL) | |
|---|---|---|---|---|---|---|---|
| Slope ª | Intercept ª | ||||||
| CBD | 1/x | 1–400 | 0.1118 ± 0.0136 | 2.9449 ± 4.0245 | 0.9980 ± 0.0014 | 1 | 1 |
| THC | 1–400 | 0.0488 ± 0.0089 | 0.6771 ± 0.5143 | 0.9990 ± 0.0008 | 1 | 1 | |
| CBN | 5–400 | 0.2865 ± 0.0275 | 0.1373 ± 0.5117 | 0.9967 ± 0.0030 | 5 | 5 | |
| 11-OH-THC | 5–400 | 0.0678 ± 0.0033 | 0.1173 ± 0.1607 | 0.9983 ± 0.0012 | 1 | 5 | |
| THC-COOH | 10–400 | 0.0588 ± 0.0065 | 0.3577 ± 0.2330 | 0.9984 ± 0.0010 | 5 | 10 | |
| Analyte | Sample Amount (mL) | LOD (ng/mL) | LLOQ (ng/mL) | Method of Detection | Reference |
|---|---|---|---|---|---|
| CBD | 1.00 | 1.00 | ns | LC-MS/MS | [38] |
| THC | 1.00 | ||||
| CBN | 1.00 | ||||
| 11-OH-THC | 1.00 | ||||
| THC-COOH | 5.00 | ||||
| CBD | 0.09 | 3.00 | 10.00 | LC-MS/MS | [39] |
| THC | 3.00 | 8.00 | |||
| CBN | 4.00 * | 9.00 | |||
| 11-OH-THC | 3.00 | 9.00 | |||
| THC-COOH | 2.00 * | 6.00 * | |||
| CBD | 1.00 | 3.00 | 9.00 | LC-MS/MS | [40] |
| THC | 2.00 | 8.00 | |||
| CBN | 4.00 * | 12.00 | |||
| 11-OH-THC | 2.00 * | 6.00 | |||
| THC-COOH | 2.00 * | 6.00 * | |||
| CBD | 2.00 | 5.00 | 16.00 | GC-MS | [41] |
| THC | 3.00 | 9.00 | |||
| CBN | 5.00 | 18.00 | |||
| 11-OH-THC | 2.60 * | 8.70 | |||
| THC-COOH | 4.50 * | 15.00 | |||
| CBD | 0.20 | 2.00 | ** | LC-MS/MS | [42] |
| THC | 1.00 | ||||
| CBN | 2.00 * | ||||
| 11-OH-THC | 2.00 | ||||
| THC-COOH | 1.00 * | ||||
| THC | 2.00 | 2.50 | 3.00 | GC-MS | [43] |
| 11-OH-THC | 1.00 | 2.00 * | |||
| THC-COOH | 1.00 * | 2.00 * | |||
| THC | 2.00 | 1.00 | 2.50 | GC-MS | [44] |
| 11-OH-THC | 2.50 | 2.50 * | |||
| THC-COOH | 2.50 * | 2.50 * | |||
| THC | 1.00 | 5.00 | 20.00 | LC-MS/MS | [33] |
| 11-OH-THC | 5.00 | 20.00 | |||
| THC-COOH | 1.00 * | 5.00 * | |||
| THC | 0.50 | 1.00 | 2.50 | LC-MS/MS | [45] |
| THC-COOH | 1.00 * | 2.50 * | |||
| THC-COOH | 0.50 | 5.00 | 10.00 | LC-MS/MS | [46] |
| THC-COOH | 0.50 | 5.00 | 10.00 | LC-MS/MS | [47] |
| CBD | 0.25 | ns | 10.00 | LC-MS/MS | [48] |
| THC | 10.00 | ||||
| CBN | 10.00 | ||||
| 11-OH-THC | 10.00 | ||||
| THC-COOH | 10.00 | ||||
| CBD | 0.025 | 0.30 | 10.00 | LC-MS/MS | [49] |
| THC | 0.30 | 10.00 | |||
| CBN | 1.40 | 10.00 | |||
