
Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
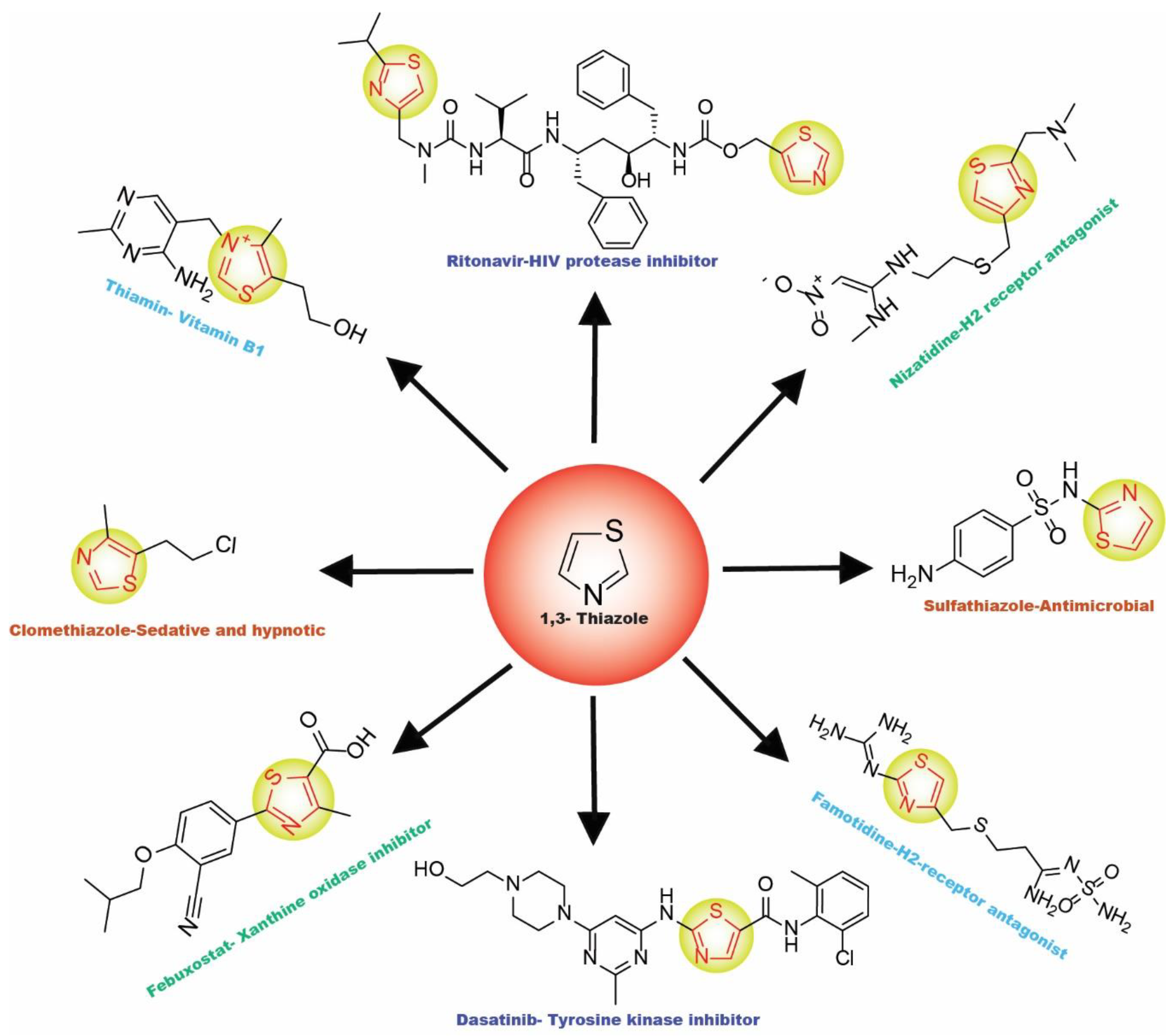
2. Thiazole-Derived Treatment Drugs
3. Thiazole Bearing Drug Candidates under Intensive Clinical Investigations
4. Thiazole-Bearing Compounds in Pre-Clinical Investigations
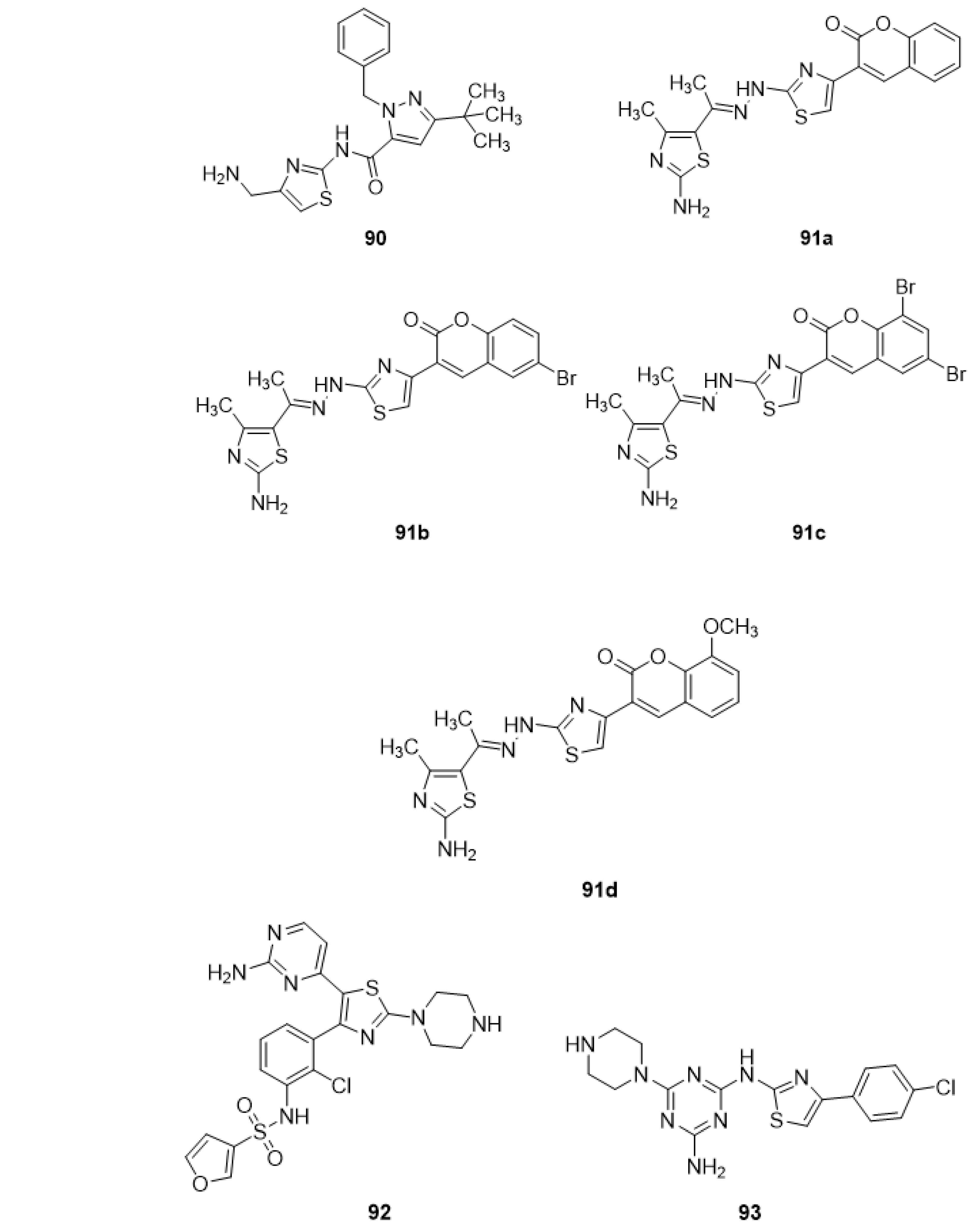
4.1. Anticonvulsant Activity
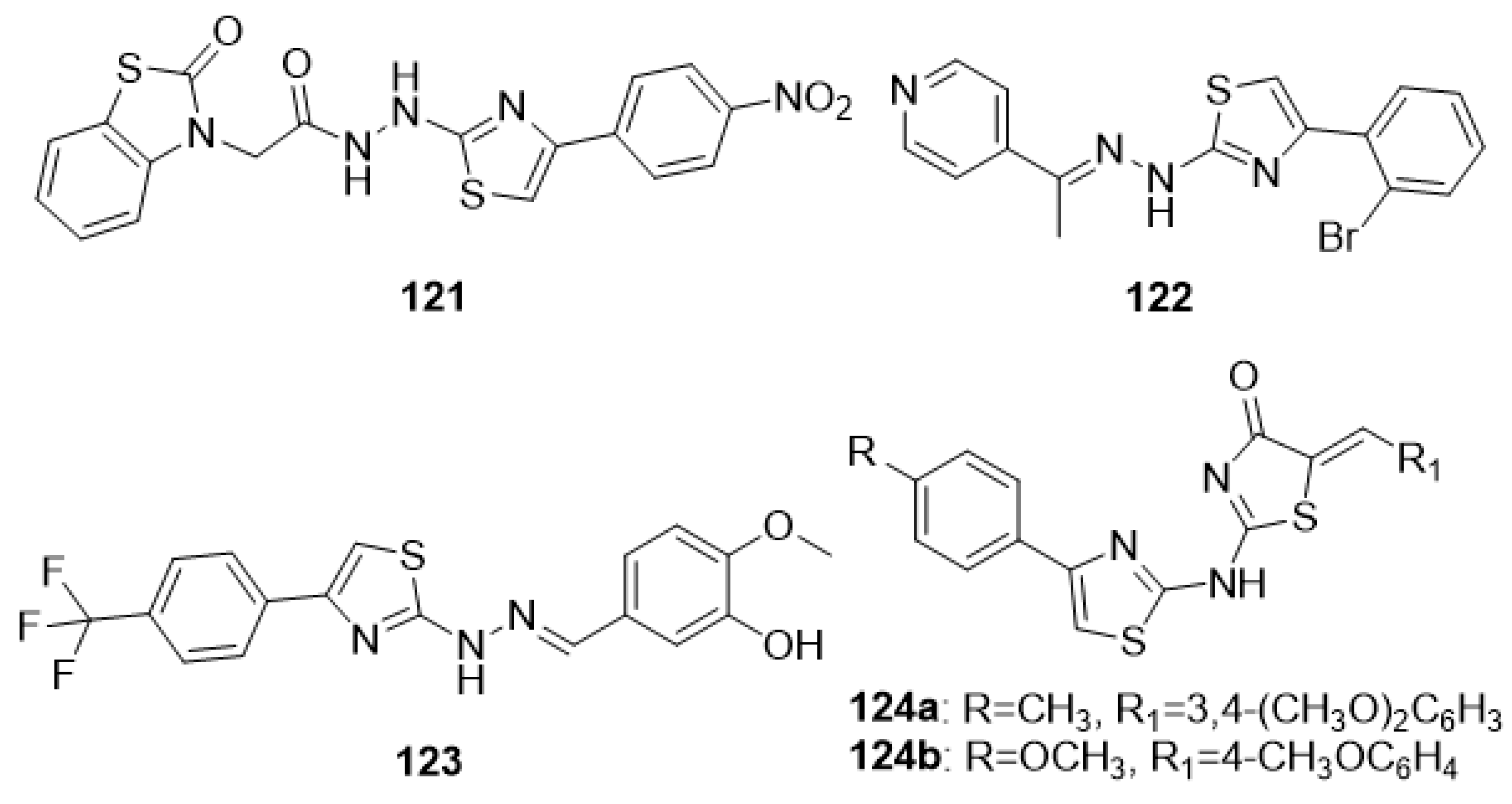
4.2. Thiazoles as Antitumor Agents
4.3. Thiazoles as Antimicrobial Agents
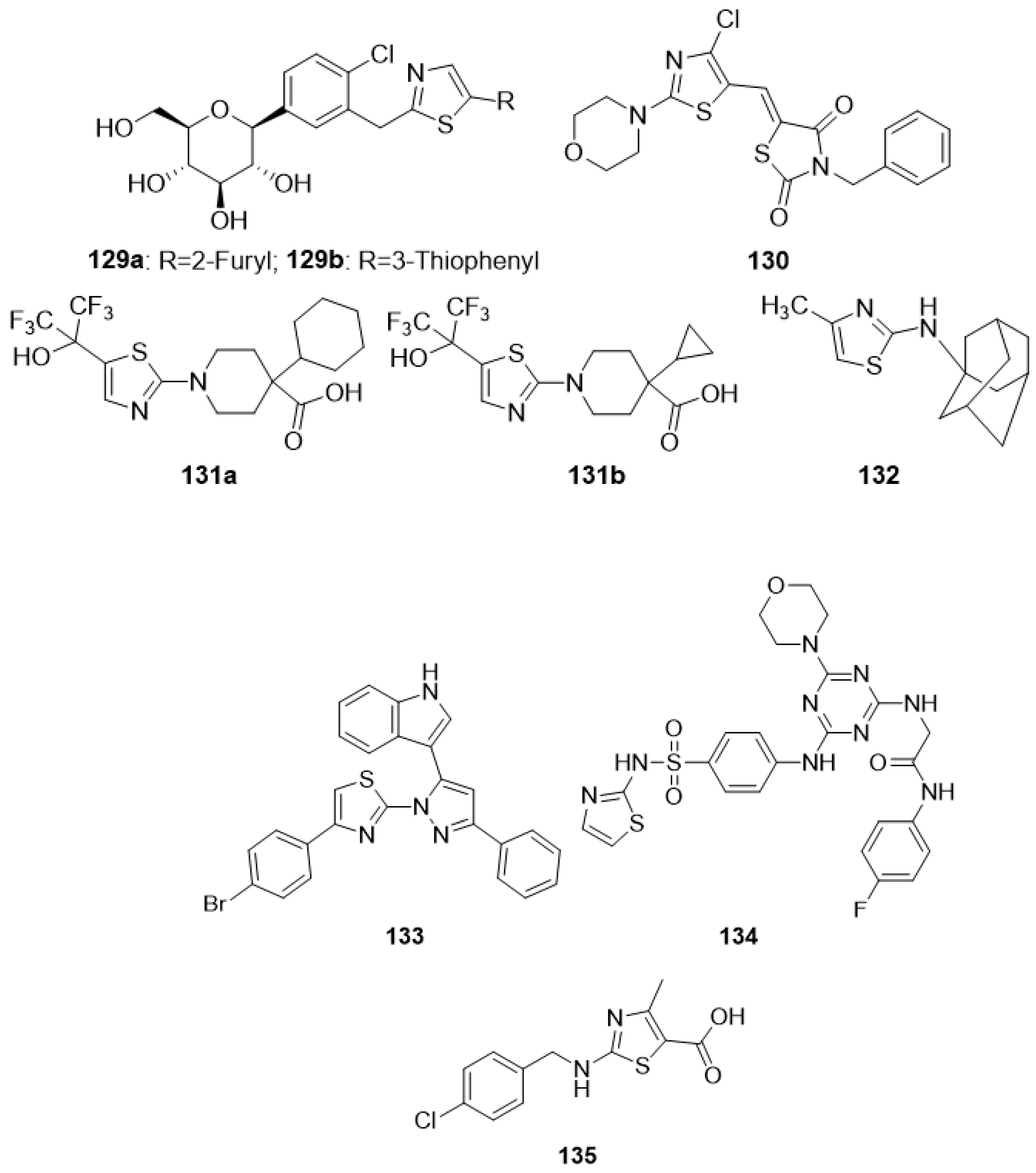
4.4. Thiazoles as Anti-Tubercular Agents
4.5. Thiazoles as Anti-Inflammatory Agents
4.6. Thiazoles as Antimalarial Agents
4.7. Thiazoles as Antiviral Agents
4.8. Thiazoles as Anti-Alzheimer Agents
4.9. Thiazoles as Anti-Diabetic Agents
4.10. Thiazoles as A1-Receptor Antagonist
4.11. Thiazoles as Bioactive (Antioxidant, Cardiotonic, Antithrombotic, Insecticide, and Anti-Repellent) Agents
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parthasarathy, A.; Borrego, E.J.; Savka, M.A.; Dobson, R.C.; Hudson, A.O. Amino acid–derived defense metabolites from plants: A potential source to facilitate novel antimicrobial development. J. Biol. Chem. 2021, 296, 100438. [Google Scholar] [CrossRef] [PubMed]
- Alajarín, M.; Cabrera, J.; Pastor, A.; Sánchez-Andrada, P.; Bautista, D. On the [2+2] Cycloaddition of 2-Aminothiazoles and Dimethyl Acetylenedicarboxylate. Experimental and Computational Evidence of a Thermal Disrotatory Ring Opening of Fused Cyclobutenes. J. Org. Chem. 2006, 71, 5328–5339. [Google Scholar] [CrossRef] [PubMed]
- Breitung, E.M.; Shu, A.C.-F.; McMahon, R.J. Thiazole and Thiophene Analogues of Donor−Acceptor Stilbenes: Molecular Hyperpolarizabilities and Structure−Property Relationships. J. Am. Chem. Soc. 2000, 122, 1154–1160. [Google Scholar] [CrossRef]
- Huang, Y.; Gan, H.; Li, S.; Xu, J.; Wu, X.; Yao, H. Oxidation of 4-carboxylate thiazolines to 4-carboxylate thiazoles by molecular oxygen. Tetrahedron Lett. 2010, 51, 1751–1753. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R.; Viggiani, L.; Zanirato, P. Photochemical Reactivity of 2-Azido-1,3-thiazole and 2-Azido-1,3-benzothiazole: A Procedure for the Aziridination of Enol Ethers. Eur. J. Org. Chem. 2009, 2009, 932–937. [Google Scholar] [CrossRef]
