
Post-Modification of Copolymers Obtained by ATRP for an Application in Heterogeneous Asymmetric Salen Catalysis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results and Discussion
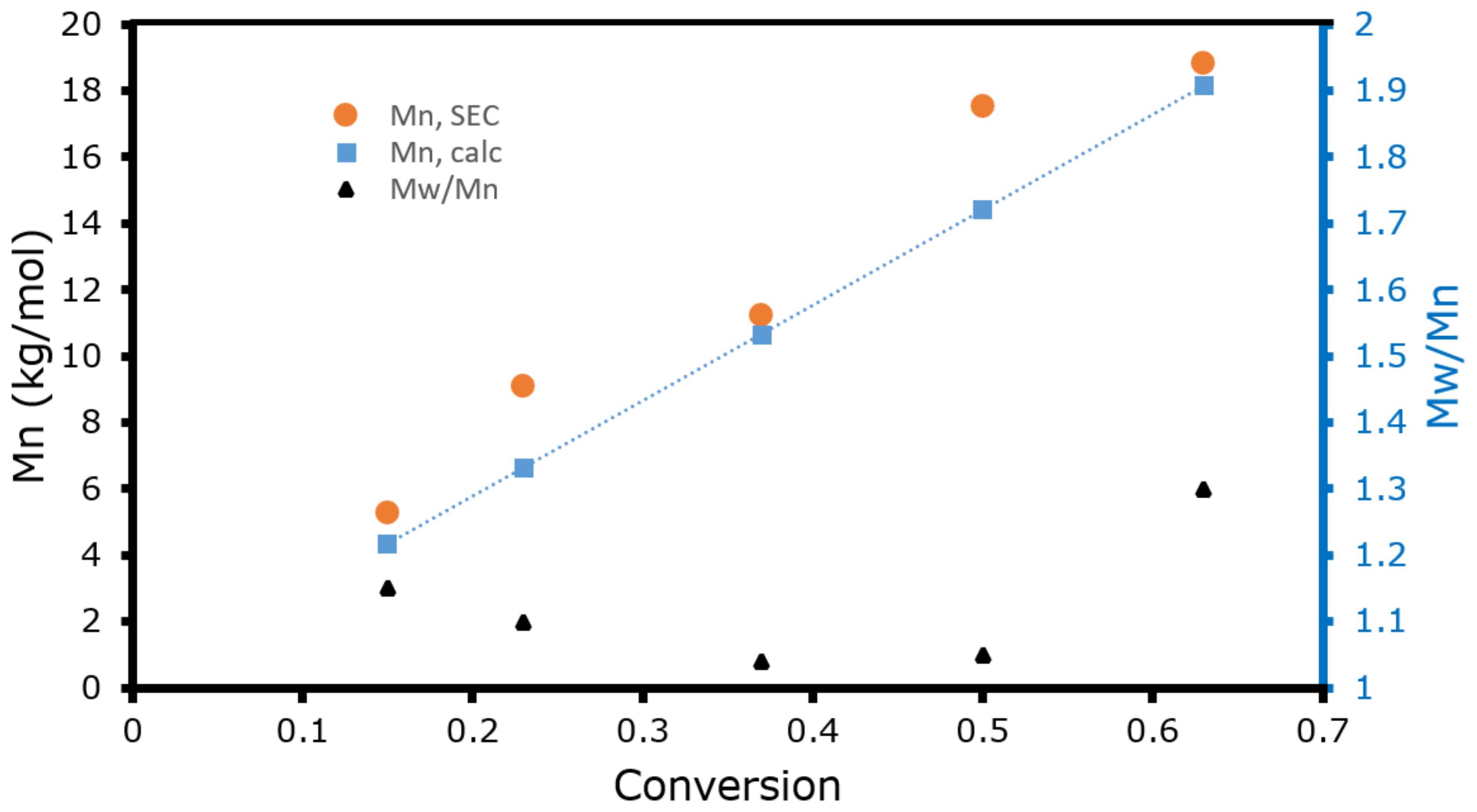
2.1. ATRP Homopolymerization
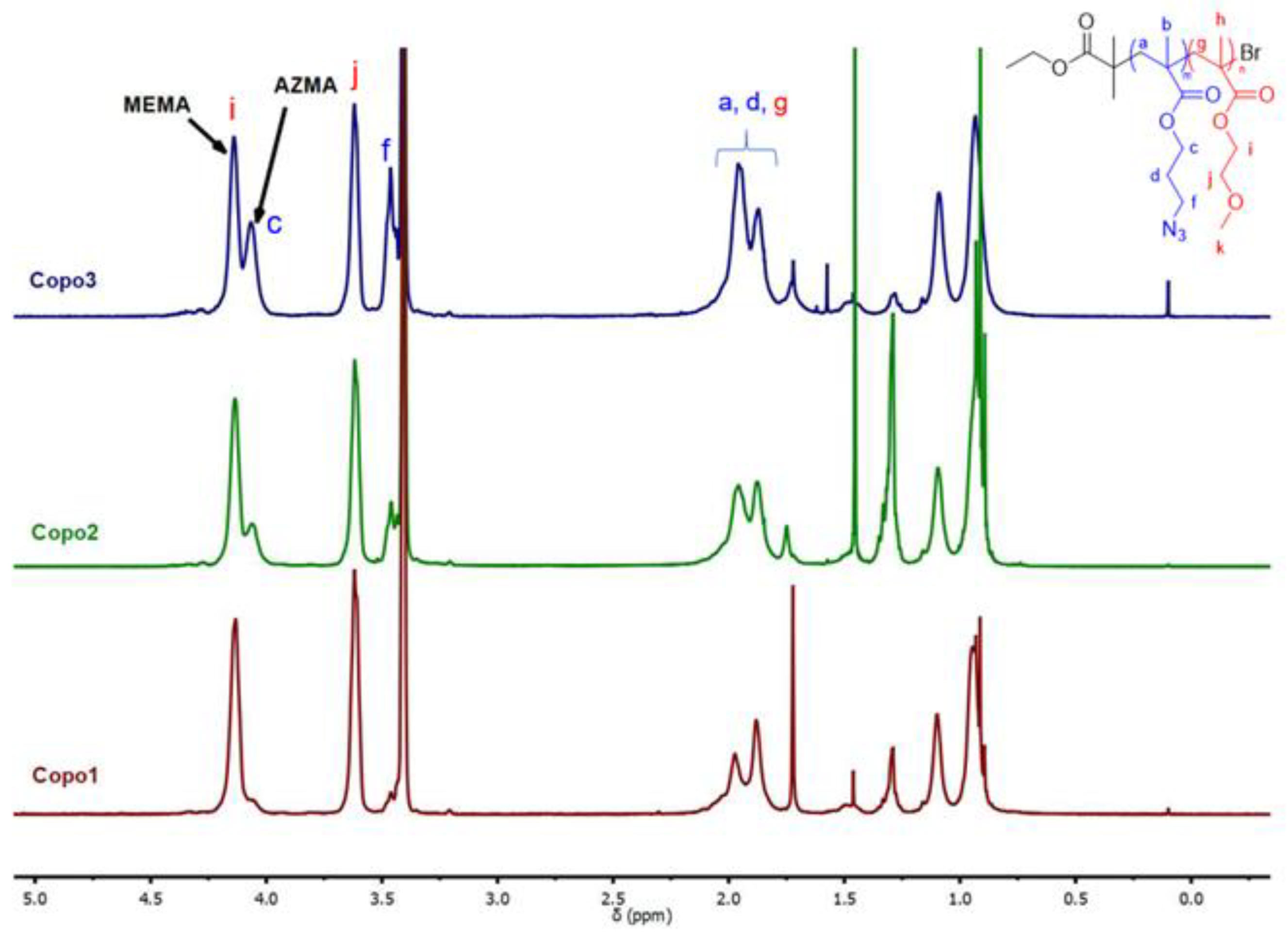
2.2. ATRP Copolymerization
2.3. DOSY NMR Experiments
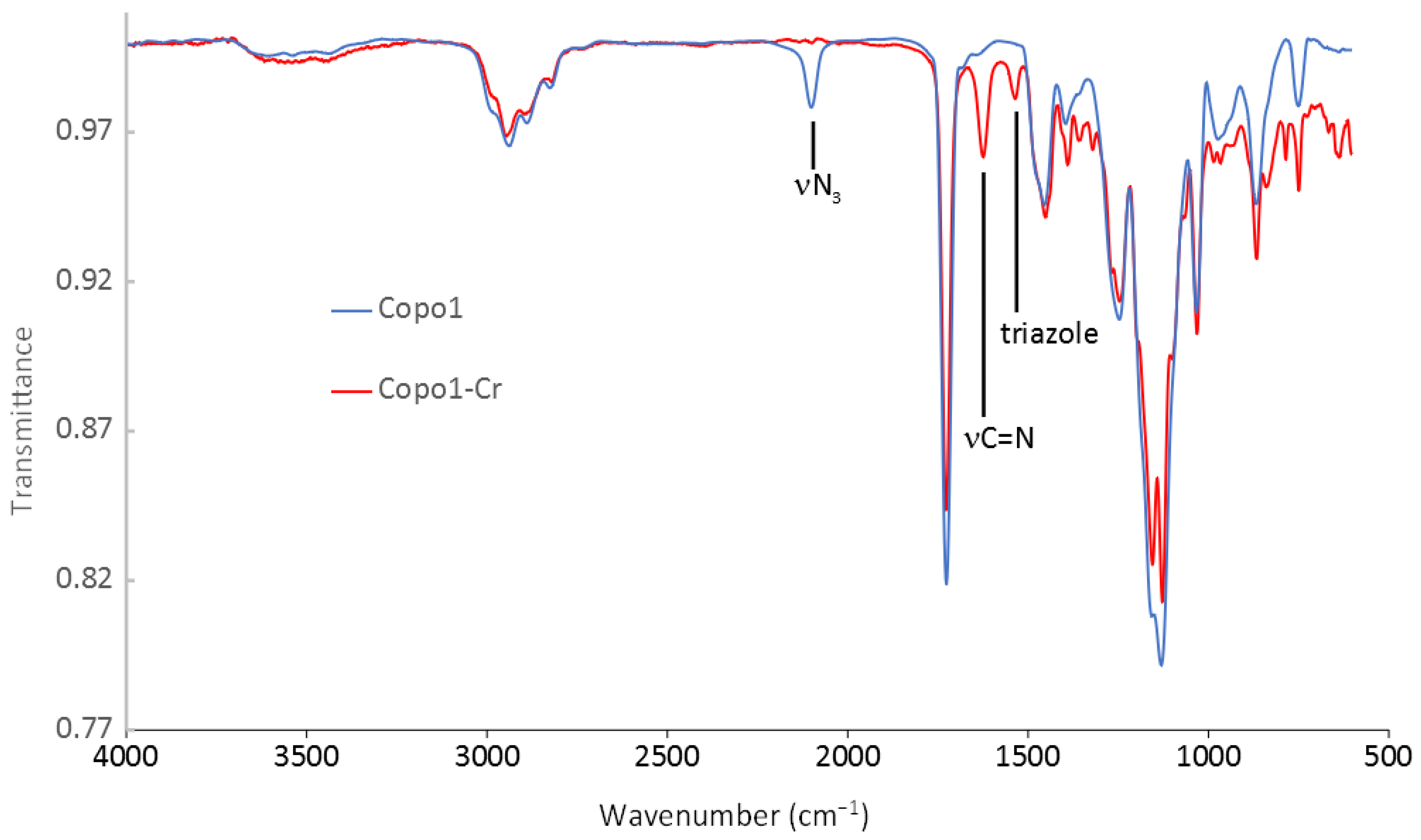
2.4. IR Characterization
2.5. Preparation of the Chromium Salen Copolymer Catalysts
2.6. Asymmetric Heterogeneous Catalysis
3. Materials and Methods
3.1. General Information
3.2. Method for the Synthesis of the Alkyne-Modified Salen Ligand and the Corresponding Chromium Complex
3.2.1. Synthesis of 3-tert-butyl-2-hydroxy-5-((prop-2-ynyloxy)methyl) Benzaldehyde
3.2.2. Synthesis of the Salen Ligand Bearing a Propargyl Moiety
3.2.3. Synthesis of the Chromium Salen Complex Bearing a Propargyl Moiety
3.3. General Procedure for the Homopolymerization: Poly (MEMA) Synthesis
3.4. General Procedure for the Copolymerization
- Copo1; MEMA90-co-AZMA10
- Copo2; MEMA70-co-AZMA30
- Copo3; MEMA50-co-AZMA50
3.5. General Procedure for the Post-Modification by Azide–Alkyne Huisgen Cycloaddition
3.6. Catalytic Procedure (ARO Reaction) in Heterogeneous Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cozzi, P.G. Metal–Salen Schiff Base Complexes in Catalysis: Practical Aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.; White, J.D. Asymmetric Catalysis Using Chiral Salen–Metal Complexes: Recent Advances. Chem. Rev. 2019, 119, 9381–9426. [Google Scholar] [CrossRef] [PubMed]
- Baleizão, C.; Garcia, H. Chiral Salen Complexes: An Overview to Recoverable and Reusable Homogeneous and Heterogeneous Catalysts. Chem. Rev. 2006, 106, 3987–4043. [Google Scholar] [CrossRef] [PubMed]
