
PI3K/mTOR Dual Inhibitor Pictilisib Stably Binds to Site I of Human Serum Albumin as Observed by Computer Simulation, Multispectroscopic, and Microscopic Studies


Abstract
:1. Introduction
2. Results and Discussion
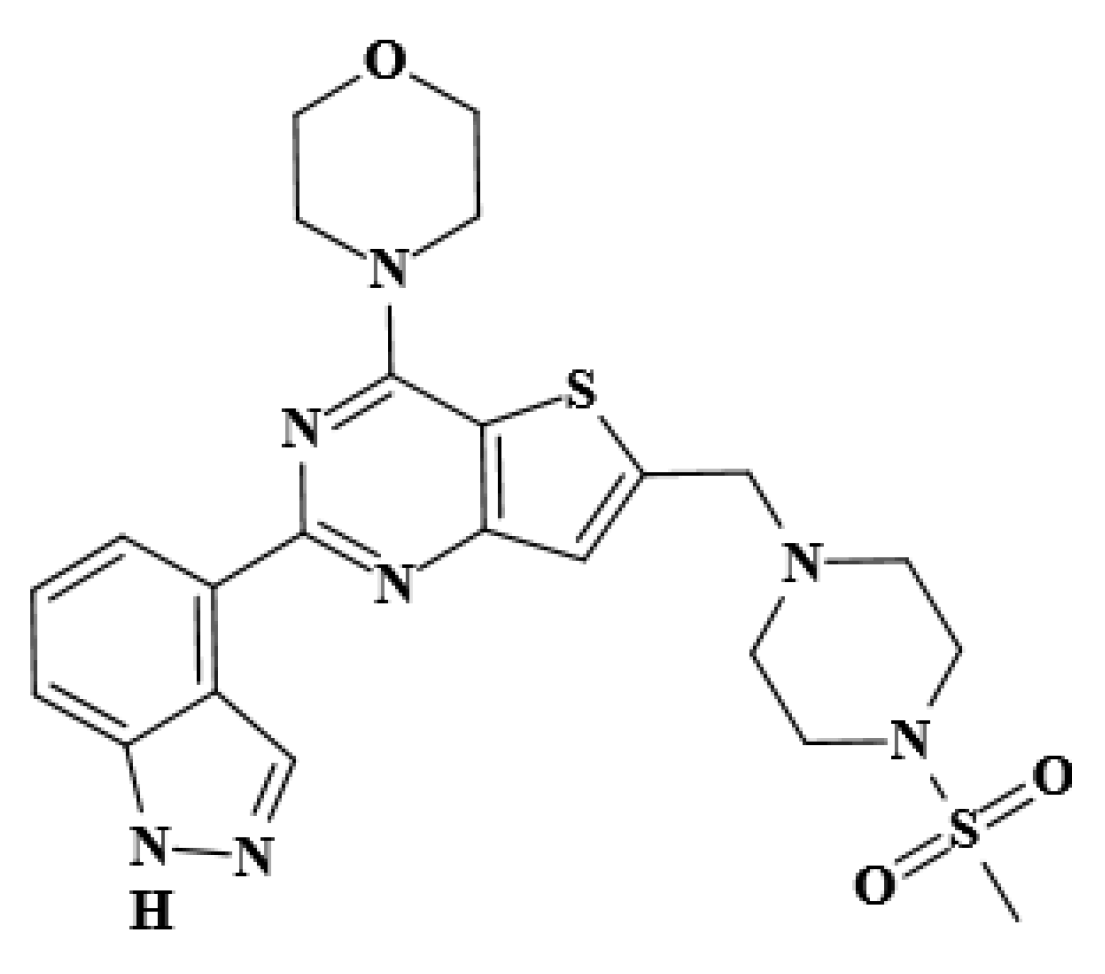
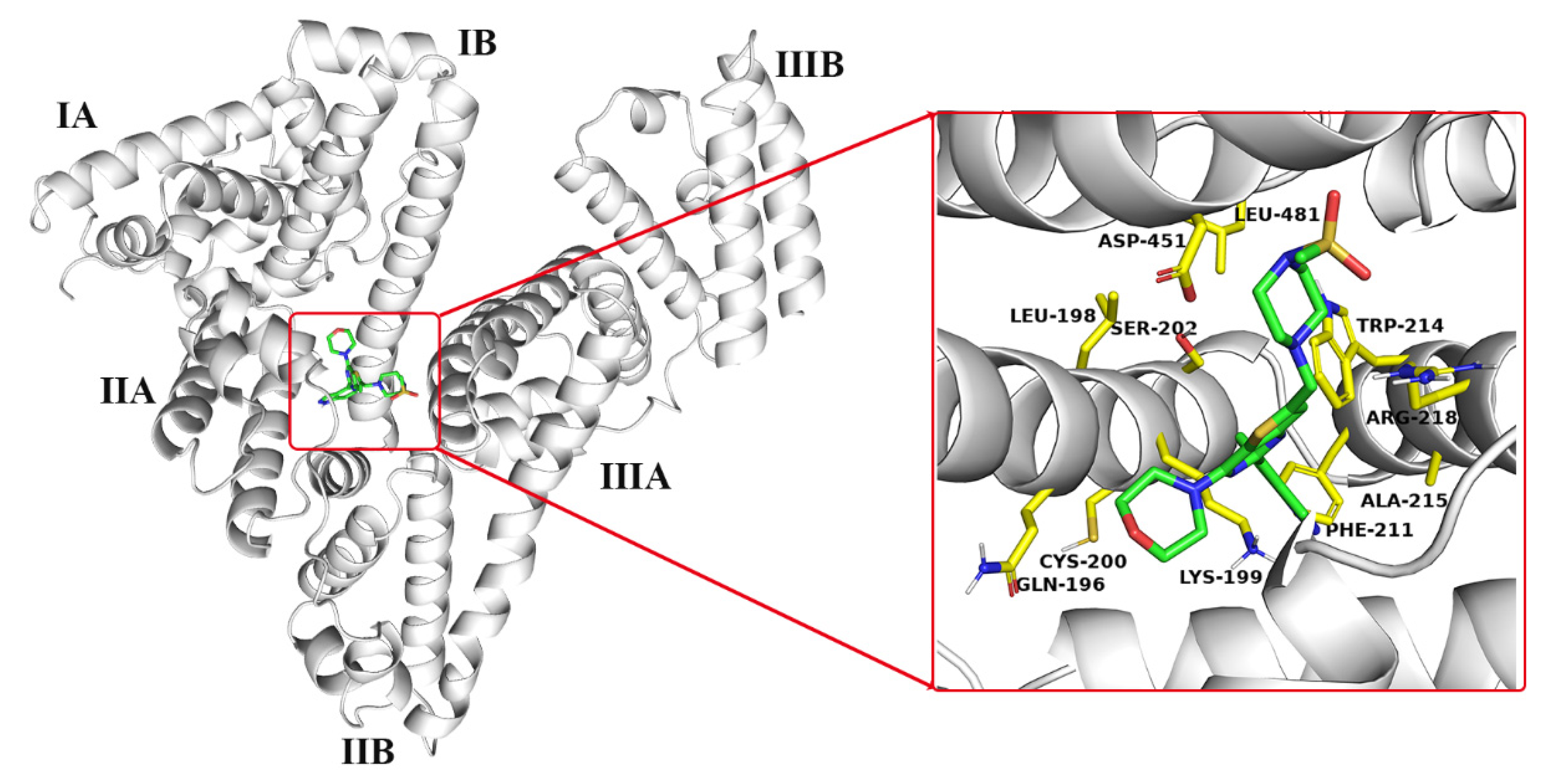
2.1. Molecular Docking Analysis
2.2. MD Trajectory Analysis
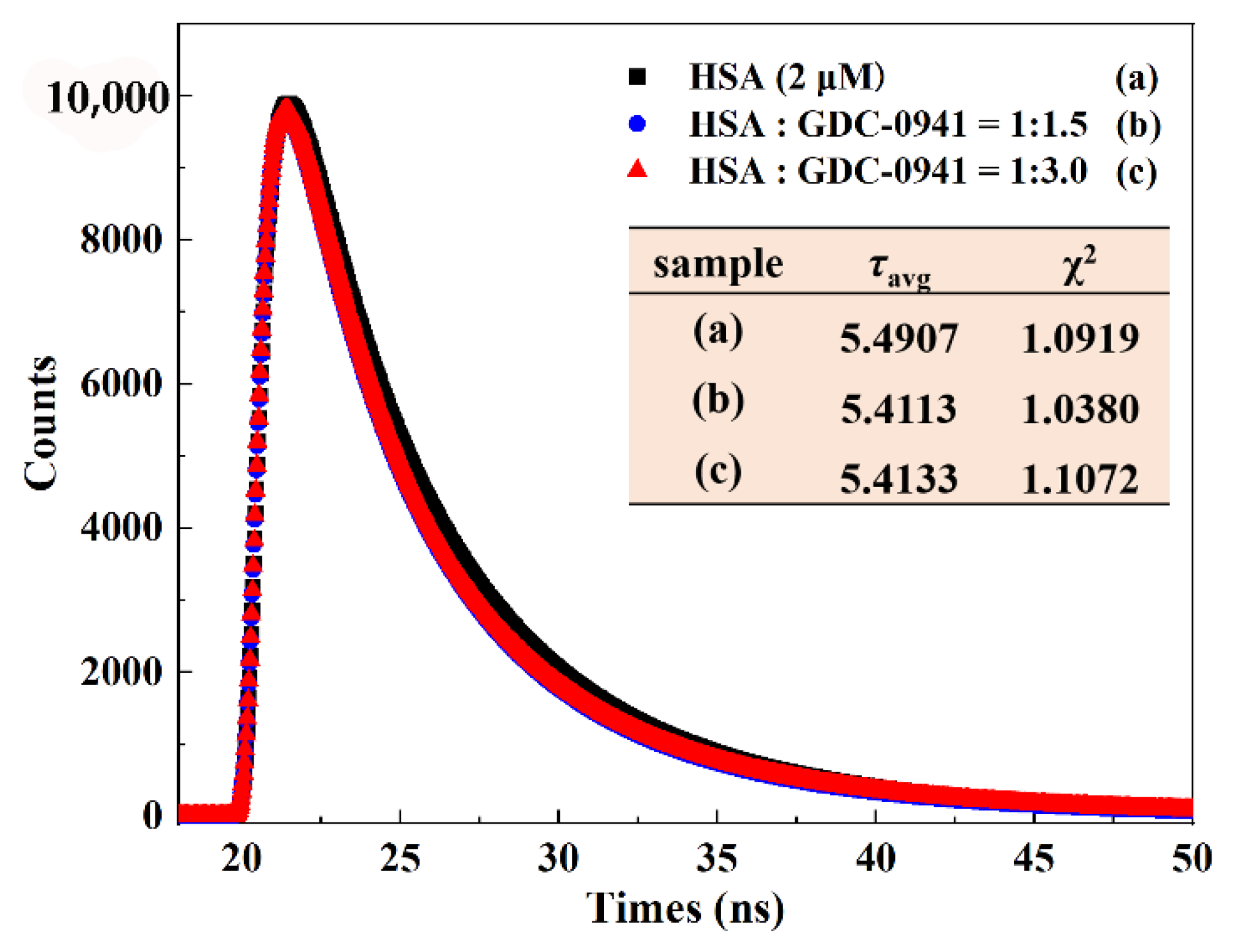
2.3. Quenching Mechanism Studies
2.4. Binding Strength and Binding Force Studies
2.5. Site Marker Displacement Studies
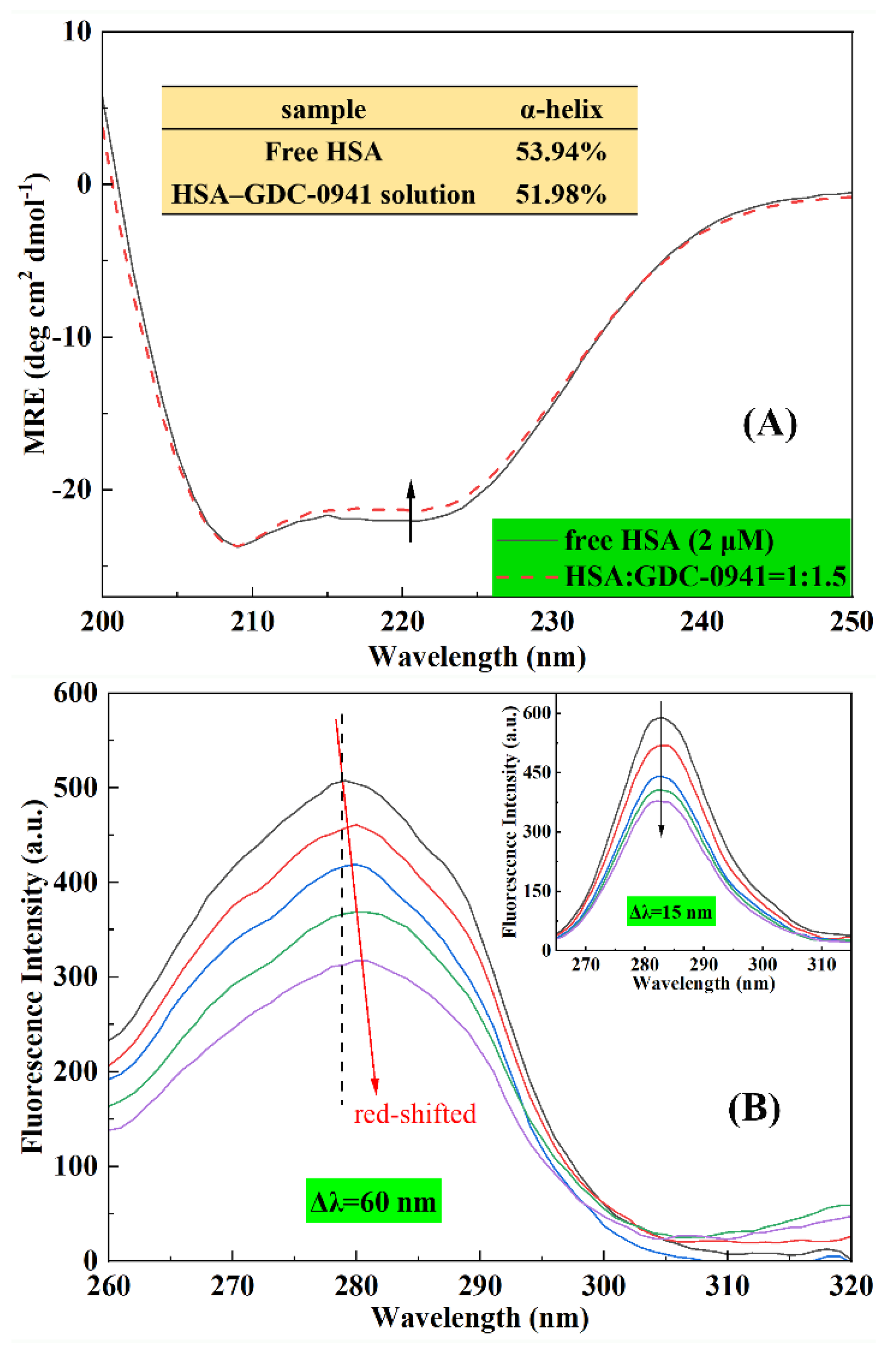
2.6. Effect of GDC-0941 on the Conformation HSA
2.6.1. CD Spectroscopy
2.6.2. Synchronous Fluorescence Spectroscopy
2.6.3. 3D Fluorescence Spectroscopy
2.7. Morphological Analysis of HSA
3. Materials and Methods
3.1. Reagents
3.2. Instruments and Operations
3.2.1. Molecular Docking
3.2.2. MD Simulation
3.2.3. Fluorescence Measurements
3.2.4. Time-Resolved Fluorescence Spectra
3.2.5. CD Spectroscopy
3.2.6. AFM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Yerena, A.; Perez, M.; Vallverdú-Queralt, A.; Escribano-Ferrer, E. Insights into the Binding of Dietary Phenolic Compounds to Human Serum Albumin and Food-Drug Interactions. Pharmaceutics 2020, 12, 1123. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Thakur, Y.; Tripathi, M.; Siddiqi, M.K.; Khan, R.H.; Pande, R. Elucidating the binding propensity of naphthyl hydroxamic acid to human serum albumin (HSA): Multi-spectroscopic and molecular modeling approach. J. Mol. Struct. 2019, 1184, 1–11. [Google Scholar] [CrossRef]
- Ribeiro, A.G.; Alves, J.E.F.; Soares, J.C.S.; dos Santos, K.L.; Jacob, Í.T.T.; da Silva Ferreira, C.J.; dos Santos, J.C.; de Azevedo, R.D.S.; de Almeida, S.M.V.; de Lima, M.d.C.A. Albumin roles in developing anticancer compounds. Med. Chem. Res. 2021, 30, 1469–1495. [Google Scholar] [CrossRef]