- D’Auria, M. Ab initio study on the photochemical isomerization of thiazole derivatives. Tetrahedron 2002, 58, 8037–8042. [Google Scholar] [CrossRef]
- Sadigova, S.E.; Magerramov, A.M.; Allakhverdiev, M.A.; Vekilova, T.M. Some Transformations of 2-Amino-4-phenyl-1,3-thiazole. Russ. J. Appl. Chem. 2004, 77, 787–789. [Google Scholar] [CrossRef]
- Shen, S.-S.; Lei, M.-Y.; Wong, Y.-X.; Tong, M.-L.; Teo, P.L.-Y.; Chiba, S.; Narasaka, K. Intramolecular nucleophilic substitution at an sp2 carbon: Synthesis of substituted thiazoles and imidazole-2-thiones. Tetrahedron Lett. 2009, 50, 3161–3163. [Google Scholar] [CrossRef]
- Pinto, M.; Takahata, Y.; Atvars, T. Photophysical properties of 2,5-diphenyl-thiazolo[5,4-d]thiazole. J. Photochem. Photobiol. A Chem. 2001, 143, 119–127. [Google Scholar] [CrossRef]
- Obushak, N.D.; Matiichuk, V.S.; Vasylyshin, R.Y.; Ostapyuk, Y.V. Heterocyclic Syntheses on the Basis of Arylation Products of Unsaturated Compounds: X. 3-Aryl-2-chloropropanals as Reagents for the Synthesis of 2-Amino-1,3-thiazole Derivatives. Russ. J. Org. Chem. 2004, 40, 383–389. [Google Scholar] [CrossRef]
- Arduengo III, A.J.; Goerlich, J.R.; Marshall, W.J. A Stable Thiazol-2-ylidene and Its Dimer. Eur. J. Org. Chem. 1997, 1997, 365–374. [Google Scholar] [CrossRef]
- Dawane, B.S.; Konda, S.G.; Kamble, V.T.; Chavan, S.A.; Bhosale, R.B.; M., S.B. Multicomponent One-Pot Synthesis of Substituted Hantzsch Thiazole Derivatives Under Solvent Free Conditions. E-J. Chem. 2009, 6, S358–S362. [Google Scholar] [CrossRef]
- Qiao, Q.; So, S.-S.; Goodnow, R.A. Stereochemical Control Factors in the Hantzsch Thiazole Synthesis: A Hammett Substitution Correlation Analysis. Org. Lett. 2001, 3, 3655–3658. [Google Scholar] [CrossRef]
- Li, J.J. Cook-Heilbron thiazole synthesis. In Name Reactions; Springer: Berlin/Heidelberg, Germany, 2003; p. 82. [Google Scholar] [CrossRef]
