
Characterizing Powdered Activated Carbon Treatment of Surface Water Samples Using Polarity-Extended Non-Target Screening Analysis


Abstract
:1. Introduction
- Bulk characterization by a non-discriminatory feature extraction workflow followed by statistical analysis [19].
2. Results and Discussion
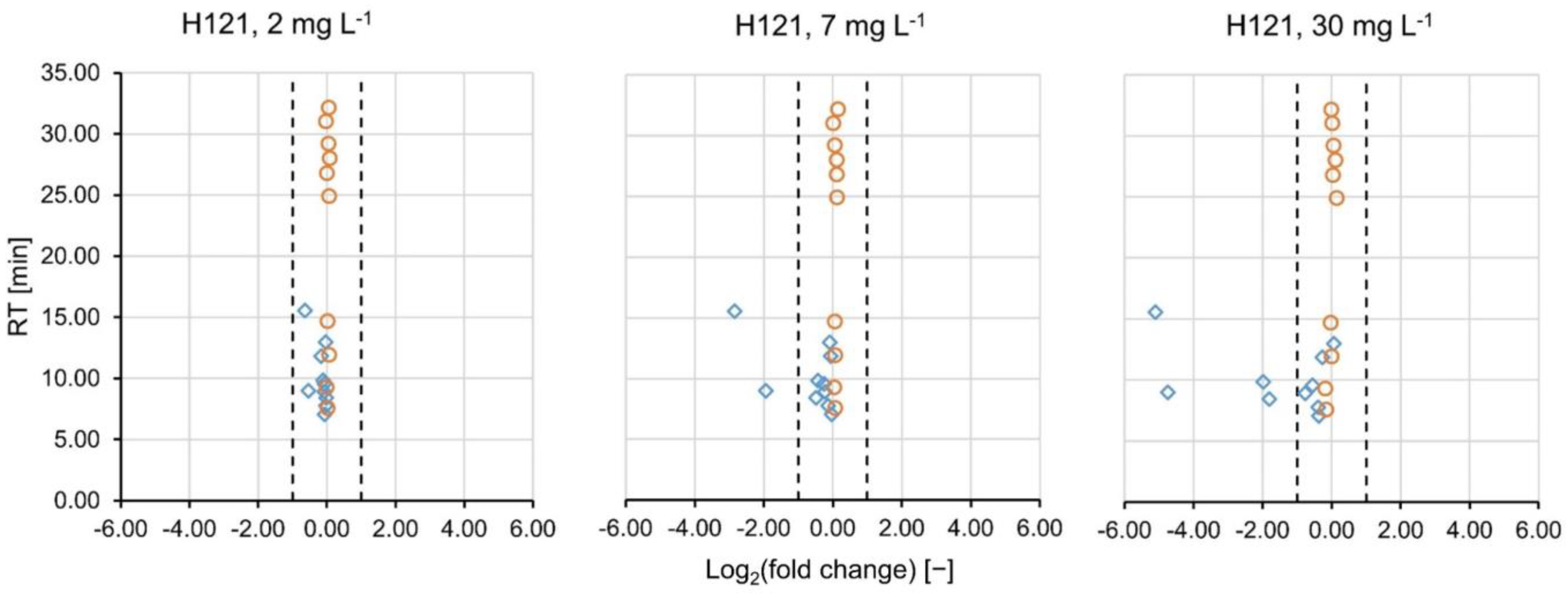
2.1. Targeted Evaluation
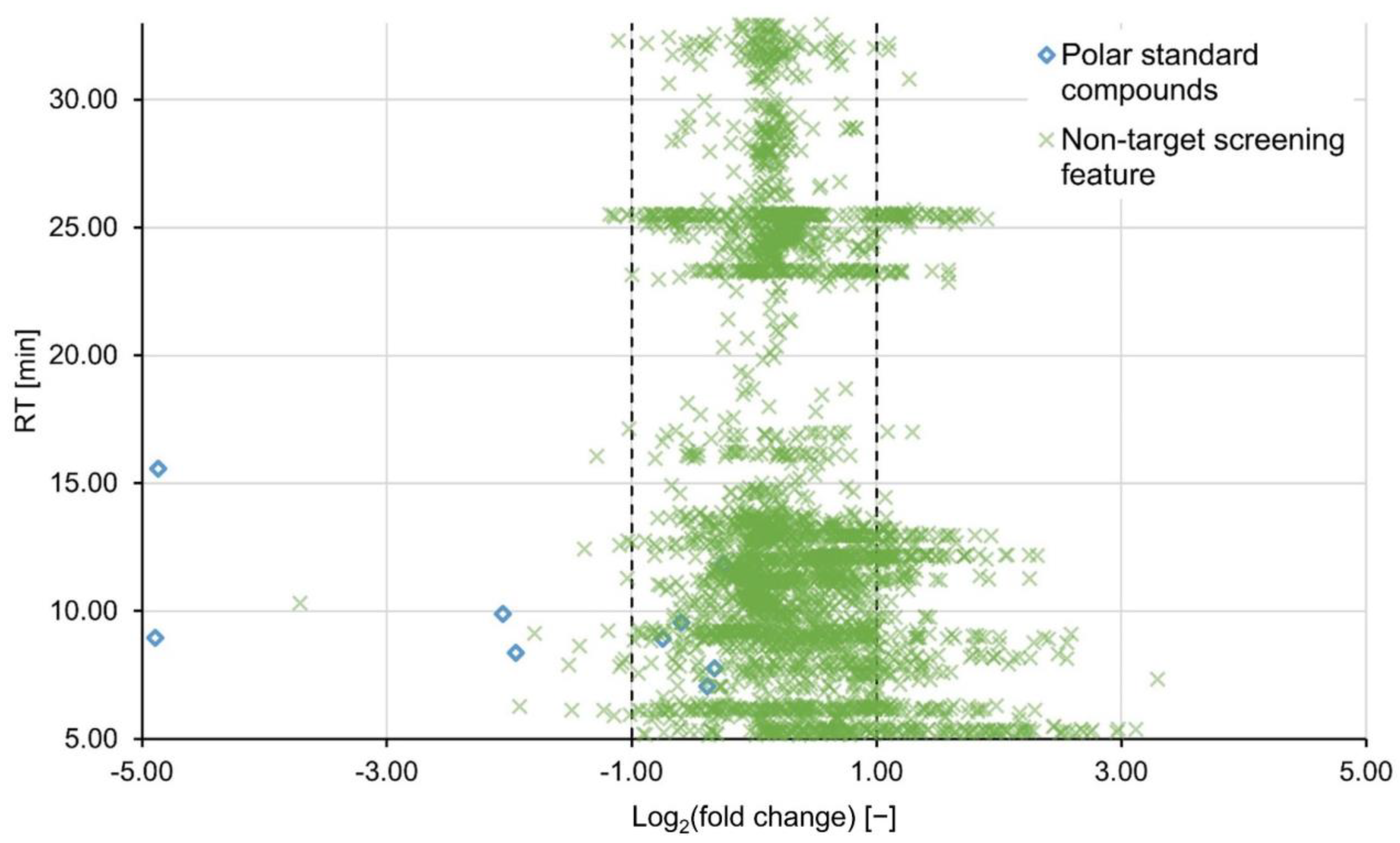