- Amoorahim, M.; Ashrafi-Kooshk, M.R.; Esmaeili, S.; Shahlaei, M.; Moradi, S.; Khodarahmi, R. Physiological changes in the albumin-bound non-esterified free fatty acids critically influence heme/bilirubin binding properties of the protein: A comparative, in vitro, spectroscopic study using the endogenous biomolecules. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 235, 118298. [Google Scholar] [CrossRef] [PubMed]
- Gan, N.; Sun, Q.; Suo, Z.; Zhang, S.; Zhao, L.; Xiang, H.; Wang, W.; Li, Z.; Liao, X.; Li, H. How hydrophilic group affects drug–protein binding modes: Differences in interaction between sirtuins inhibitors Tenovin-1/Tenovin-6 and human serum albumin. J. Pharm. Biomed. Anal. 2021, 201, 114121. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, L.D.; Sun, Q.M.; Gan, N.; Zhang, Q.; Yang, J.; Yi, B.; Liao, X.X.; Zhu, D.; Li, H. Study on the interaction between 2,6-dihydroxybenzoic acid nicotine salt and human serum albumin by multi-spectroscopy and molecular dynamics simulation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 270, 120868. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Experimental and Computational Investigation on the Interaction of Anticancer Drug Gemcitabine with Human Plasma Protein: Effect of Copresence of Ibuprofen on the Binding. Molecules 2022, 27, 1635. [Google Scholar] [CrossRef]
- Kasparek, A.; Smyk, B. Spectroscopic demonstration of sinapic acid methyl ester complexes with serum albumins. RSC Adv. 2020, 10, 8810–8820. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, Y.; Li, Y.; Zhang, X. Interactions between Human Serum Albumin and Sulfadimethoxine Determined Using Spectroscopy and Molecular Docking. Molecules 2022, 27, 1526. [Google Scholar] [CrossRef]