- Warburton, W.K. Arylthiazathiolium Salts And o-Aminoaryl Thiols—The Herz Reaction. Chem. Rev. 1957, 57, 1011–1020. [Google Scholar] [CrossRef]
- Aguilar, E.; Meyers, A. Reinvestigation of a modified Hantzsch thiazole synthesis. Tetrahedron Lett. 1994, 35, 2473–2476. [Google Scholar] [CrossRef]
- Venugopala, K.N. Design, Synthesis and Characterization of Benzothiazole Analogues as Promising Pharmacological Agents. J. Young- Pharm. 2017, 9, 158–161. [Google Scholar] [CrossRef] [Green Version]
- Abdu-Rahem, L.R.; Ahmad, A.K.; Abachi, F.T. Synthesis And Medicinal Attributes Of Thiazole Derivatives: A Review. Sys. Rev. Pharm. 2021, 12, 290–295. [Google Scholar]
- Ali, S.H.; Sayed, A.R. Review of the synthesis and biological activity of thiazoles. Synth. Commun. 2020, 51, 670–700. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Gümüş, M.; Yakan, M.; Koca, İ. Recent advances of thiazole hybrids in biological applications. Future Med. Chem. 2019, 11, 1979–1998. [Google Scholar] [CrossRef]
- Kashyap, S.J.; Garg, V.K.; Sharma, P.K.; Kumar, N.; Dudhe, R.; Gupta, J.K. Thiazoles: Having diverse biological activities. Med. Chem. Res. 2011, 21, 2123–2132. [Google Scholar] [CrossRef]
- Mishra, C.B.; Kumari, S.; Tiwari, M. Thiazole: A promising heterocycle for the development of potent CNS active agents. Eur. J. Med. Chem. 2015, 92, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Chhabria, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Siddiqui, N.; Arshad, M.F.; Ahsan, W.; Alam, M.S. Thiazoles: A valuable insight into the recent advances and biological activities. Int. J. Pharm. Sci. Drug Res. 2009, 1, 136–143. [Google Scholar]
- Bennett, J.P.; Piercey, M.F. Pramipexole—A new dopamine agonist for the treatment of Parkinson’s disease. J. Neurol. Sci. 1999, 163, 25–31. [Google Scholar] [CrossRef]
- Storch, A.; Burkhardt, K.; Ludolph, A.C.; Schwarz, J. Protective effects of riluzole on dopamine neurons: Involvement of oxidative stress and cellular energy metabolism. J. Neurochem. 2008, 75, 2259–2269. [Google Scholar] [CrossRef]
- Henry, J.; Hill, I. Fatal interaction between ritonavir and MDMA. Lancet 1998, 352, 1751–1752. [Google Scholar] [CrossRef]