- Zulauf, A.; Mellah, M.; Hong, X.; Schulz, E. Recoverable Chiral Salen Complexes for Asymmetric Catalysis: Recent Progress. Dalton Trans. 2010, 39, 6911–6935. [Google Scholar] [CrossRef]
- Abd El Sater, M.; Jaber, N.; Schulz, E. Chiral Salen Complexes for Asymmetric Heterogeneous Catalysis: Recent Examples for Recycling and Cooperativity. ChemCatChem 2019, 11, 3662–3687. [Google Scholar] [CrossRef] [Green Version]
- Konsler, R.G.; Karl, K.; Jacobsen, E.N. Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc. 1998, 120, 10780–10781. [Google Scholar] [CrossRef]
- Belser, T.; Jacobsen, E.N. Cooperative Catalysis in the Hydrolytic Kinetic Resolution of Epoxides by Chiral [(salen)Co(III)] Complexes Immobilized on Gold Colloids. Adv. Synth. Catal. 2008, 350, 967–971. [Google Scholar] [CrossRef]
- Loy, R.N.; Jacobsen, E.N. Enantioselective Intramolecular Openings of Oxetanes Catalyzed by (Salen)Co(III) Complexes: Access to Enantioenriched Tetrahydrofurans. J. Am. Chem. Soc. 2009, 131, 2786–2787. [Google Scholar] [CrossRef] [Green Version]
- Haak, R.M.; Martinez Belmonte, M.; Escudero-Adan, E.C.; Benet-Buchholz, J.; Kleij, A.W. Olefin Metathesis as a Tool for Multinuclear Co(III)salen Catalyst Construction: Access to Cooperative Catalysts. Dalton Trans. 2010, 39, 593–602. [Google Scholar] [CrossRef]
- Nielsen, L.P.C.; Stevenson, C.P.; Blackmond, D.G.; Jacobsen, E.N. Mechanistic Investigation Leads to a Synthetic Improvement in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc. 2004, 126, 1360–1362. [Google