
Sensing the ortho Positions in C6Cl6 and C6H4Cl2 from Cl2− Formation upon Molecular Reduction

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Experimental Setup
4.2. Computational Methods
- (a)
- Energy thresholds for the dissociation of Cl, Cl2 and their anions from C6Cl6 and C6H4Cl2 were calculated by means of the quantum thermochemical extrapolation methods G4MP2 [40,41] and CBS-QB3 [42,43]. These semi-empirical compound schemes are known to accurately predict the energy differences between reactants and products. For comparison, straightforward density functional calculations with the B3LYP-GD3 functional [44] with dispersion corrections [45] and the aug-cc-pVTZ basis-set [46] were also performed and were in accordance with the expected tolerances. (Tables S1–S3). Since the three model chemistries were based on independent assumptions, this lent credibility to the reported values.
- (b)
- Energy thresholds contain information about reaction thermodynamics but not about kinetics. Therefore, we covered possible reaction pathways by means of transition state searches using the Berny algorithm [47] to move from reactants to products. Specifically, a lowest-energy pathway where one Cl atom moved toward another one until Cl2 abstraction was possible was calculated. The transition states along the pathways had to have one vibrational mode corresponding to the shift from reactants to intermediates (of reactants) with an imaginary frequency, while all other vibrational modes indicated the local energy minima of their normal coordinates. The intermediates were isomers with all vibrational frequencies being real numbers. We verified that these conditions had been fulfilled. The pathway calculations were performed with the M062X functional [48] and the 6-31G (d,p) basis set [49,50]. Transition state energies are not well covered with many standard functionals, and M062X was specifically developed to obtain accurate barriers. This relatively small basis set is often used together with the M062X functional, since it allows for the large number of single-point calculations and optimizations imposed by the search procedure. No geometrical restraints were imposed in the reaction pathway calculations.
- (c)