- Romero, M.; Franzosi, M.G. Nizatidine. Medicina (Florence Italy) 1989, 9, 93–96. [Google Scholar]
- Meşeli, T.; Doğan, D.; Gündüz, M.G.; Kökbudak, Z.; Bogojevic, S.S.; Noonan, T.; Vojnovic, S.; Wolber, G.; Nikodinovic-Runic, J. Design, synthesis, antibacterial activity evaluation and molecular modeling studies of new sulfonamides containing a sulfathiazole moiety. New J. Chem. 2021, 45, 8166–8177. [Google Scholar] [CrossRef]
- Howard, J.; Chremos, A.; Collen, M.; Mcarthur, K.; Cherner, J.; Maton, P.; Ciarleglio, C.; Cornelius, M.; Gardner, J.; Jensen, R. Famotidine, a New, Potent, Long-Acting Histamine H2-Receptor Antagonist: Comparison With Cimetidine and Ranitidine in the Treatment of Zollinger-Ellison Syndrome. Gastroenterology 1985, 88, 1026–1033. [Google Scholar] [CrossRef]
- Hochhaus, A.; Kantarjian, H. The development of dasatinib as a treatment for chronic myeloid leukemia (CML): From initial studies to application in newly diagnosed patients. J. Cancer Res. Clin. Oncol. 2013, 139, 1971–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Valladares, I.; Khan, T.; Espinoza, L.R. Efficacy and safety of febuxostat in patients with hyperuricemia and gout. Ther. Adv. Musculoskelet. Dis. 2011, 3, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gann, H.; Hartig, K.; Feige, B.; Brueck, R.; Hohagen, F.; Weske, G.; Van Calker, D.; Riemann, D. The effects of clomethiazole on polysomnographically recorded sleep in healthy subjects. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 284–290. [Google Scholar] [CrossRef]
- Tracy, J.W.; Catto, A.B.; Webster, L.T. Reductive metabolism of niridazole by adult Schistosoma mansoni. Correlation with covalent drug binding to parasite macromolecules. Mol. Pharmacol. 1983, 24, 291–299. [Google Scholar]
- Barat, R.; Srinatha, A.; Pandit, J.K.; Ridhurkar, D.; Balasubramaniam, J.; Mittal, N.; Mishra, D.N. Niridazole Biodegradable Inserts for Local Long-Term Treatment of Periodontitis: Possible New Life for an Orphan Drug. Drug Deliv. 2006, 13, 365–373. [Google Scholar] [CrossRef]