Scholar] [CrossRef]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashiruma, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/“Living” Radical Polymerization. Atom Transfer Radical Polymerization in the Presence of Transition-Metal Complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Zidelmal, N.; Aubry-Barroca, N.; Lepoittevin, B.; Mellah, M.; Costa, L.; Ozanam, F.; Gouget-Laemmel, A.-C.; Schulz, E.; Roger, P. Synthesis, Characterization and Catalytic Properties of Salen-Containing Polymers Obtained by Atom Transfer Radical Polymerization. Polymer 2018, 135, 261–270. [Google Scholar] [CrossRef]
- Martínez, L.E.; Leighton, J.L.; Carsten, D.H.; Jacobsen, E.N. Highly Enantioselective Ring Opening of Epoxides Catalyzed by (salen)Cr(III) Complexes. J. Am. Chem. Soc. 1995, 117, 5897–5898. [Google Scholar] [CrossRef]
- Schaus, S.E.; Larrow, J.F.; Jacobsen, E.N. Practical Synthesis of Enantiopure Cyclic 1,2-Amino Alcohols via Catalytic Asymmetric Ring Opening of Meso Epoxides. J. Org. Chem. 1997, 62, 4197–4199. [Google Scholar] [CrossRef]
- Bandini, M.; Cozzi, P.G.; Umani-Ronchi, A. [Cr(Salen)] as a ‘Bridge’ between Asymmetric Catalysis, Lewis Acids and Redox Processes. Chem. Commun. 2002, 9, 919–927. [Google Scholar] [CrossRef]
- Dioos, B.M.L.; Jacobs, P.A. Heterogenisation of Dimeric Cr(salen) with Supported Ionic Liquids. J. Catal. 2006, 243, 217–219. [Google Scholar] [CrossRef]
- Zulauf, A.; Mellah, M.; Schulz, E. Original Use of the Same Heterogeneous Chiral Catalyst Batch to Promote Different Asymmetric Reactions. Chem. Commun. 2009, 43, 6574–6576. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Hobbs, C.; Chayanant, C. Polyolefin-Supported Recoverable/Reusable Cr(III)-Salen Catalysts. J. Org. Chem. 2011, 76, 523–533. [Google Scholar] [CrossRef]
