
Three New Stigmatellin Derivatives Reveal Biosynthetic Insights of Its Side Chain Decoration


Abstract
:1. Introduction
2. Results and Discussion
2.1. Discovery of Stigmatellic Acid (1), Iso-methoxy-stigmatellin A (2), and Stigmatellin C (3)
2.2. Bioactivity Testing of 1–4
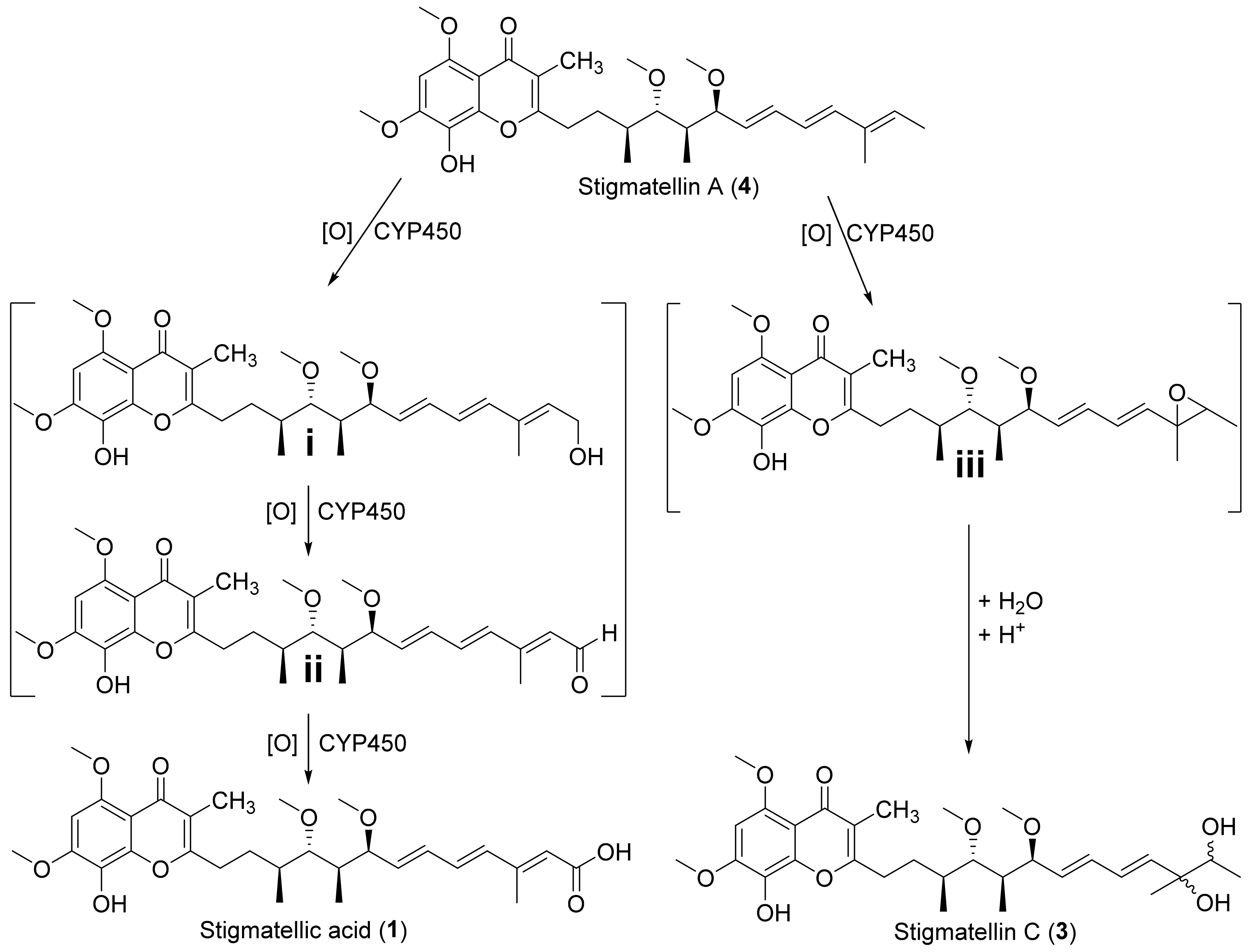
2.3. Biosynthesis of 1–3
3. Materials and Methods
3.1. Maintenance of Myxobacterial Cultures
3.2. Analysis of Secondary Metabolism of Broth Extracts
3.3. Isolation of 1–4 by Semi-Preparative HPLC
- Stigmatellic acid (1):
- Iso-methoxy-stigmatellin A (2):
- Stigmatellin C (3):
- Stigmatellin A (4):
3.4. NMR Based Structure Elucidation and Chiroptical Measurement
3.5. Bioactivity Profiling
3.6. Applied Software, DNA Sequence Analysis, and Bioinformatics Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. Privileged Structures Revisited. Angew. Chem. Int. Ed. Engl. 2017, 56, 7971–7974. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Reymond, J.-L.; Awale, M. Exploring Chemical Space for Drug Discovery Using the Chemical Universe Database. ACS Chem. Neurosci. 2012, 3, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Dejong, C.A.; Franczak, B.C.; McNicholas, P.D.; Magarvey, N.A. Comparative analysis of chemical similarity methods for modular natural products with a hypothetical structure enumeration algorithm. J. Cheminform. 2017, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldívar-González, F.I.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Chemical space of naturally occurring compounds. Phys. Sci. Rev. 2018, 4, 20180103. [Google Scholar] [CrossRef]
- Hug, J.J.; Bader, C.D.; Remškar, M.; Cirnski, K.; Müller, R. Concepts and Methods to Access Novel Antibiotics from Actinomycetes. Antibiotics 2018, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Challinor, L.V.; Helge, B. Bode. Bioactive natural products from novel microbial sources. Ann. N. Y. Acad. Sci. 2015, 1354, 82–97. [Google Scholar] [CrossRef]
- Munoz-Dorado, J.; Marcos-Torres, F.J.; Garcia-Bravo, E.; Moraleda-Munoz, A.; Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [Green Version]
- Wolgemuth, C.; Hoiczyk, E.; Kaiser, D.; Oster, G. How myxobacteria glide. Curr. Biol. 2002, 12, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Berleman, J.E.; Kirby, J.R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol. Rev. 2009, 33, 942–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfle, G.; Kunze, B.; Zorzin, C.; Reichenbach, H. Antibiotika aus Gleitenden Bakterien, XXIII: Stigmatellin A und B—Zwei neue Antibiotika aus Stigmatella aurantiaca (Myxobacterales). Liebigs Ann. Chem. 1984, 8, 1883–1904. [Google Scholar] [CrossRef]
- Kunze, B.; Kemmer, T.; Höfle, G.; Reichenbach, H. Stigmatellin, a new antibiotic from Stigmatella aurantiaca (Myxobacterales). I. Production, physico-chemical and biological properties. J. Antibiot. 1984, 37, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Thierbach, G.; Kunze, B.; Reichenbach, H.; Höfle, G. The mode of action of stigmatellin, a new inhibitor of the cytochorme b-c1 segment of the respiratory chain. Biochim. Biophys. Acta 1984, 765, 227–235. [Google Scholar] [CrossRef]