- Xiong, X.; Gan, R.; Suo, Z.; Tang, P.; Zhang, S.; Zhu, Y.; Sun, Q.; Hui, L. Interactions between the antiviral drug telaprevir and human serum albumin: A combined study with spectroscopic methods and molecular modeling. New J. Chem. 2018, 42, 9791–9800. [Google Scholar] [CrossRef]
- Wynendaele, E.; Ma, G.J.; Xu, X.; Cho, N.-J.; De Spiegeleer, B. Conformational stability as a quality attribute for the cell therapy raw material human serum albumin. RSC Adv. 2021, 11, 15332–15339. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.; Wade, D. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharm. 1975, 11, 824–832. [Google Scholar]
- Sudlow, G.; Birkett, D.; Wade, D. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharm. 1976, 12, 1052–1061. [Google Scholar]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.; Garrett, J. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 93. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers 2021, 13, 3517. [Google Scholar] [CrossRef]
- Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend, C.M., Jr.; Anthony, L.; Wasilchenko, C.; Melton, M.L.; et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [Google Scholar] [CrossRef]
- Zou, Z.-Q.; Zhang, L.-N.; Wang, F.; Bellenger, J.; Shen, Y.-Z.; Zhang, X.-H. The novel dual PI3K/mTOR inhibitor GDC-0941 synergizes with the MEK inhibitor U0126 in non-small cell lung cancer cells. Mol. Med. Rep. 2012, 5, 503–508. [Google Scholar]