- Xia, Q.; Liu, Y.; Li, Z.; Gong, W.; Cui, Y. A Cr(salen)-Based Metal–Organic Framework as a Versatile Catalyst for Efficient Asymmetric Transformations. Chem. Commun. 2016, 52, 13167–13170. [Google Scholar] [CrossRef]
- Jiao, J.; Tan, C.; Li, Z.; Liu, Y.; Han, X.; Cui, Y. Design and Assembly of Chiral Coordination Cages for Asymmetric Sequential Reactions. J. Am. Chem. Soc. 2018, 140, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Abd El Sater, M.; Mellah, M.; Dragoe, D.; Kolodziej, E.; Jaber, N.; Schulz, E. Chiral Chromium Salen@rGO as Multipurpose and Recyclable Heterogeneous Catalyst. Chem. Eur. J. 2021, 27, 9454–9460. [Google Scholar]
- Gill, C.S.; Venkatasubbaiah, K.; Phan, N.T.S.; Weck, M.; Jones, C.W. Enhanced Cooperativity Through Design: Pendant Co(III)-salen Polymer Brush Catalysts for the Hydrolytic Kinetic Resolution of Epichlorohydrin (salen = N, N’-bis(salicylidene)ethylenediamine dianion). Chem. Eur. J. 2008, 14, 7306–7313. [Google Scholar] [CrossRef]
- Gill, C.S.; Long, W.; Jones, C.W. Magnetic Nanoparticle Polymer Brush Catalysts: Alternative Hybrid Organic/Inorganic Structures to Obtain High, Local Catalyst Loadings for Use in Organic Transformations. Catal. Lett. 2009, 131, 425–431. [Google Scholar] [CrossRef]
- Salunke, S.B.; Babu, N.S.; Chen, C.-T. Asymmetric Aerobic Oxidation of α-Hydroxy Acid Derivatives Catalyzed by Reusable, Polystyrene-Supported Chiral N-Salicylidene Oxidovanadium tert-Leucinates. Adv. Synth. Catal. 2011, 353, 1234–1240. [Google Scholar] [CrossRef]