- Marmo, E. Experimental and clinical pharmacology of fentiazac, a new, non-steroidal anti-inflammatory agent. Curr. Med. Res. Opin. 1979, 6, 53–63. [Google Scholar] [CrossRef]
- Dotevall, G.; Herner, B. Treatment of Acute Primidone Poisoning with Bemegride and Amiphenazole. Br. Med. J. 1957, 2, 451–452. [Google Scholar] [CrossRef] [Green Version]
- Borelli, C.; Schaller, M.; Niewerth, M.; Nocker, K.; Baasner, B.; Berg, D.; Tiemann, R.; Tietjen, K.; Fugmann, B.; Lang-Fugmann, S.; et al. Modes of Action of the New Arylguanidine Abafungin beyond Interference with Ergosterol Biosynthesis and in vitro Activity against Medically Important Fungi. Chemotherapy 2008, 54, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Hawtin, R.E.; Stockett, D.E.; Byl, J.A.W.; McDowell, R.S.; Tan, N.; Arkin, M.R.; Conroy, A.; Yang, W.; Osheroff, N.; Fox, J.A. Voreloxin Is an Anticancer Quinolone Derivative that Intercalates DNA and Poisons Topoisomerase II. PLoS ONE 2010, 5, e10186. [Google Scholar] [CrossRef]
- Hogan, M.; Bridgeman, M.B.; Min, G.H.; Dixit, D.; Bridgeman, P.J.; Narayanan, N. Effectiveness of empiric aztreonam compared to other beta-lactams for treatment of Pseudomonas aeruginosa infections. Infect. Drug. Resist. 2008, 11, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portugal, R.; Schaffel, R.; Almeida, L.; Spector, N.; Nucci, M. Thiabendazole for the prophylaxis of strongyloidiasis in immunosuppressed patients with hematological diseases: A randomized double-blind placebo-controlled study. Haematologica 2002, 87, 663–664. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, T.; Yan, H.; Guo, K.; Liu, Z.; Wei, L.; Lu, W.; Qiu, C.; Jiang, J. Fatostatin reverses progesterone resistance by inhibiting the SREBP1-NF-κB pathway in endometrial carcinoma. Cell Death Dis. 2021, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- NIH. Study to Evaluate the Efficacy, Safety, and Tolerability of Mirabegron in Older Adult Subjects with Overactive Bladder (OAB) (PILLAR). 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02216214 (accessed on 1 June 2021).