- Shi, F.; Zhang, J.Y.; Liu, H.Y.; Wu, L.L.; Jiang, H.Y.; Wu, Q.Y.; Liu, T.Y.; Lou, M.Q.; Wu, H. The dual PI3K/mTOR inhibitor dactolisib elicits anti-tumor activity in vitro and in vivo. Oncotarget 2018, 9, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Walana, W.; Zhao, F.; Li, F.; Luo, F. GDC-0941 and CXCL8 (3-72) K11R/G31P combination therapy confers enhanced efficacy against breast cancer. Future Oncol. 2020, 16, 911–921. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, S.J.; Ryu, J.H.; Kim, S.H.; Kwon, I.C.; Roberts, T.M. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J. Control. Release 2020, 318, 98–108. [Google Scholar] [CrossRef]
- Patel, R.P.; Thomas, J.R.; Curt, K.M.; Fitzsimmons, C.M.; Batista, P.J.; Bates, S.E.; Gottesman, M.M.; Robey, R.W. Dual Inhibition of Histone Deacetylases and the Mechanistic Target of Rapamycin Promotes Apoptosis in Cell Line Models of Uveal Melanoma. Invest. Ophth. Vis. Sci. 2021, 62, 16. [Google Scholar] [CrossRef] [PubMed]
- Munugalavadla, V.; Mariathasan, S.; Slaga, D.; Du, C.; Berry, L.; Rosario, G.D.; Yan, Y.; Boe, M.; Sun, L.; Friedman, L.S.J.O. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene 2014, 33, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan–Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sio, T.T.; Oberoi, R.K.; Grams, M.P.; Furutani, K.M.; Gupta, S.K.; Wilson, Z.C.; Pokorny, J.L.; Bakken, K.K.; Schroeder, M.A.; Carlson, B.L.; et al. The Impact of Hemi-brain Irradiation on Accumulation of PI3K/mTOR Inhibitors with Limited (GDC-0980) and Robust (GNE-317) Blood-Brain Barrier Penetration. Int. J. Radiat. Oncol. 2014, 90, S197. [Google Scholar] [CrossRef]
- Salphati, L.; Shahidi-Latham, S.; Quiason, C.; Barck, K.; Nishimura, M.; Alicke, B.; Pang, J.; Carano, R.A.; Olivero, A.G.; Phillips, H.S.J.D.M. Disposition, Distribution of the Phosphatidylinositol 3-Kinase Inhibitors Pictilisib (GDC-0941) and GNE-317 in U87 and GS2 Intracranial Glioblastoma Models--Assessment by Matrix-Assisted Laser Desorption Ionization Imaging. Drug Metab. Dispos. 