- Vibrational frequencies were also calculated at the B3LYP-GD3/aug-cc-pVTZ level, as were the various orbital energies and charge distributions (Tables S4 and S5). The same functional and basis set were used in the pointwise relaxed scan of the potential energy curve for the symmetric detachment of Cl2−. (Figure 5, lower panel and Figure S2 in the Supplementary Materials). This functional augmented with corrections for exchange contributions, together with a reasonably large basis set including diffuse functions, is well established for energies and structures, including those of anionic systems.
- (d)
- B3LYP-GD3/aug-cc-pVTZ method and basis set were also used in studying the symmetric direct abstraction of Cl2−. (Supplementary Materials Section 4 and Figure 6). Calculations of the anion started from trial wavefunctions, where the Cl2 fragment already carried a negative charge and the C6Cl4 fragment was neutral. In this way, we circumvented the use of multiconfigurational methods that would otherwise be necessary to describe smooth multiple bond breakings and bond formations. The geometrical details are given in the Supplementary Materials, Section 4.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bailey, R.E. Global hexachlorobenzene emissions. Chemosphere 2001, 43, 167–182. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Environmental Toxicology Branch. Toxicologic Profile of Hexachlorobenzene; U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Environmental Toxicology Branch: Atlanta, GA, USA, 2015. [Google Scholar]
- Brubaker, W.W.; Hites, R.A. Experimental Section. Environ. Sci. Technol. 1998, 32, 766–769. [Google Scholar] [CrossRef]
- Ormad, M.P.; Miguel, N.; Claver, A.; Matesanz, J.M.; Ovelleiro, J.L. Pesticides removal in the process of drinking water production. Chemosphere 2008, 71, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.; Prados, M. Removal of Pesticides by Use of Ozone or Hydrogen Peroxide/Ozone. Ozone Sci. Eng. 1995, 17, 657–672. [Google Scholar] [CrossRef]
- Calaminus, B.; Trouvé, G.; Delfosse, L. Experimental study of the quantitative conversion of hexachlorobenzene during high temperature pyrolysis. J. Anal. Appl. Pyrolysis 1993, 27, 281–292. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Su, C.J.; Chang, B.V. Microbial Dechlorination of Hexach;lorobenzene in Anaerobic Sewage Sludge. Chemosphere 1999, 38, 1015–1023. [Google Scholar] [CrossRef]