| 11-OH-THC | 1.90 | 10.00 | |||
| THC-COOH | 1.40 | 10.00 | |||
| THC-COOH | 0.50 * | 1.00 * | GC-MS/MS | [50] | |
| CBD | 0.25 | 0.10 * | 0.30 * | GC-MS/MS | [51] |
| THC | 0.15 * | 0.30 * | |||
| CBN | 0.15 * | 0.20 * | |||
| 11-OH-THC | 0.15 * | 0.30 * | |||
| THC-COOH | 1.00 * | 3.00 * | |||
| CBD | 1.00 | 0.20 * | 0.30 * | GC-MS/MS | [52] |
| THC | 0.20 * | 0.30 * | |||
| CBN | 0.20 * | 0.30 * | |||
| 11-OH-THC | 0.20 * | 0.30 * | |||
| THC-COOH | 2.00 * | 3.00 * | |||
| THC-COOH | ns | 1.00 * | 5.00 * | GC-MS/MS | [53] |
| THC | 1.50 | 0.78 * | ** | UHPLC-MS/MS | [54] |
| 11-OH-THC | 0.78 * | ||||
| THC-COOH | 0.78 * | ||||
| CBD | 0.20 | 0.50 * | ** | LC-MS/MS | [55] |
| THC | 0.50 * | ||||
| CBN | 0.50 * | ||||
| 11-OH-THC | 0.50 * | ||||
| THC-COOH | 0.50 * | ||||
| THC-COOH | 0.12 | 0.20 * | 0.70 * | LC-MS/MS | [56] |
| THC-COOH | 1.00 | 0.20 * | 5.00 * | LC-MS/MS | [59] |
| THC | 0.10–1.00 | 0.16 * | 0.27 * | LC-MS/MS | [58] |
| 11-OH-THC | 0.17 * | 0.28 * | |||
| THC-COOH | 0.14 * | 0.23 * | |||
| THC-COOH | 0.20 | 0.50 * | 7.50 * | LC-MS/MS | [57] |
| CBD | 10.00 | 0.20 * | 1.00 | HPLC-UV | [60] |
| THC | 0.50 * | 1.00 | |||
| CBN | 0.10 * | 1.00 * |
| Analyte | Concentration (ng/mL) | Inter-Day (n = 5) | Intra-Day (n = 5) | Intermediate (n = 15) | |||
|---|---|---|---|---|---|---|---|
| CV (%) | RE (%) | CV (%) | RE (%) | CV (%) | RE (%) | ||
| CBD | 1 | 10.27 | 6.68 | 7.67 | 7.38 | ||
| 5 | 14.26 | 1.80 | 10.50 | 9.06 | |||
| 10 | 9.02 | 6.34 | |||||
| 15 | 10.08 | 3.04 | 2.80 | 1.46 | |||
| 50 | 1.04 | 1.04 | |||||
| 100 | 3.13 | 3.13 | 9.10 | 9.55 | |||
| 200 | 1.68 | 1.68 | |||||
| 240 | 9.12 | 0.43 | |||||
| 300 | 3.51 | 3.51 | |||||
| 360 | 7.84 | 4.87 | |||||
| 400 | 0.49 | 0.49 | 9.81 | 6.98 | |||
| THC | 1 | 8.71 | 0.88 | 7.27 | 3.94 | ||
| 5 | 9.23 | 0.76 | 5.00 | 7.51 | |||
| 10 | 9.61 | 1.39 | |||||
| 15 | 9.03 | 0.76 | 7.01 | 3.91 | |||
| 50 | 10.00 | 2.89 | |||||
| 100 | 4.49 | 1.39 | 9.56 | 8.52 | |||
| 200 | 2.89 | 0.79 | |||||
| 240 | 7.14 | 1.35 | |||||
| 300 | 1.75 | 1.10 | |||||
| 360 | 5.84 | 3.53 | |||||
| 400 | 1.45 | 0.79 | 5.08 | 7.39 | |||
| CBN | 5 | 9.39 | 4.17 | 2.02 | 11.37 | ||