2014, 42, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J.F.; et al. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salphati, L.; Alicke, B.; Heffron, T.P.; Shahidi-Latham, S.; Nishimura, M.; Cao, T.; Carano, R.A.; Cheong, J.; Greve, J.; Koeppen, H.; et al. Brain Distribution and Efficacy of the Brain Penetrant PI3K Inhibitor GDC-0084 in Orthotopic Mouse Models of Human Glioblastoma. Drug Metab. Dispos. 2016, 44, 1881–1889. [Google Scholar] [CrossRef] [Green Version]
- Salphati, L.; Pang, J.; Plise, E.G.; Lee, L.B.; Olivero, A.G.; Prior, W.W.; Sampath, D.; Wong, S.; Zhang, X. Preclinical Assessment of the Absorption and Disposition of the Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor GDC-0980 and Prediction of Its Pharmacokinetics and Efficacy in Human. Drug Metab. Dispos. 2012, 40, 1785. [Google Scholar] [CrossRef] [Green Version]
- Salphati, L.; Heffron, T.P.; Alicke, B.; Nishimura, M.; Barck, K.; Carano, R.A.; Cheong, J.; Edgar, K.A.; Greve, J.; Kharbanda, S.; et al. Targeting the PI3K pathway in the brain--efficacy of a PI3K inhibitor optimized to cross the blood-brain barrier. Clin. Cancer Res. 2012, 18, 6239–6248. [Google Scholar] [CrossRef] [Green Version]
- Altine, B.; Gai, Y.; Han, N.; Jiang, Y.; Ji, H.; Fang, H.; Niyonkuru, A.; Bakari, K.H.; Rajab Arnous, M.M.; Liu, Q.; et al. Preclinical Evaluation of a Fluorine-18 ((18)F)-Labeled Phosphatidylinositol 3-Kinase Inhibitor for Breast Cancer Imaging. Mol. Pharm. 2019, 16, 4563–4571. [Google Scholar] [CrossRef]