- Von Jagow, G.; Ohnishi, T. The chromone inhibitor stigmatellin—Binding to the ubiquinol oxidation center at the C-side of the mitochondrial membrane. FEBS Lett. 1985, 185, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Gurung, B.; Linda, Y.; Yu, C.A. Stigmatellin induces iron-sulfur protein reduction in cytochrome bc1 complex in the absence of apparent electron source. JBC 2008, 283, 28087–28094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, L.; Shulmeister, V.M.; Chi, Y.I.; Kim, K.K.; Hung, L.W.; Crofts, A.R.; Berry, E.A.; Kim, S.H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar] [CrossRef]
- Enders, D.; Geibel, G.; Osborne, S. Diastereo- and Enantioselective Total Synthesis of Stigmatellin, A. Chem. Eur. J. 2000, 6, 1302–1309. [Google Scholar] [CrossRef]
- Beyer, S.; Kunze, B.; Silakowski, B.; Müller, R. Metabolic diversity in myxobacteria: Identification of the myxalamid and the stigmatellin biosynthetic gene cluster of Stigmatella aurantiaca Sg a15 and a combined polyketide-(poly)peptide gene cluster from the epothilone producing strain Sorangium cellulosum So ce90. Biochim. Biophys. Acta 1999, 1445, 185–195. [Google Scholar]
- Gaitatzis, N.; Silakowski, B.; Kunze, B.; Nordsiek, G.; Blöcker, H.; Höfle, G.; Müller, R. The biosynthesis of the aromatic myxobacterial electron transport inhibitor stigmatellin is directed by a novel type of modular polyketide synthase. J. Biol. Chem. 2002, 277, 13082–13090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathi, S.; Vashisth, R.; Manoharan, P.; Kandasamy, R.; Sivakumar, N. Stigmatellin Y—An anti-biofilm compound from Bacillus subtilis BR4 possibly interferes in PQS–PqsR mediated quorum sensing system in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2017, 27, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Brandt, U.; von Jagow, G. Studies on the effect of stigmatellin derivatives on cytochrome b and the Rieske iron-sulfur cluster of cytochrome c reductase from bovine heart mitochondria. Eur. J. Biochem. 1988, 176, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Skiba, M.A.; Bivins, M.M.; Schultz, J.R.; Bernard, S.M.; Fiers, W.D.; Dan, Q.; Kulkarni, S.; Wipf, P.; Gerwick, W.H.; Sherman, D.H.; et al. Structural Basis of Polyketide Synthase O-Methylation. ACS Chem. Biol. 2018, 13, 3221–3228. [Google Scholar] [CrossRef]
- Phillips, A.J.; Henderson, J.A.; Jackson, K.L. Pyrans and their Benzo Derivatives: Structure and Reactivity. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 337–418. ISBN 9780080449920. [Google Scholar]
- Enders, D.; Osborne, S. Determination of the relative and absolute configuration of Stigmatellin A by chemical correlation. J. Chem. Soc. Chem. Commun. 1993, 424–426. [Google Scholar] [CrossRef]