- Ramanand, K.; Balba, M.T.; Duffy, J. Reductive dehalogenation of chlorinated benzenes and toluenes under methanogenic conditions. Appl. Environ. Microbiol. 1993, 59, 3266–3272. [Google Scholar] [CrossRef] [Green Version]
- Bosma, T.N.P.; van der Meer, J.R.; Schraa, G.; Tros, M.E.; Zehnder, A.J.B. Reductive dechlorination of all trichloro- and dichlorobenzene isomers. FEMS Microbiol. Ecol. 1988, 53, 223–229. [Google Scholar]
- Izadi, F.; Arthur-Baidoo, E.; Strover, L.T.; Yu, L.J.; Coote, M.L.; Moad, G.; Denifl, S. Selective Bond Cleavage in RAFT Agents Promoted by Low-Energy Electron Attachment. Angew. Chem. Int. Ed. 2021, 60, 19128–19132. [Google Scholar] [CrossRef]
- Ptasińska, S.; Denifl, S.; Gohlke, S.; Scheier, P.; Illenberger, E.; Märk, T.D. Decomposition of thymidine by low-energy electrons: Implications for the molecular mechanisms of single-strand breaks in DNA. Angew. Chem. Int. Ed. 2006, 45, 1893–1896. [Google Scholar] [CrossRef]
- Ptasinska, S.; Denifl, S.; Scheier, P.; Illenberger, E.; Märk, T.D. Bond- and site-selective loss of H atoms from nucleobases by very-low-energy electrons (<3 eV). Angew. Chem. Int. Ed. 2005, 44, 6941–6943. [Google Scholar]
- Denifl, S.; Sulzer, P.; Huber, D.; Zappa, F.; Probst, M.; Märk, T.D.; Scheier, P.; Injan, N.; Limtrakul, J.; Abouaf, R.; et al. Influence of functional groups on the site-selective dissociation of adenine upon low-energy electron attachment. Angew. Chem. Int. Ed. 2007, 46, 5238–5241. [Google Scholar] [CrossRef]
- Da Silva, F.F.; Matias, C.; Almeida, D.; García, G.; Ingólfsson, O.; Flosadóttir, H.D.; Ómarsson, B.; Ptasinska, S.; Puschnigg, B.; Scheier, P.; et al. NCO-, a key fragment upon dissociative electron attachment and electron transfer to pyrimidine bases: Site selectivity for a slow decay process. J. Am. Soc. Mass Spectrom. 2013, 24, 1787–1797. [Google Scholar] [CrossRef]
- Almeida, D.; Antunes, R.; Martins, G.; Eden, S.; Ferreira Da Silva, F.; Nunes, Y.; Garcia, G.; Limão-Vieira, P. Electron transfer-induced fragmentation of thymine and uracil in atom-molecule collisions. Phys. Chem. Chem. Phys. 2011, 13, 15657–15665. [Google Scholar] [CrossRef]
- Almeida, D.; Kinzel, D.; Ferreira da Silva, F.; Puschnigg, B.; Gschliesser, D.; Scheier, P.; Denifl, S.; García, G.; González, L.; Limão-Vieira, P. N-site de-methylation in pyrimidine bases as studied by low energy electrons and ab initio calculations. Phys. Chem. Chem. Phys. 2013, 15, 11431–11440. [Google Scholar] [CrossRef] [Green Version]