- Yang, H.Q.; Zeng, Q.L.; He, Z.; Wu, D.; Li, H. Interaction of novel Aurora kinase inhibitor MK-0457 with human serum albumin: Insights into the dynamic behavior, binding mechanism, conformation and esterase activity of human serum albumin. J. Pharm. Biomed. Anal. 2020, 178, 112962. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yi, Z.; Lu, D.; Zhang, A.J.E.T. Pharmacology, Molecular simulation study of the specific combination between four kinds of phthalic acid esters and human serum albumin. Environ. Toxicol. Pharm. 2016, 41, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Tang, P.; He, J.; Yang, H.; Li, H. Characterization of the binding of a novel antitumor drug ibrutinib with human serum albumin: Insights from spectroscopic, calorimetric and docking studies. J. Photoch. Photobio. B 2018, 184, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.Z.; Tee, W.V.; Mohamad, S.B.; Alias, Z.; Tayyab, S.J.R.A. Interaction of an anticancer drug, gefitinib with human serum albumin: Insights from fluorescence spectroscopy and computational modeling analysis. RSC Adv. 2016, 6, 91756–91767. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Sun, Q.; Gan, N.; Zhang, S.; Zhao, L.; Tang, P.; Pu, H.; Zhai, Y.; Gan, R.; Li, H. Insights into protein recognition for γ-lactone essences and the effect of side chains on interaction via microscopic, spectroscopic, and simulative technologies. Food Chem. 2019, 278, 127–135. [Google Scholar] [CrossRef]
- Khan, S.; Zafar, A.; Naseem, I. Probing the interaction of a coumarin-di(2-picolyl)amine hybrid drug-like molecular entity with human serum albumin: Multiple spectroscopic and molecular modeling techniques. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 223, 117330. [Google Scholar] [CrossRef]
- Maurya, N.; Maurya, J.K.; Singh, U.K.; Dohare, R.; Zafaryab, M.; Moshahid Alam Rizvi, M.; Kumari, M.; Patel, R. In Vitro Cytotoxicity and Interaction of Noscapine with Human Serum Albumin: Effect on Structure and Esterase Activity of HSA. Mol. Pharm. 2019, 16, 952–966. [Google Scholar] [CrossRef]