- Yadav, J.S.; Revathi, G.; Reddy, B.S. Formal total synthesis of stigmatellin A. Tetrahedron Lett. 2017, 58, 3943–3946. [Google Scholar] [CrossRef]
- Hedden, P. The Current Status of Research on Gibberellin Biosynthesis. Plant Cell Physiol. 2020, 61, 1832–1849. [Google Scholar] [CrossRef]
- Rudolf, J.D.; Chang, C.-Y.; Ma, M.; Shen, B. Cytochromes P450 for natural product biosynthesis in Streptomyces: Sequence, structure, and function. Nat. Prod. Rep. 2017, 34, 1141–1172. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, mechanism, and biochemistry. In Ortiz de Montellano, 3rd ed.; Paul, R., Ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005; ISBN 0306483246. [Google Scholar]
- Cryle, M.J.; Stok, J.E.; de Voss, J.J. Reactions Catalyzed by Bacterial Cytochromes P450. Aust. J. Chem. 2003, 56, 749. [Google Scholar] [CrossRef]
- Heneghan, M.N.; Yakasai, A.A.; Williams, K.; Kadir, K.A.; Wasil, Z.; Bakeer, W.; Fisch, K.M.; Bailey, A.M.; Simpson, T.J.; Cox, R.J.; et al. The programming role of trans-acting enoyl reductases during the biosynthesis of highly reduced fungal polyketides. Chem. Sci. 2011, 2, 972–979. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, H.; Li, S.; Zhu, Y.; Zhang, G.; Zhang, H.; Zhang, W.; Shi, R.; Zhang, C. Carboxyl formation from methyl via triple hydroxylations by XiaM in xiamycin A biosynthesis. Org. Lett. 2012, 14, 6142–6145. [Google Scholar] [CrossRef] [PubMed]
- Awal, R.P.; Garcia, R.; Gemperlein, K.; Wink, J.; Kunwar, B.; Parajuli, N.; Müller, R. Vitiosangium cumulatum gen. nov., sp. nov. and Vitiosangium subalbum sp. nov., soil myxobacteria, and emended descriptions of the genera Archangium and Angiococcus, and of the family Cystobacteraceae. Int. J. Syst. Evol. Microbiol. 2017, 67, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Okoth Dorothy, A.; Hug, J.J.; Garcia, R.; Spröer, C.; Overmann, J.; Müller, R. 2-Hydroxysorangiadenosine: Structure and Biosynthesis of a Myxobacterial Sesquiterpene–Nucleoside. Molecules 2020, 25, 2676. [Google Scholar] [CrossRef] [PubMed]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef]
- Cole, R.B. Electrospray and MALDI Mass Spectrometry; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010; ISBN 9780470588901. [Google Scholar]
- Hug, J.J.; Kjaerulff, L.; Garcia, R.; Müller, R. New Deoxyenhygrolides from Plesiocystis pacifica Provide Insights into Butenolide Core Biosynthesis. Mar. Drugs 2022, 20, 72. [Google Scholar] [CrossRef]
- Hug, J.J.; Panter, F.; Krug, D.; Müller, R. Genome mining reveals uncommon alkylpyrones as type III PKS products from myxobacteria. J. Ind. Microbiol. Biotechnol. 2019, 46, 319–334. [Google Scholar] [CrossRef]
- Okoth, D.A.; Hug, J.J.; Mándi, A.; Kurtán, T.; Garcia, R.; Müller, R. Structure and biosynthesis of sorangipyranone—A new γ-dihydropyrone from the myxobacterial strain MSr12020. J. Ind. Microbiol. 2021, 48, kuab029. [Google Scholar] [CrossRef]