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.-M.; et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl] piperazin-1-yl]-8-methylimidazo [1, 2-a] pyridin-3-yl] methylamino]-4-(4-fluorophenyl) thiazole-5-carbonitrile (GLPG1690), a first-in-class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2017, 60, 3580–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. A Clinical Study to Test How Effective and Safe GLPG1690 is for Participants with Systemic Sclerosis (NOVESA). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03798366 (accessed on 1 June 2021).
- Lessene, G.; Czabotar, P.; Sleebs, B.; Zobel, K.; Lowes, K.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-XL inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef]
- Cheng, X.; Yoshida, H.; Raoofi, D.; Saleh, S.; Alborzinia, H.; Wenke, F.; Göhring, A.; Reuter, S.; Mah, N.; Fuchs, H.; et al. Ethyl 2-((4-Chlorophenyl)amino)thiazole-4-carboxylate and Derivatives Are Potent Inducers of Oct3/4. J. Med. Chem. 2015, 58, 5742–5750. [Google Scholar] [CrossRef]
- Naruse, T.; Aoki, M.; Fujimoto, N.; Arase, S.; Oura, H.; Ueda, Y.; Ikeda, A. Novel ALK5 inhibitor TP0427736 reduces TGF-β induced growth inhibition in human outer root sheath cells and elongates anagen phase in mouse hair follicles. Pharmacol. Rep. 2017, 69, 485–491. [Google Scholar] [CrossRef]
- NIH. Study of the Clinical Activity, Safety, and Tolerability of SRT2104 in Subjects with Moderate to Severe Plaque-Type Psoriasis. 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01154101 (accessed on 1 June 2021).
- NIH. A Clinical Trial to Assess the Safety of Oral SRT2104 and Its Effects on Vascular Dysfunction in Otherwise Healthy Cigarette Smokers and Subjects with Type 2 Diabetes Mellitus. 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT01031108 (accessed on 1 June 2021).
- Umar, S.; Palasiewicz, K.; Van Raemdonck, K.; Volin, M.V.; Romay, B.; Amin, M.A.; Zomorrodi, R.K.; Arami, S.; Gonzalez, M.; Rao, V.; et al. IRAK4 inhibition: A promising strategy for treating RA joint inflammation and bone erosion. Cell. Mol. Immunol. 2020, 18, 2199–2210. [Google Scholar] [CrossRef]
- Haasbach, E.; Reiling, S.J.; Ehrhardt, C.; Droebner, K.; Rückle, A.; Hrincius, E.R.; Leban, J.; Strobl, S.; Vitt, D.; Ludwig, S.; et al. The NF-kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza A virus. Antivir. Res. 2013, 99, 336–344. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Deater, M.; Schubert, K.; Marquez-Loza, L.; Pelz, C.; Sinclair, D.A.; Grompe, M. The Sirt1 activator SRT3025 expands hematopoietic stem and progenitor cells and improves hematopoiesis in Fanconi anemia mice. Stem Cell Res. 2015, 15, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Ndagi, U.; Abdullahi, M.; Hamza, A.N.; Soliman, M.E. An analogue of a kinase inhibitor exhibits subjective characteristics that contribute to its inhibitory activities as a potential anti-cancer candidate: Insights through computational biomolecular modelling of UM-164 binding with lyn protein. RSC Adv. 2019, 10, 145–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Otaibi, F. An overview of structurally diversified anticonvulsant agents. Acta Pharm. 2019, 69, 321–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissdorfer, M.; Venugopala, K.N.; Venugopala, R.; ILIŞ, M.; Badea, F.D. Lipophilicity of schiff bases of substituted thiazolyl bromocoumarins by chembiodraw–perkinelmer and alogps 2.1. UPB Sci. Bull. B Chem. Mater. Sci. 2014, 76, 113–124. [Google Scholar]
- Farag, A.A.; Abd-Alrahman, S.N.; Ahmed, G.F.; Ammar, R.M.; Ammar, Y.A.; Abbas, S.Y. Synthesis of Some Azoles Incorporating a Sulfonamide Moiety as Anticonvulsant Agents. Arch. Der Pharm. 2012, 345, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Ghabbour, H.A.; Kadi, A.; Eltahir, K.E.H.; Angawi, R.F.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular docking studies of thiazole-based pyrrolidinones and isoindolinediones as anticonvulsant agents. Med. Chem. Res. 2015, 24, 3194–3211. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Sałat, K.; Misiura, K.; Podkowa, A.; Malikowska, N. Synthesis and anticonvulsant activities of novel 2-(cyclopentylmethylene)hydrazinyl-1,3-thiazoles in mouse models of seizures. J. Enzym. Inhib. Med. Chem. 2016, 31, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Mishchenko, M.; Shtrygol, S.; Kaminskyy, D.; Lesyk, R. Thiazole-Bearing 4-Thiazolidinones as New Anticonvulsant Agents. Sci. Pharm. 2020, 88, 16. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, A.A.; Partap, S.; Khisal, S.; Yar, M.S.; Mishra, R. Synthesis, anti-convulsant activity and molecular docking study of novel thiazole pyridazinone hybrid analogues. Bioorganic Chem. 2020, 99, 103584. [Google Scholar] [CrossRef]