- Chaves, O.A.; Menezes, L.B.; Iglesias, B.A. Multiple spectroscopic and theoretical investigation of meso-tetra-(4-pyridyl)porphyrin-ruthenium(II) complexes in HSA-binding studies. Effect of Zn(II) in protein binding. J. Mol. Liq. 2019, 294, 111581. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, J.; Wang, Y.; Sun, H. Investigating the Effect of Tyrosine Kinase Inhibitors on the Interaction between Human Serum Albumin by Atomic Force Microscopy. Biomolecules 2022, 12, 819. [Google Scholar] [CrossRef]
- Rimac, H.; Tandarić, T.; Vianello, R.; Bojić, M. Indomethacin Increases Quercetin Affinity for Human Serum Albumin: A Combined Experimental and Computational Study and Its Broader Implications. Int. J. Mol. Sci. 2020, 21, 5740. [Google Scholar] [CrossRef] [PubMed]
- Wichapong, K.; Rohe, A.; Platzer, C.; Slynko, I.; Erdmann, F.; Schmidt, M.; Sippl, W. Application of Docking and QM/MM-GBSA Rescoring to Screen for Novel Myt1 Kinase Inhibitors. J. Chem. Inf. Model. 2014, 54, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Samsonov, S.A.; Pisabarro, M.T. Computational analysis of interactions in structurally available protein–glycosaminoglycan complexes. Glycobiology 2016, 26, 850–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSA–GDC-0941 Complex | |||||
|---|---|---|---|---|---|
| Residue Name | Residue # | Contribution to ∆Gbind | Residue Name | Residue # | Contribution to ∆Gbind |
| SER | 192 | –0.37 | LEU | 219 | –1.68 |
| LYS | 195 | –1.94 | GLN | 221 | –0.21 |
| GLN | 196 | –1.76 | ARG | 222 | –1.41 |
| LEU | 198 | –0.49 | VAL | 235 | –0.30 |
| LYS | 199 | –1.09 | LEU | 238 | –1.39 |