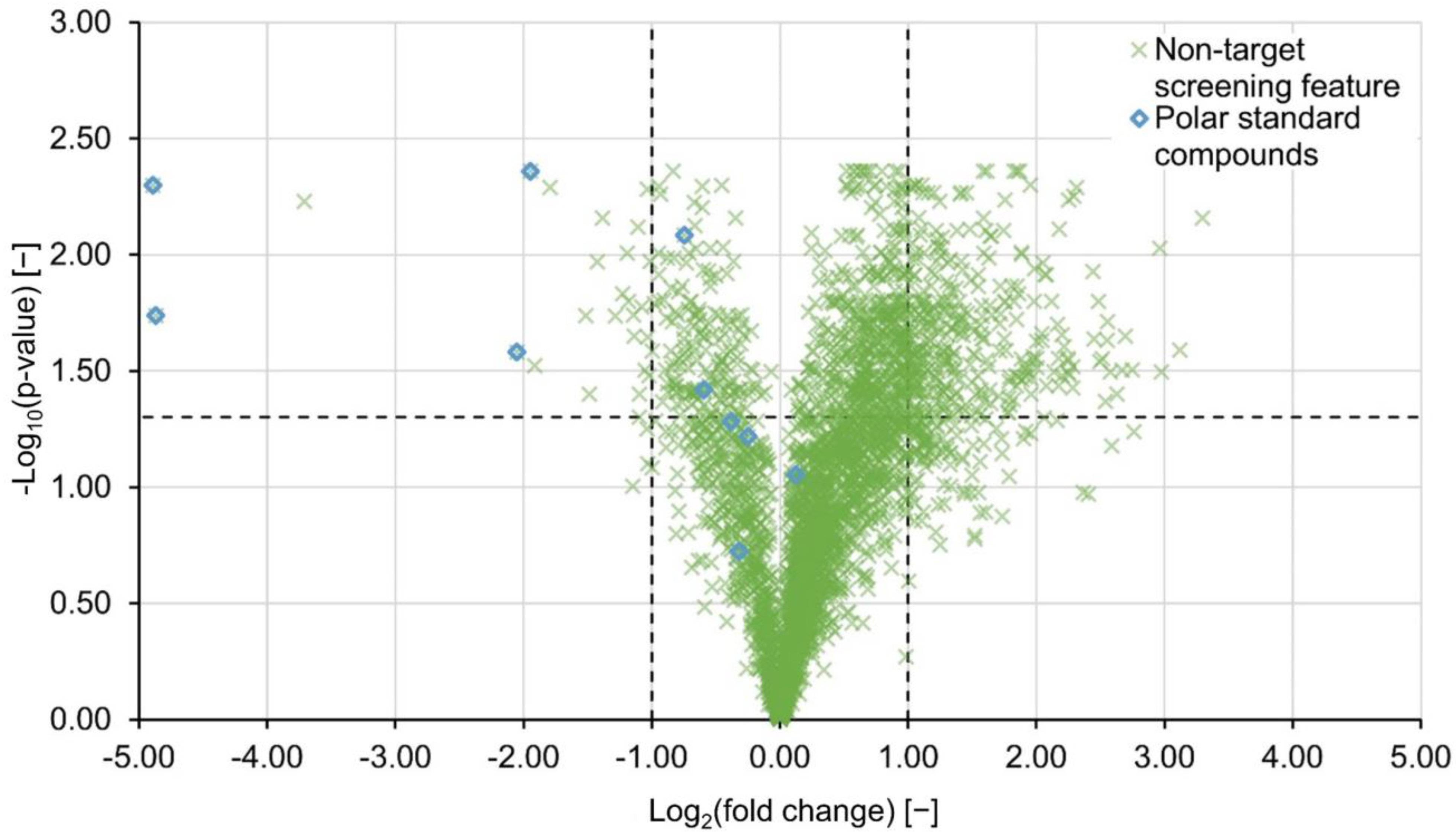
2.2. Non-Targeted Evaluation
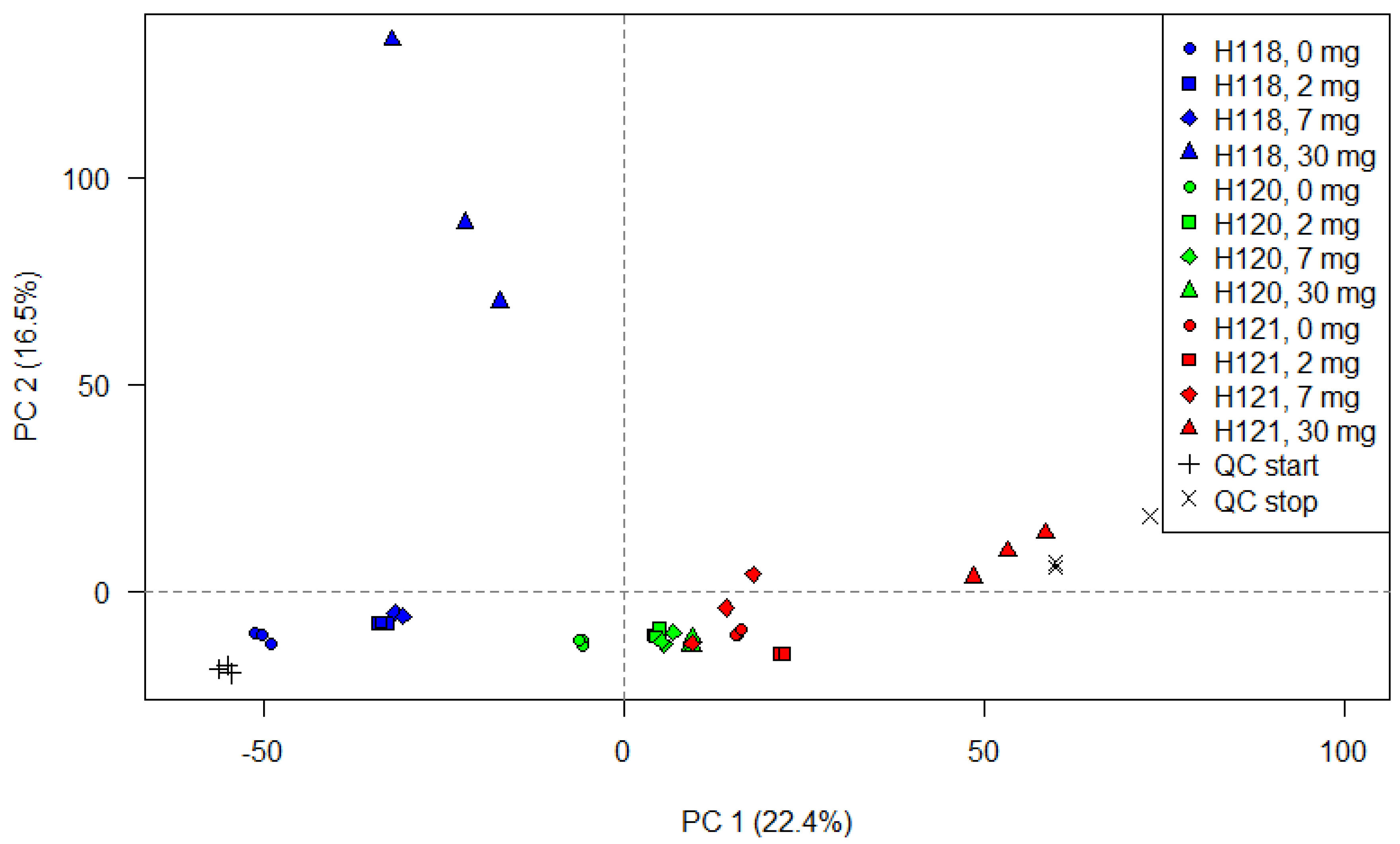
2.3. Further Statistical Analysis
3. Material and Methods
3.1. Chemicals
3.2. Samples
3.3. LC–MS Analysis
3.4. Data Analysis
3.4.1. Extracting Target Compounds
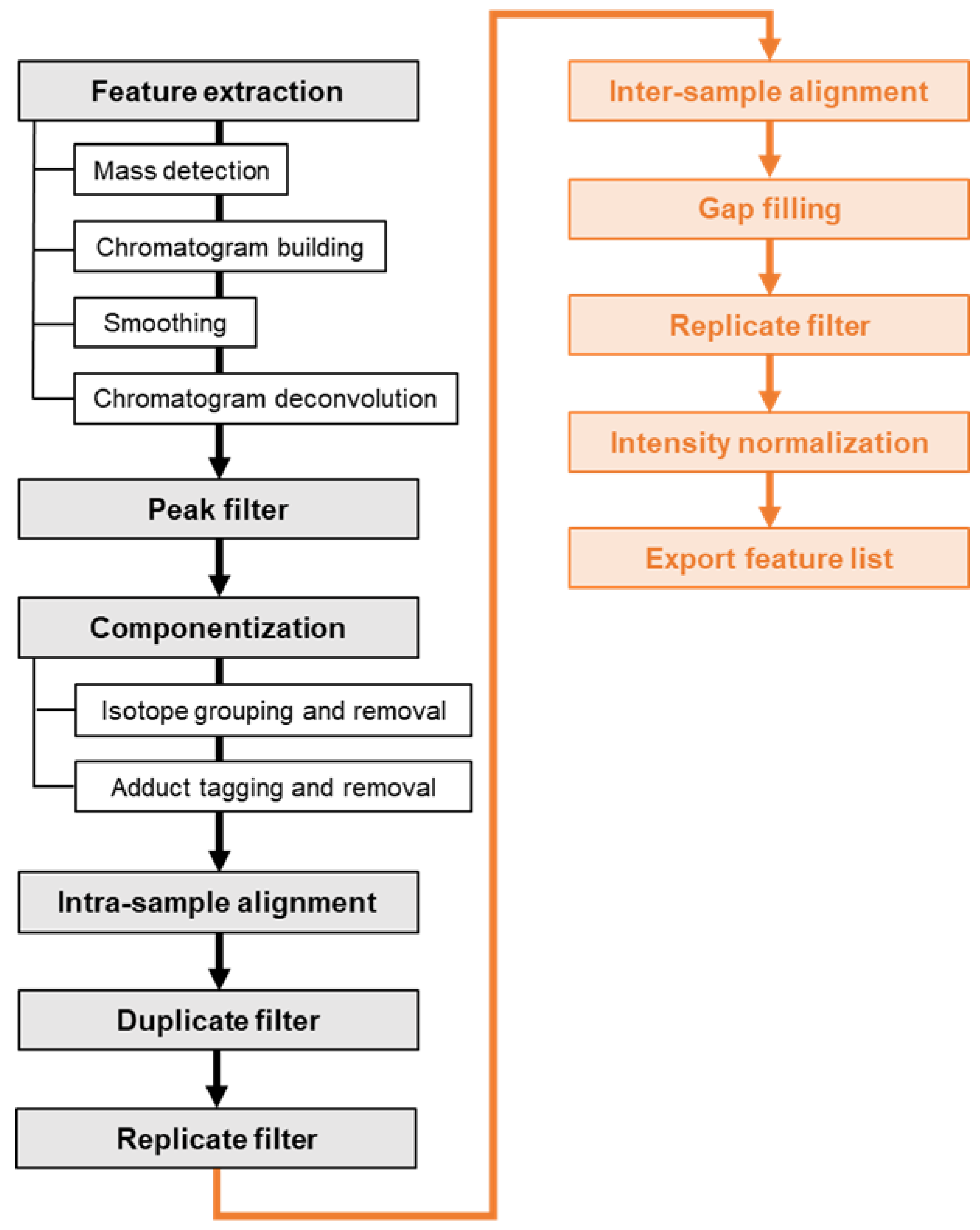
3.4.2. Extracting aligned non-target feature lists
3.4.3. Further Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Luo, Y.; Guo, W.; Ngo, H.H.; Nghiem, L.D.; Hai, F.I.; Zhang, J.; Liang, S.; Wang, X.C. A review on the occurrence of micropollutants in the aquatic environment and their fate and removal during wastewater treatment. Sci. Total. Env. 2014, 473–474, 619–641. [Google Scholar] [CrossRef] [PubMed]
- Mompelat, S.; Bot, B.L.; Thomas, O. Occurrence and fate of pharmaceutical products and by-products, from resource to drinking water. Env. Int. 2009, 35, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Karakurt, S.; Schmid, L.; Hu, U.; Drewes, J.E. Dynamics of Wastewater Effluent Contributions in Streams and Impacts on Drinking Water Supply via Riverbank Filtration in Germany-A National Reconnaissance. Env. Sci. Technol. 2019, 53, 6154–6161. [Google Scholar] [CrossRef] [PubMed]
- Eggen, R.I.L.; Hollender, J.; Joss, A.; Schärer, M.; Stamm, C. Reducing the discharge of micropollutants in the aquatic environment: The benefits of upgrading wastewater treatment plants. Env. Sci. Technol. 2014, 48, 7683–7689. [Google Scholar] [CrossRef]
- 814.201 Gewässerschutzverordnung (GSchV) of 10/28/1998, status of 01/01/2021. Der Schweizerische Bundesrat. Available online: https://www.fedlex.admin.ch/eli/cc/1998/2863_2863_2863/de. (accessed on 10 August 2022).