- Ahangar, N.; Ayati, A.; Alipour, E.; Pashapour, A.; Foroumadi, A.; Emami, S. 1-[(2-Arylthiazol-4-yl)methyl]azoles as a New Class of Anticonvulsants: Design, Synthesis, In vivo Screening, and In silico Drug-like Properties. Chem. Biol. Drug Des. 2011, 78, 844–852. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Konklewska, N.; Biernasiuk, A.; Malm, A.; Sałat, K.; Furgała, A.; Dzitko, K.; Bekier, A.; Baranowska-Łączkowska, A.; Paneth, A. Thiazoles with cyclopropyl fragment as antifungal, anticonvulsant, and anti-Toxoplasma gondii agents: Synthesis, toxicity evaluation, and molecular docking study. Med. Chem. Res. 2018, 27, 2125–2140. [Google Scholar] [CrossRef] [Green Version]
- Gomha, S.M.; Abdelaziz, M.R.; Kheder, N.A.; Abdel-Aziz, H.M.; Alterary, S.; Mabkhot, Y.N. A facile access and evaluation of some novel thiazole and 1,3,4-thiadiazole derivatives incorporating thiazole moiety as potent anticancer agents. Chem. Central J. 2017, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S.; Edrees, M.M.; Salem, H.H.; Kheder, N.A.; Gomha, S.M.; Abdelaziz, M.R. Synthesis and Biological Evaluation of Some Novel Thiazole-Based Heterocycles as Potential Anticancer and Antimicrobial Agents. Molecules 2019, 24, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anuradha; Patel, S.K.; Patle , R.K.; Parameswaran, P.; Jain, A.; Shard, A. Design, computational studies, synthesis and biological evaluation of thiazole-based molecules as anticancer agents. Eur. J. Pharm. Sci. 2019, 134, 20–30. [Google Scholar] [CrossRef]
- Evren, A.E.; Yurttas, L.; Ekselli, B.; Akalin-Ciftci, G. Synthesis and biological evaluation of 5-methyl-4-phenyl thiazole derivatives as anticancer agents. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 820–828. [Google Scholar] [CrossRef]
- Finiuk, N.S.; Hreniuh, V.P.; Ostapiuk, Y.V.; Matiychuk, V.S.; Frolov, D.A.; Obushak, M.; Stoika, R.S.; Babsky, A.M. Antineoplastic activity of novel thiazole derivatives. Biopolym. Cell 2017, 33, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Vaddula, B.R.; Tantak, M.P.; Sadana, R.; Gonzalez, M.; Kumar, D. One-pot synthesis and in-vitro anticancer evaluation of 5-(2′-indolyl)thiazoles. Sci. Rep. 2016, 6, 23401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-H.; Wu, H.-M.; Deng, S.-N.; Cai, X.-Y.; Yao, Y.; Mwenda, M.C.; Wang, J.-Y.; Cai, D.; Chen, Y. Design, Synthesis, and Anticancer Activities of Novel 2-Amino-4-phenylthiazole Scaffold Containing Amide Moieties. J. Chem. 2018, 2018, 4301910. [Google Scholar] [CrossRef]
- Bayazeed, A.A.; Alnoman, R.B. Synthesis of New Thiazole-Pyridine Hybrids and Their Anticancer Activity. Russ. J. Gen. Chem. 2020, 90, 2004–2011. [Google Scholar] [CrossRef]
- Aly, A.A.; Mohamed, A.H.; Ramadan, M. Synthesis and colon anticancer activity of some novel thiazole/-2-quinolone derivatives. J. Mol. Struct. 2020, 1207, 127798. [Google Scholar] [CrossRef]
- Sayed, A.R.; Gomha, S.M.; Taher, A.E.; Muhammad, A.Z.; El-Seedi, H.R.; Gaber, H.M.; Ahmed, M.M. One-Pot Synthesis of Novel Thiazoles as Potential Anti-Cancer Agents. Drug Des. Dev. Ther. 2020, 14, 1363–1375. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Xu, Q.; Xu, J.; Wu, Y.; Wang, Y.; Zuo, D.; Guan, Q.; Bao, K.; Wang, J.; Wu, Y.; et al. Synthesis and bioevaluation of N,4-diaryl-1,3-thiazole-2-amines as tubulin inhibitors with potent antiproliferative activity. PLoS ONE 2017, 12, e0174006. [Google Scholar] [CrossRef]