| PHE | 206 | –0.95 | THR | 239 | –0.38 |
| TRP | 214 | –1.20 | VAL | 241 | –0.29 |
| ALA | 215 | –0.73 | HIS | 242 | –1.06 |
| ALA | 217 | –0.23 | CYS | 245 | –0.35 |
| ARG | 218 | –2.68 | CYS | 246 | –0.31 |
| Interaction Forces | Residue | Distance | Protein Charged? | Ligand Group/Atoms | Protein Atom |
|---|---|---|---|---|---|
| Hydrophobic Interactions | LYS195 | 3.94 | Yes | C15–H | C |
| LYS199 | 3.73 | Yes | C15–H | C | |
| PHE206 | 3.59 | No | C19 | Ar–H | |
| TRP214 | 3.61 | No | C8 | Ar–H | |
| π–Cation Interactions | LYS199 | 4.95 | Yes | Aromatic (N3/5, C9/10/11/12) | N–H |
| T (K) | Ksv (×104 M−1) | Kq (×1012 L mol−1 s−1) | n | Ka (×105 M−1) | ΔG (kJ mol−1) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| 298 | 7.85 | 7.85 | 1.14 | 4.09 | −31.92 | −118.75 | −291.38 |
| 304 | 7.31 | 7.31 | 1.06 | 1.41 | −30.17 | ||
| 310 | 6.18 | 6.18 | 1.00 | 0.64 | −28.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Ma, Y.; Zhang, H.; Ma, J. PI3K/mTOR Dual Inhibitor Pictilisib Stably Binds to Site I of Human Serum Albumin as Observed by Computer Simulation, Multispectroscopic, and Microscopic Studies. Molecules 2022, 27, 5071. https://doi.org/10.3390/molecules27165071
Yang H, Ma Y, Zhang H, Ma J. PI3K/mTOR Dual Inhibitor Pictilisib Stably Binds to Site I of Human Serum Albumin as Observed by Computer Simulation, Multispectroscopic, and Microscopic Studies. Molecules. 2022; 27(16):5071. https://doi.org/10.3390/molecules27165071
Chicago/Turabian StyleYang, Hongqin, Yanjun Ma, Hongjie Zhang, and Junyi Ma. 2022. "PI3K/mTOR Dual Inhibitor Pictilisib Stably Binds to Site I of Human Serum Albumin as Observed by Computer Simulation, Multispectroscopic, and Microscopic Studies" Molecules 27, no. 16: 5071. https://doi.org/10.3390/molecules27165071