- Hernández-Leal, L.; Temmink, H.; Zeeman, G.; Buisman, C.J.N. Removal of micropollutants from aerobically treated grey water via ozone and activated carbon. Water Res. 2011, 45, 2887–2896. [Google Scholar] [CrossRef]
- Leal, L.H.; Vieno, N.; Temmink, H.; Zeeman, G.; Buisman, C.J.N. Occurrence of xenobiotics in gray water and removal in three biological treatment systems. Env. Sci. Technol. 2010, 44, 6835–6842. [Google Scholar] [CrossRef]
- Kovalova, L.; Siegrist, H.; Gunten, U.V.; Eugster, J.; Hagenbuch, M.; Wittmer, A.; Moser, R.; McArdell, C.S. Elimination of micropollutants during post-treatment of hospital wastewater with powdered activated carbon, ozone, and UV. Env. Sci. Technol. 2013, 47, 7899–7908. [Google Scholar] [CrossRef]
- Kovalova, L.; Knappe, D.R.U.; Lehnberg, K.; Kazner, C.; Hollender, J. Removal of highly polar micropollutants from wastewater by powdered activated carbon. Environ. Sci. Pollut. Res. 2013, 20, 3607–3615. [Google Scholar] [CrossRef]
- Boxall, A.; Sinclair, C.; Fenner, K.; Koplin, D.; Maund, S. When synthetic chemicals degrade in the environment. Env. Sci. Technol. 2004, 38, 368–375. [Google Scholar] [CrossRef]
- Schmitt-Jansen, M.; Bartels, P.; Adler, N.; Altenburger, R. Phytotoxicity assessment of diclofenac and its phototransformation products. Anal. Bioanal. Chem. 2007, 387, 1389–1396. [Google Scholar] [CrossRef]
- Petrie, B.; Barden, R.; Kasprzyk-Hordern, B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015, 72, 3–27. [Google Scholar] [CrossRef]
- Gómez, M.J.; Sirtori, C.; Mezcua, M.; Fernández-Alba, A.R.; Agüera, A. Photodegradation study of three dipyrone metabolites in various water systems: Identification and toxicity of their photodegradation products. Water Res. 2008, 42, 2698–2706. [Google Scholar] [CrossRef]
- Krauss, M.; Singer, H.; Hollender, J. LC-high resolution MS in environmental analysis: From target screening to the identification of unknowns. Anal. Bioanal. Chem. 2010, 397, 943–951. [Google Scholar] [CrossRef]
- Schulz, W.; Lucke, T. Non-Target Screening in Water Analysis—Guideline for the Application of LC-ESI-HRMS for Screening; German Water Chemistry Society: Mülheim an der Ruhr, Germany, 2019; Available online: https://www.wasserchemische-gesellschaft.de/images/HAIII/NTS-Guidline_EN_s.pdf (accessed on 10 August 2022).