- Groß, S.; Panter, F.; Pogorevc, D.; Seyfert, C.E.; Deckarm, S.; Bader, C.D.; Herrmann, J.; Müller, R. Improved broad-spectrum antibiotics against Gram-negative pathogens via darobactin biosynthetic pathway engineering. Chem. Sci. 2021, 12, 11882–11893. [Google Scholar] [CrossRef]
- Bader, C.D.; Panter, F.; Garcia, R.; Tchesnokov, E.P.; Haid, S.; Walt, C.; Spröer, C.; Kiefer, A.F.; Götte, M.; Overmann, J.; et al. Sandacrabins—Structurally Unique Antiviral RNA Polymerase Inhibitors from a Rare Myxobacterium. Chem. Eur. J. 2022, 28, e202104484. [Google Scholar] [CrossRef]
- Herrmann, J.; Elnakady, Y.A.; Wiedmann, R.M.; Ullrich, A.; Rohde, M.; Kazmaier, U.; Vollmar, A.M.; Müller, R. Pretubulysin: From hypothetical biosynthetic intermediate to potential lead in tumor therapy. PLoS ONE 2012, 7, e37416. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.C.; Müller, R. Myxobacteria—Unique microbial secondary metabolite factories. In Comprehensive Natural Products Chemistry II, Vol 2: Structural Diversity II—Secondary Metabolite Sources, Evolution and Selected Molecular Structures; Moore, B.S., Ed.; Elsevier: Oxford, UK, 2010; pp. 189–222. [Google Scholar]
- Keatinge-Clay, A.T. A Tylosin Ketoreductase Reveals How Chirality is Determined in Polyketides. Chem. Biol. 2007, 14, 898–908. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC Values of 1–4 in μg/mL | |||||
|---|---|---|---|---|---|
| Microorganism | 1 | 2 | 3 | 4 | Ctr. |
| Acinetobacter baumanii DSM 30007 | >128 | 128 | >128 | 128 | 1.00 a |
| Mucor hiemalis DSM 2656 | 64 | 32 | 128 | 16 | 0.25 c |
| Staphylococcus aureus Newman | 128 | 128 | >128 | 128 | 1.00 b |
| Pseudomonas aeruginosa PA14 (DSM 19882) | >128 | >128 | >128 | >128 | 0.13 a |
| Escherichia coli acrB JW0451-2 | >128 | >128 | >128 | >128 | <0.01 a |
| E. coli wild-type (DSM 1116) | >128 | >128 | >128 | >128 | 0.03 a |
| Bacillus subtilis DSM 10 | >128 | >128 | >128 | 128 | 0.50 b |
| Candida albicans DSM 1665 | 128 | 128 | 128 | 16 | 0.25 c |
| Pichia anomala DSM 6766 | 64 | 128 | 128 | 16 | 0.25 c |
| Citrobacter freundii DSM 30039 | >128 | >128 | >128 | >128 | 0.03 a |
| IC50 Values of 1–4 in μg/mL | |||||
|---|---|---|---|---|---|
| Cancer Cell Line | 1 | 2 | 3 | 4 | Ctr. |
| HCT-116 | 0.35 | 0.25 | 1.16 | 0.09 | 0.02 |
| KB-3-1 | 0.95 | 0.67 | 3.15 | 0.14 | 0.19 |
| U2OS | 5.36 | 3.34 | 18.80 | 0.50 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okoth, D.A.; Hug, J.J.; Garcia, R.; Müller, R. Three New Stigmatellin Derivatives Reveal Biosynthetic Insights of Its Side Chain Decoration. Molecules 2022, 27, 4656. https://doi.org/10.3390/molecules27144656
Okoth DA, Hug JJ, Garcia R, Müller R. Three New Stigmatellin Derivatives Reveal Biosynthetic Insights of Its Side Chain Decoration. Molecules. 2022; 27(14):4656. https://doi.org/10.3390/molecules27144656
Chicago/Turabian StyleOkoth, Dorothy A., Joachim J. Hug, Ronald Garcia, and Rolf Müller. 2022. "Three New Stigmatellin Derivatives Reveal Biosynthetic Insights of Its Side Chain Decoration" Molecules 27, no. 14: 4656. https://doi.org/10.3390/molecules27144656