- Almeida, D.; Ferreira da Silva, F.; García, G.; Limão-Vieira, P. Selective bond cleavage in potassium collisions with pyrimidine bases of DNA. Phys. Rev. Lett. 2013, 110, 023201. [Google Scholar] [CrossRef]
- Scharping, H.; Zetzsch, C.; Dessouki, H.A. The UV absorption spectra of the trichlorobenzenes and the higher chlorinated benzenes in the gas phase and in n-hexane solution. J. Mol. Spectrosc. 1987, 123, 382–391. [Google Scholar] [CrossRef]
- Scherer, J.R.; Evans, J.C. Vibrational spectra and assignments for sixteen chlorobenzenes. Spectrochim. Acta 1963, 19, 1739–1775. [Google Scholar] [CrossRef]
- Kato, T.; Yamabe, T. Vibronic interactions and charge transfer in negatively charged chloroacenes. Chem. Phys. 2006, 321, 149–158. [Google Scholar] [CrossRef]
- Wiley, J.R.; Chen, E.C.M.; Chen, E.S.D.; Richardson, P.; Reed, W.R.; Wentworth, W.E. The determination of absolute electron affinities of chlorobenzenes, chloronaphthalenes and chlorinated biphenyls from reduction potentials. J. Electroanal. Chem. Interfacial Electrochem. 1991, 307, 169–182. [Google Scholar] [CrossRef]
- Voora, V.K. Molecular Electron Affinities Using the Generalized Kohn-Sham Semicanonical Projected Random Phase Approximation. J. Phys. Chem. Lett. 2021, 12, 433–439. [Google Scholar] [CrossRef]
- Xu, W.; Gao, A. DFT study on the electron affinities of the chlorinated benzenes. J. Mol. Struct. THEOCHEM 2005, 732, 63–70. [Google Scholar] [CrossRef]
- Meißner, R.; Kočišek, J.; Feketeová, L.; Fedor, J.; Fárník, M.; Limão-Vieira, P.; Illenberger, E.; Denifl, S. Low-energy electrons transform the nimorazole molecule into a radiosensitiser. Nat. Commun. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Millar, T.J.; Walsh, C.; Field, T.A. Negative ions in space. Chem. Rev. 2017, 117, 1765–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonich, S.L.; Hites, R.A. Global distribution of persistent organochlorine compounds. Science 1995, 269, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kilich, T.; Łabuda, M.; García, G.; Limão-Vieira, P. Anionic states of C6Cl6 probed in electron transfer experiments. Phys. Chem. Chem. Phys. 2022, 24, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Regeta, K.; Kumar, S.; Cunha, T.; Mendes, M.; Lozano, A.I.; Pereira, P.J.S.; García, G.; Moutinho, A.M.C.; Bacchus-Montabonel, M.C.; Limão-Vieira, P. Combined Experimental and Theoretical Studies on Electron Transfer in Potassium Collisions with CCl4. J. Phys. Chem. A 2020, 124, 3220–3227. [Google Scholar] [CrossRef]