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go? Env. Sci. Technol. 2017, 51, 11505–11512. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.P.; Longrée, P.; Loos, M.; Ruff, M.; Stravs, M.A.; Vidal, C.R.; Hollender, J. Strategies to characterize polar organic contamination in wastewater: Exploring the capability of high resolution mass spectrometry. Env. Sci. Technol. 2014, 48, 1811–1818. [Google Scholar] [CrossRef]
- Minkus, S.; Bieber, S.; Letzel, T. (Very) polar organic compounds in the Danube river basin: Non-target screening workflow and prioritization strategy for extracting highly confident features. Anal. Methods. 2021, 13, 2044–2054. [Google Scholar] [CrossRef]
- Schollée, J.E.; Schymanski, E.L.; Hollender, J. Statistical Approaches for LC-HRMS Data to Characterize, Prioritize, and Identify Transformation Products from Water Treatment Processes. ACS Symp. Ser. 2016, 4, 45–65. [Google Scholar]
- Brunner, A.M.; Bertelkamp, C.; Dingemansm, M.M.L.; Kolkman, A.; Wols, B.; Harmsen, D.; Siegers, W.; Martijn, B.J.; Oorthuizen, W.A.; ter Laak, T.L. Integration of target analyses, non-target screening and effect-based monitoring to assess OMP related water quality changes in drinking water treatment. Sci. Total. Env. 2020, 705, 135779. [Google Scholar] [CrossRef]
- Schollée, J.E.; Hollender, J.; McArdell, C.S. Characterization of advanced wastewater treatment with ozone and activated carbon using LC-HRMS based non-target screening with automated trend assignment. Water Res. 2021, 200, 117209. [Google Scholar] [CrossRef]
- Bader, T.; Schulz, W.; Kümmerer, K.; Winzenbacher, R. LC-HRMS Data Processing Strategy for Reliable Sample Comparison Exemplified by the Assessment of Water Treatment Processes. Anal. Chem. 2017, 89, 13219–13226. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef]
- Fischler, M.A.; Bolles, R.C. Random Sample Consensus: A Paradigm for Model Fitting with Applications to Image Analysis and Automated Cartography. Commun. ACM 1981, 24, 381–395. [Google Scholar] [CrossRef]
- Guida, R.D.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 1–14. [Google Scholar] [CrossRef]
- Reineke, A.; Liesener, A.; Betz, L. Anwendung von Nontarget-Screening und Wirkungsbezogener Analytik in der Charakterisierung von Aktivkohle zur Aufbereitung von Trinkwasser. Vom Wasser. 2021, 119, 78–81. [Google Scholar] [CrossRef]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef]
- Chen, S.Y.; Feng, Z.; Yi, X. A general introduction to adjustment for multiple comparisons. J. Thorac. Dis. 2017, 9, 1725–1729. [Google Scholar] [CrossRef]
- Bender, R.; Lange, S. Adjusting for multiple testing—when and how? Ralf. J Clin. Epidemiol. 2001, 54, 343–349. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Greco, G.; Grosse, S.; Letzel, T. Serial coupling of reversed-phase and zwitterionic hydrophilic interaction LC/MS for the analysis of polar and nonpolar phenols in wine. J. Sep. Sci. 2013, 36, 1379–1388. [Google Scholar] [CrossRef]
- Bieber, S.; Greco, G.; Grosse, S.; Letzel, T. RPLC-HILIC and SFC with Mass Spectrometry: Polarity-Extended Organic Molecule Screening in Environmental (Water) Samples. Anal. Chem. 2017, 89, 7907–7914. [Google Scholar] [CrossRef] [PubMed]
- Minkus, S.; Grosse, S.; Bieber, S.; Veloutsou, S.; Letzel, T. Optimized hidden target screening for very polar molecules in surface waters including a compound database inquiry. Anal. Bioanal. Chem. 2020, 412, 4953–4966. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- European MassBank Mass Spectral Database of the NORMAN Network. Available online: https://massbank.eu/MassBank/ (accessed on 2 February 2022).
- MassBank of North America. Available online: https://mona.fiehnlab.ucdavis.edu/ (accessed on 2 February 2022).