- Sumerlin, B.S.; Tsarevsky, N.V.; Louche, G.; Lee, R.Y.; Matyjaszewski, K. Highly Efficient “Click” Functionalization of Poly(3-azidopropyl methacrylate) Prepared by ATRP. Macromolecules 2005, 38, 7540–7545. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, X.; Bian, R.; Tong, M.; Tang, D.; Zhou, X.; Zhao, Y. Facile Synthesis of A2mB2n-Type Starlike Copolymers with Two Types of V-Shaped Arms by Combination of RAFT, ATRP and ROP Processes. Polymer 2015, 64, 249–259. [Google Scholar] [CrossRef]
- Gelbrich, T.; Marten, G.U.; Schmidt, A.M. Reversible Thermoflocculation of Magnetic Core-shell Particles Induced by Remote Magnetic Heating. Polymer 2010, 51, 2818–2824. [Google Scholar] [CrossRef]
- Holbach, M.; Zheng, X.; Burd, C.; Jones, C.W.; Weck, M. A Practical One-Pot Synthesis of Enantiopure Unsymmetrical Salen Ligands. J. Org. Chem. 2006, 71, 2903–2906. [Google Scholar] [CrossRef]
- Schaus, S.E.; Branalt, J.; Jacobsen, E.N. Asymmetric Hetero-Diels−Alder Reactions Catalyzed by Chiral (Salen)Chromium(III) Complexes. J. Org. Chem. 1998, 63, 403–405. [Google Scholar] [CrossRef]
- Gökce, H.; Sen, F.; Sert, Y.; Abdel-Wahab, B.F.; Kariuki, B.; El-Hiti, G.A. Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N’-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide. Molecules 2022, 27, 2193. [Google Scholar] [CrossRef] [PubMed]
- Altava, B.; Burguete, M.I.; Garcia-Verdugo, E.; Luis, S.V. Chiral Catalysts Immobilized on Achiral Polymers: Effect of the Polymer Support on the Performance of the Catalyst. Chem. Soc. Rev. 2018, 47, 2722–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stejskal, J.; Janca, J.; Kratochvil, P. Solution Properties of Poly(2-Methoxyethyl Methacrylate). Polymer J. 1976, 8, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H. Metal(II) d-Tartrates Catalyzed Asymmetric Ring Opening of Oxiranes with Various Nucleophiles. Bull. Chem. Soc. Jpn. 1988, 61, 1213–1220. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comonomer Feed MEMA/AZMA (%/%) | Conversions a pMEMA/pAZMA (%/%) | Mn calc b (g/mol) | MnSEC c (g/mol) | ᴆ c | Composition d MEMA/AZMA (%/%) | |
|---|---|---|---|---|---|---|
| Copo1 | 90/10 | 73/71 | 21,300 | 22,500 | 1.05 | 93/7 |
| Copo2 | 70/30 | 64/61 | 19,200 | 25,700 | 1.20 | 78/22 |
| Copo3 | 50/50 | 66/63 | 21,200 | 30,800 | 1.28 | 60/40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakangura, E.; Roger, P.; Soares, R.S.B.; Mellah, M.; Barroca-Aubry, N.; Gouget-Laemmel, A.-C.; Ozanam, F.; Costa, L.; Baltaze, J.-P.; Schulz, E. Post-Modification of Copolymers Obtained by ATRP for an Application in Heterogeneous Asymmetric Salen Catalysis. Molecules 2022, 27, 4654. https://doi.org/10.3390/molecules27144654
Bakangura E, Roger P, Soares RSB, Mellah M, Barroca-Aubry N, Gouget-Laemmel A-C, Ozanam F, Costa L, Baltaze J-P, Schulz E. Post-Modification of Copolymers Obtained by ATRP for an Application in Heterogeneous Asymmetric Salen Catalysis. Molecules. 2022; 27(14):4654. https://doi.org/10.3390/molecules27144654
Chicago/Turabian StyleBakangura, Erigene, Philippe Roger, Rafaela S. B. Soares, Mohamed Mellah, Nadine Barroca-Aubry, Anne-Chantal Gouget-Laemmel, François Ozanam, Ludovic Costa, Jean-Pierre Baltaze, and Emmanuelle Schulz. 2022. "Post-Modification of Copolymers Obtained by ATRP for an Application in Heterogeneous Asymmetric Salen Catalysis" Molecules 27, no. 14: 4654. https://doi.org/10.3390/molecules27144654