| 10 | 7.37 | 0.74 | |||||
| 15 | 14.57 | 6.36 | 11.71 | 1.78 | |||
| 50 | 0.40 | 14.69 | |||||
| 100 | 0.21 | 5.01 | 8.57 | 9.38 | |||
| 200 | 6.08 | 0.15 | |||||
| 240 | 8.08 | 6.95 | |||||
| 300 | 6.44 | 2.87 | |||||
| 360 | 9.52 | 9.21 | |||||
| 400 | 2.55 | 2.61 | 14.72 | 5.05 | |||
| 11-OH-THC | 5 | 4.54 | 9.68 | 3.21 | 9.83 | ||
| 10 | 5.60 | 1.41 | |||||
| 15 | 5.62 | 2.68 | 12.11 | 1.39 | |||
| 50 | 6.33 | 8.67 | |||||
| 100 | 2.86 | 13.80 | 2.24 | 0.81 | |||
| 200 | 1.91 | 0.93 | |||||
| 240 | 9.99 | 2.61 | |||||
| 300 | 1.01 | 0.23 | |||||
| 360 | 10.52 | 3.77 | |||||
| 400 | 2.06 | 1.34 | 12.92 | 1.24 | |||
| THC-COOH | 10 | 10.15 | 3.07 | 1.48 | 14.00 | ||
| 15 | 12.80 | 1.77 | 8.15 | 4.74 | |||
| 50 | 13.26 | 13.26 | |||||
| 100 | 14.41 | 14.41 | 3.55 | 0.08 | |||
| 200 | 1.09 | 1.15 | |||||
| 240 | 10.95 | 0.86 | |||||
| 300 | 1.71 | 0.37 | |||||
| 360 | 7.92 | 5.95 | |||||
| 400 | 1.45 | 0.36 | 13.53 | 0.94 | |||
| Analyte | Recovery (%) ª | ||
|---|---|---|---|
| 50 ng/mL | 100 ng/mL | 400 ng/mL | |
| CBD | 37.9 ± 3.8 | 42.6 ± 8.1 | 51.1 ± 7.6 |
| THC | 28.0 ± 2.9 | 30.6 ± 5.8 | 26.7 ± 3.8 |
| CBN | 30.7 ± 3.5 | 33.7 ± 5.7 | 57.3 ± 4.7 |
| 11-OH-THC | 47.7 ± 6.3 | 66.5 ± 7.2 | 74.7 ± 12.6 |
| THC-COOH | 63.4 ± 12.1 | 85.8 ± 11.5 | 82.6 ± 14.9 |
| Analyte | Concentration (ng/mL) | Processed Samples (n = 3) | Shor-Term Stability (n = 3) | Freeze/Thaw Stability (n = 3) | |||
|---|---|---|---|---|---|---|---|
| CV (%) | RE (%) | CV (%) | RE (%) | CV (%) | RE (%) | ||
| CBD | 15 | 1.64 | 5.01 | 1.83 | 10.62 | 1.69 | 11.82 |
| 240 | 13.36 | 13.32 | 1.85 | 0.27 | 1.00 | 14.02 | |
| 360 | 5.48 | 0.04 | 9.22 | 0.89 | 3.14 | 12.45 | |
| THC | 15 | 0.33 | 13.40 | 2.28 | 12.27 | 1.95 | 5.93 |
| 240 | 2.09 | 9.09 | 9.06 | 6.91 | 6.45 | 5.70 | |
| 360 | 3.91 | 2.29 | 6.97 | 14.36 | 1.55 | 3.46 | |
| CBN | 15 | 12.06 | 3.07 | 0.86 | 13.71 | 0.41 | 4.89 |
| 240 | 9,87 | 1.59 | 0.50 | 13.54 | 6.36 | 9.39 | |
| 360 | 7.29 | 2.26 | 0.18 | 14.52 | 3.45 | 8.74 | |
| 11-OH-THC | 15 | 5.14 | 4.29 | 0.42 | 11.98 | 5.69 | 6.15 |
| 240 | 6.65 | 5.69 | 6.03 | 7.62 | 1.81 | 13.23 | |
| 360 | 5.45 | 2.86 | 5.67 | 5.05 | 1.83 | 2.27 | |