- Mendes, M.; García, G.; Bacchus-Montabonel, M.C.; Limão-Vieira, P. Electron transfer induced decomposition in potassium–nitroimidazoles collisions: An experimental and theoretical work. Int. J. Mol. Sci. 2019, 20, 6170. [Google Scholar] [CrossRef] [Green Version]
- Mendes, M.; Probst, M.; Maihom, T.; García, G.; Limão-Vieira, P. Selective Bond Excision in Nitroimidazoles by Electron Transfer Experiments. J. Phys. Chem. A 2019, 123, 4068–4073. [Google Scholar] [CrossRef]
- Kumar, S.; Izadi, F.; Ončák, M.; Limão-Vieira, P.; Denifl, S. Hexachlorobenzene-negative ion formation in electron attachment experiments. Phys. Chem. Chem. Phys. 2022, 24, 13335–13342. [Google Scholar] [CrossRef]
- Wenthold, P.G.; Squires, R.R.; Lineberger, W.C. Ultraviolet photoelectron spectroscopy of the o-, m-, and p-benzyne negative ions. Electron affinities and singlet-triplet splittings for o-, m-, and p-benzyne. J. Am. Chem. Soc. 1998, 120, 5279–5290. [Google Scholar] [CrossRef]
- Martin, J.D.D.; Hepburn, J.W. Determination of bond dissociation energies by threshold ion-pair production spectroscopy: An improved D0(HCl). J. Chem. Phys. 1998, 109, 8139–8142. [Google Scholar] [CrossRef]
- Polanyi, J.C.; Zewail, A.H. Direct observation of the transition state. Acc. Chem. Res. 1995, 28, 119–132. [Google Scholar] [CrossRef]
- Almeida, D.; Ferreira da Silva, F.; García, G.; Limão-Vieira, P. Dynamic of negative ions in potassium-D-ribose collisions. J. Chem. Phys. 2013, 139, 114304. [Google Scholar] [CrossRef] [Green Version]
- Mendes, M.; Pamplona, B.; Kumar, S.; da Silva, F.F.; Aguilar, A.; García, G.; Bacchus-Montabonel, M.C.; Limão-Vieira, P. Ion-pair formation in neutral potassium-neutral pyrimidine collisions: Electron transfer experiments. Front. Chem. 2019, 7, 264. [Google Scholar] [CrossRef]
- Lozano, A.I.; Maioli, L.S.; Pamplona, B.; Romero, J.; Mendes, M.; Ferreira da Silva, F.; Kossoski, F.; Probst, M.; Süβ, D.; Bettega, M.H.F.; et al. Selective bond breaking of halothane induced by electron transfer in potassium collisions. Phys. Chem. Chem. Phys. 2020, 22, 23837–23846. [Google Scholar] [CrossRef]