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal. Chem. 2017, 89, 8696–8703. [Google Scholar] [CrossRef] [PubMed]
- Katajamaa, M.; Orešič, M. Processing methods for differential analysis of LC/MS profile data. BMC Bioinform. 2005, 6, 1–12. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; p. 201. [Google Scholar]
- Team, R.S. RStudio: Integrated Development for R. RStudio, PBC. Boston MA 2020, 770, 165–171. [Google Scholar]
- Welch, B.L. The Generalization of “Student’s” Problem when Several Different Population Variances are Involved. Biometrika 1947, 34, 28–35. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration [mg L−1] | H118 | H120 | H121 |
|---|---|---|---|
| log2(fc) | log2(fc) | log2(fc) | |
| Internal standards | n = 10 | n = 10 | n = 10 |
| 2 | 0.09 ± 0.05 | −0.20 ± 0.07 | 0.03 ± 0.04 |
| 7 | −0.04 ± 0.09 | −0.25 ± 0.14 | 0.08 ± 0.04 |
| 30 | −0.03 ± 0.10 | −0.21 ± 0.16 | −0.01 ± 0.10 |
| Polar standard compounds | n = 10 | n = 10 | n = 10 |
| 2 | −0.40 ± 0.49 | −0.21 ± 0.49 | −0.17 ± 0.22 |
| 7 | −1.22 ± 1.46 | −0.70 ± 1.30 | −0.65 ± 0.95 |
| 30 | −2.31 ± 2.21 | −2.07 ± 2.45 | −1.59 ± 1.88 |
| Concentration [mg L−1] | Number of Features | Mean log2(fc) | Increasing/Decreasing Features [%] | Significant Features |
|---|---|---|---|---|
| H118 | ||||
| 2 | 2981 | −0.16 ± 0.38 | 0.6/2.7 | 38 |
| 7 | 3366 | −0.26 ± 0.43 | 0.4/5.1 | 0 |
| 30 | 3000 | 0.07 ± 0.57 | 4.5/2.7 | 13 |
| H120 | ||||
| 2 | 2941 | 0.11 ± 0.39 | 2.1/0.8 | 40 |
| 7 | 2856 | 0.11 ± 0.39 | 1.7/1.2 | 29 |
| 30 | 3058 | 0.17 ± 0.42 | 3.0/0.7 | 95 |
| H121 | ||||
| 2 | 2842 | −0.04 ± 0.37 | 1.1/1.8 | 28 |
| 7 | 2886 | 0.17 ± 0.41 | 3.5/0.7 | 2 |
| 30 | 3099 | 0.36 ± 0.62 | 13.4/0.8 | 336 |
| Laboratory Name | H118 | H120 | H121 |
|---|---|---|---|
| Manufacturer | Supplier A | Supplier A | Supplier B |
| Water content | 8.1 % | 1.5 % | 2.0 % |
| Ash content | 6.7 % | 13.6 % | 10.2 % |
| Contact pH | 10.8 | 9.9 | 10.1 |
| Iodine number | 1088 mg g−1 | 1019 mg g−1 | 944 mg g−1 |
| Particle size distribution (wet sieving) | |||
| <150 µm | 99.1 | 99.1 | 99.7 |
| <50 µm | 72.0 | 88.6 | 70.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minkus, S.; Bieber, S.; Letzel, T. Characterizing Powdered Activated Carbon Treatment of Surface Water Samples Using Polarity-Extended Non-Target Screening Analysis. Molecules 2022, 27, 5214. https://doi.org/10.3390/molecules27165214
Minkus S, Bieber S, Letzel T. Characterizing Powdered Activated Carbon Treatment of Surface Water Samples Using Polarity-Extended Non-Target Screening Analysis. Molecules. 2022; 27(16):5214. https://doi.org/10.3390/molecules27165214
Chicago/Turabian StyleMinkus, Susanne, Stefan Bieber, and Thomas Letzel. 2022. "Characterizing Powdered Activated Carbon Treatment of Surface Water Samples Using Polarity-Extended Non-Target Screening Analysis" Molecules 27, no. 16: 5214. https://doi.org/10.3390/molecules27165214