| THC-COOH | 15 | 14.20 | 2.70 | 2.41 | 12.66 | 0.21 | 13.55 |
| 240 | 0.12 | 13.10 | 0.05 | 5.90 | 9.84 | 2.06 | |
| 360 | 1.43 | 0.96 | 1.43 | 13.69 | 6.42 | 7.92 | |
| Sample Number | Concentration (ng/mL) | ||||
|---|---|---|---|---|---|
| CBD | THC | CBN | 11-OH-THC | THC-COOH | |
| 1 | Negative | Negative | 4.84 | 0.04 | 115.61 |
| 2 | Negative | Negative | 6.58 | 0.30 | 90.32 |
| 3 | 0.29 | Negative | 5.01 | 0.53 | 63.56 |
| 4 | 8.36 | Negative | 5.11 | 0.60 | 21.89 |
| 5 | 1.86 | Negative | 4.70 | 0.43 | 29.59 |
| 6 | 1.96 | Negative | 2.03 | 5.54 | 556.18 |
| 7 | 0.90 | Negative | 1.00 | 3.58 | 6.66 |
| Analyte | Retention Time (min) | Ions (m/z) | Dwell Time (µs) |
| CBD | 390 | 100 | |
| 8.84 | 337 | 100 | |
| 301 | 100 | ||
| THC | 9.57 | 371 | 100 |
| 386 | 100 | ||
| 315 | 100 | ||
| THC-d3 ª | 9.53 | 374 | 50 |
| CBN | 10.20 | 367 | 100 |
| 382 | 100 | ||
| 310 | 100 | ||
| 11-THC-OH | 11.56 | 371 | 100 |
| 449 | 100 | ||
| 403 | 100 | ||
| 11-THC-OH-d3 ª | 11.54 | 374 | 50 |
| THC-COOH | 13.20 | 371 | 100 |
| 488 | 100 | ||
| 473 | 100 | ||
| THC-COOH-d3 ª | 13.17 | 374 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosendo, L.M.; Rosado, T.; Oliveira, P.; Simão, A.Y.; Margalho, C.; Costa, S.; Passarinha, L.A.; Barroso, M.; Gallardo, E. The Determination of Cannabinoids in Urine Samples Using Microextraction by Packed Sorbent and Gas Chromatography-Mass Spectrometry. Molecules 2022, 27, 5503. https://doi.org/10.3390/molecules27175503
Rosendo LM, Rosado T, Oliveira P, Simão AY, Margalho C, Costa S, Passarinha LA, Barroso M, Gallardo E. The Determination of Cannabinoids in Urine Samples Using Microextraction by Packed Sorbent and Gas Chromatography-Mass Spectrometry. Molecules. 2022; 27(17):5503. https://doi.org/10.3390/molecules27175503
Chicago/Turabian StyleRosendo, Luana M., Tiago Rosado, Patrik Oliveira, Ana Y. Simão, Cláudia Margalho, Suzel Costa, Luís A. Passarinha, Mário Barroso, and Eugenia Gallardo. 2022. "The Determination of Cannabinoids in Urine Samples Using Microextraction by Packed Sorbent and Gas Chromatography-Mass Spectrometry" Molecules 27, no. 17: 5503. https://doi.org/10.3390/molecules27175503