- Molski, M.J.; Khanfar, M.A.; Shorafa, H.; Seppelt, K. Halogenated benzene cation radicals. Eur. J. Org. Chem. 2013, 2013, 3131–3136. [Google Scholar] [CrossRef]
- Antunes, R.; Almeida, D.; Martins, G.; Mason, N.J.; Garcia, G.; Maneira, M.J.P.; Nunes, Y.; Limão-Vieira, P. Negative ion formation in potassium–nitromethane collisions. Phys. Chem. Chem. Phys. 2010, 12, 12513–12519. [Google Scholar] [CrossRef] [Green Version]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 2000, 112, 6532–6542. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, H.B. Optimization of Equilibrium Geometries and Transition Structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]







{kind=link}
{kind=link}
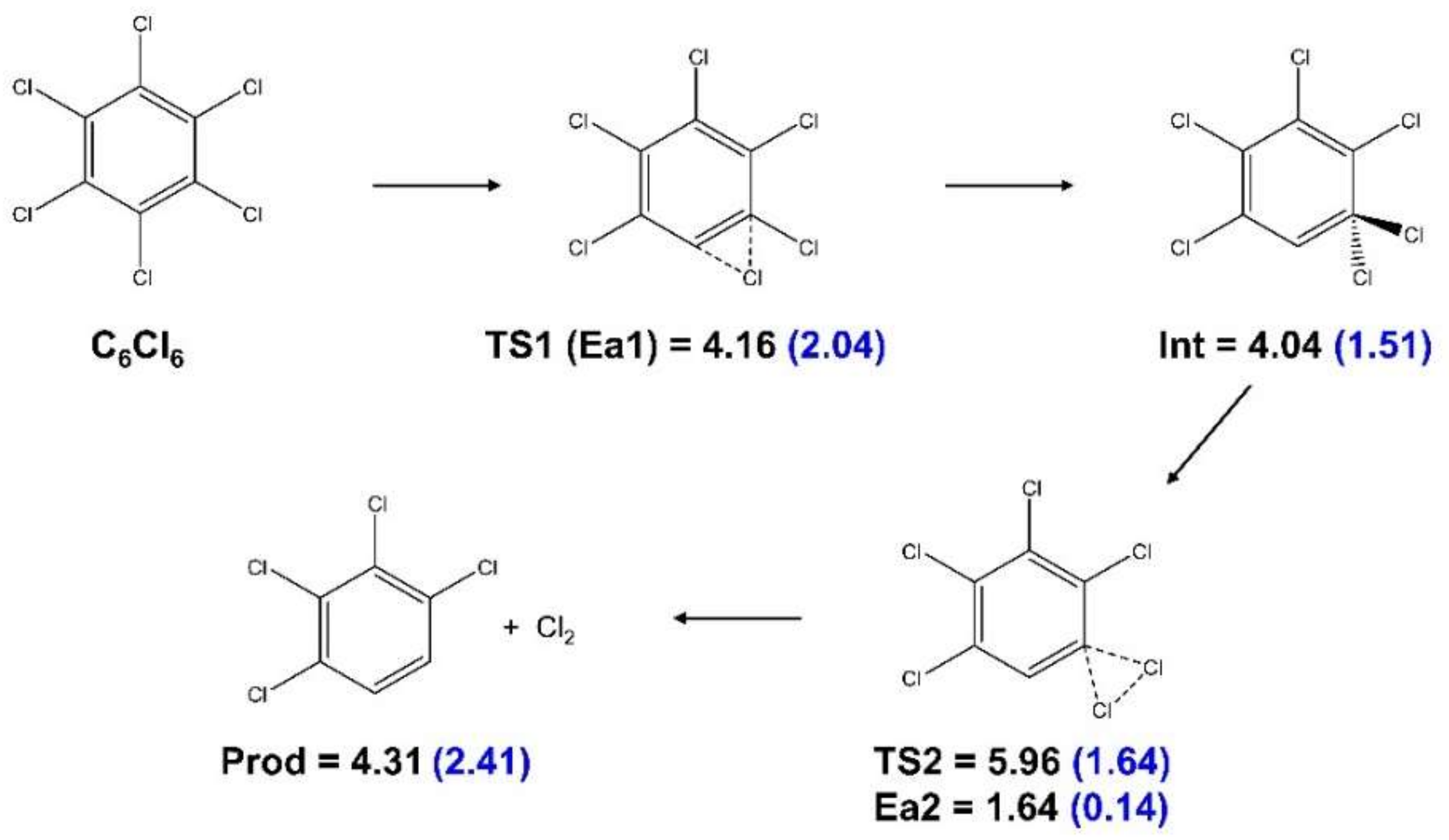{kind=link}
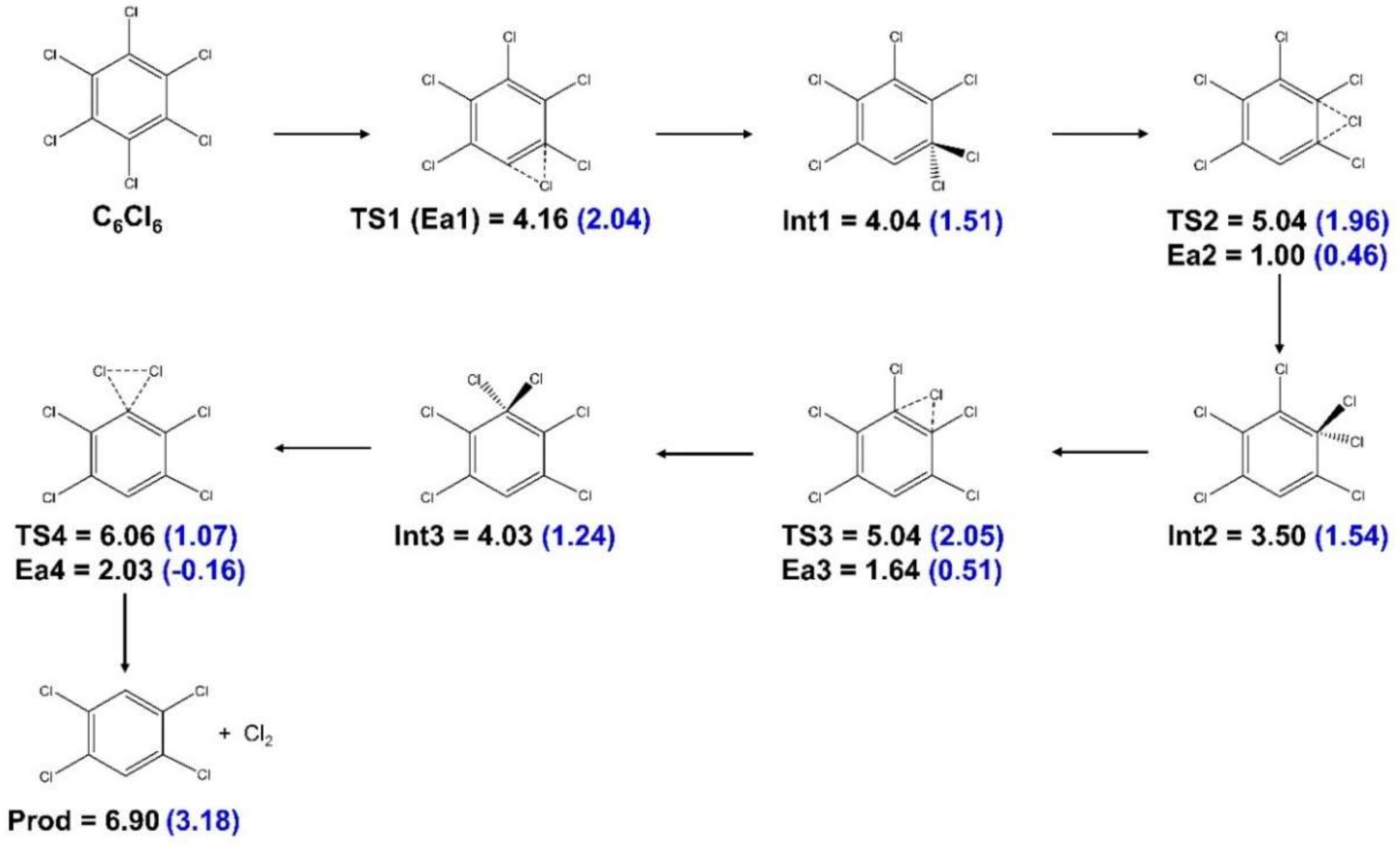{kind=link}
{kind=link}
{kind=link}
| CBS-QB3 (eV) | G4MP2 (eV) | |
|---|---|---|
| AE(Cl2−),o-C6Cl6 | 1.97 | 1.96 |
| AE(Cl2−),m-C6Cl6 | 2.39 | 2.39 |
| AE(Cl2−),p-C6Cl6 | 3.66 | 3.65 |
| D(C6Cl5-Cl) | 4.38 | 4.18 |
| D(o-C6Cl4-Cl) | 2.66 | 2.64 |
| D(m-C6Cl4-Cl) | 3.07 | 3.08 |
| D(p-C6Cl4-Cl) | 4.35 | 4.34 |
| D(Cl–Cl) | 2.57 | 2.51 |
| EA(Cl2) | 2.50 | 2.36 |
| Title 1 | CBS-QB3 (eV) | G4MP2 (eV) |
|---|---|---|
| AE(Cl2−),o-C6H4Cl2 | 2.08 | 2.07 |
| AE(Cl2−),m-C6H4Cl2 | 2.78 | 2.79 |
| AE(Cl2−),p-C6H4Cl2 | 3.29 | 3.29 |
| D(o-C6H4Cl-Cl) | 4.41 | 4.23 |
| D(m-C6H4Cl-Cl) | 4.38 | 4.22 |
| D(p-C6H4Cl-Cl) | 4.41 | 4.25 |
| D(o-C6H4-Cl) | 2.74 | 2.71 |
| D(m-C6H4-Cl) | 3.46 | 3.44 |
| D(p-C6H4-Cl) | 3.95 | 3.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Romero, J.; Probst, M.; Maihom, T.; García, G.; Limão-Vieira, P. Sensing the ortho Positions in C6Cl6 and C6H4Cl2 from Cl2− Formation upon Molecular Reduction. Molecules 2022, 27, 4820. https://doi.org/10.3390/molecules27154820
Kumar S, Romero J, Probst M, Maihom T, García G, Limão-Vieira P. Sensing the ortho Positions in C6Cl6 and C6H4Cl2 from Cl2− Formation upon Molecular Reduction. Molecules. 2022; 27(15):4820. https://doi.org/10.3390/molecules27154820
Chicago/Turabian StyleKumar, Sarvesh, José Romero, Michael Probst, Thana Maihom, Gustavo García, and Paulo Limão-Vieira. 2022. "Sensing the ortho Positions in C6Cl6 and C6H4Cl2 from Cl2− Formation upon Molecular Reduction" Molecules 27, no. 15: 4820. https://doi.org/10.3390/molecules27154820