- Sayed, A.R.; Gomha, S.M.; Abdelrazek, F.M.; Farghaly, M.S.; Hassan, S.A.; Metz, P. Design, efficient synthesis and molecular docking of some novel thiazolyl-pyrazole derivatives as anticancer agents. BMC Chem. 2019, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, T.A.R.; da Silva, A.C.; Silva, E.B.; Gomes, P.A.T.D.M.; Espíndola, J.W.P.; Cardoso, M.V.D.O.; Moreira, D.R.; Leite, A.C.L.; Pereira, V.R. Antitumor and immunomodulatory activities of thiosemicarbazones and 1,3-Thiazoles in Jurkat and HT-29 cells. Biomed. Pharmacother. 2016, 82, 555–560. [Google Scholar] [CrossRef]
- Becan, L.; Pyra, A.; Rembiałkowska, N.; Bryndal, I. Synthesis, Structural Characterization and Anticancer Activity of New 5-Trifluoromethyl-2-thioxo-thiazolo[4,5-d]pyrimidine Derivatives. Pharmaceuticals 2022, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.M.; Bayazeed, A.A. Synthesis and antiproliferative activity studies of new functionalized pyridine linked thiazole derivatives. Arab. J. Chem. 2020, 14, 102914. [Google Scholar] [CrossRef]
- Al-Jaidi, B.A.; Deb, P.K.; Telfah, S.T.; Dakkah, A.N.; Bataineh, Y.A.; Aga, Q.A.A.K.; Al-Dhoun, M.A.; Al-Subeihi, A.A.A.; Odetallah, H.M.; Bardaweel, S.K.; et al. Synthesis and evaluation of 2,4,5-trisubstitutedthiazoles as carbonic anhydrase-III inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1483–1490. [Google Scholar] [CrossRef]
- Arshad, M.F.; Al-Otaibi, F.; Kumar, S.; Nagarethinam, S.; Elkerdasy, A.; Upmanyu, N. Discovery of novel β-Lactum Amalgamated 4-Methylthaizole-5-Carboxylic acid derivatives as potential agents. Acta Pol. Pharm. 2017, 74, 1699–1709. [Google Scholar]
- Bikobo, D.S.N.; Vodnar, D.C.; Stana, A.; Tiperciuc, B.; Nastasă, C.; Douchet, M.; Oniga, O. Synthesis of 2-phenylamino-thiazole derivatives as antimicrobial agents. J. Saudi Chem. Soc. 2017, 21, 861–868. [Google Scholar] [CrossRef]
- Liaras, K.; Geronikaki, A.; Glamočlija, J.; Ćirić, A.; Soković, M. Thiazole-based aminopyrimidines and N-phenylpyrazolines as potent antimicrobial agents: Synthesis and biological evaluation. MedChemComm 2014, 5, 915–922. [Google Scholar] [CrossRef]
- El-Sayed, E.H.; Fadda, A.A. Synthesis and Antimicrobial Activity of Some Novel bis Polyfunctional Pyridine, Pyran, and Thiazole Derivatives. J. Heterocycl. Chem. 2018, 55, 2251–2260. [Google Scholar] [CrossRef]
- Mohammad, H.; Cushman, M.; Seleem, M. Antibacterial Evaluation of Synthetic Thiazole Compounds In Vitro and In Vivo in a Methicillin-Resistant Staphylococcus aureus (MRSA) Skin Infection Mouse Model. PLoS ONE 2015, 10, e0142321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlThagafi, I.; El-Metwaly, N.; Farghaly, T.A. New Series of Thiazole Derivatives: Synthesis, Structural Elucidation, Antimicrobial Activity, Molecular Modeling and MOE Docking. Molecules 2019, 24, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Kumar, S.; Narasimhan, B.; Ramasamy, K.; Lim, S.M.; Shah, S.A.A.; Mani, V. 4-(4-Bromophenyl)-thiazol-2-amine derivatives: Synthesis, biological activity and molecular docking study with ADME profile. BMC Chem. 2019, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Desai, N.; Makwana, A.H.; Rajpara, K. Synthesis and study of 1,3,5-triazine based thiazole derivatives as antimicrobial agents. J. Saudi Chem. Soc. 2016, 20, S334–S341. [Google Scholar] [CrossRef] [Green Version]
- Khidre, R.E.; Radini, I.A.M. Design, synthesis and docking studies of novel thiazole derivatives incorporating pyridine moiety and assessment as antimicrobial agents. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Li, Y.; Sun, N.; Ser, H.-L.; Long, W.; Li, Y.; Chen, C.; Zheng, B.; Huang, X.; Liu, Z.; Lu, Y.-J. Antibacterial activity evaluation and mode of action study of novel thiazole-quinolinium derivatives. RSC Adv. 2020, 10, 15000–15014. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Kolokotroni, A.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Kostic, M.; Sokovic, M.; Carazo, A.; Mladěnka, P.; et al. 5-Benzyliden-2-(5-methylthiazol-2-ylimino)thiazolidin-4-ones as Antimicrobial Agents. Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Antibiotics 2021, 10, 309. [Google Scholar] [CrossRef]
- Morsy, M.A.; Ali, E.M.; Kandeel, M.; Venugopala, K.N.; Nair, A.B.; Greish, K.; El-Daly, M. Screening and Molecular Docking of Novel Benzothiazole Derivatives as Potential Antimicrobial Agents. Antibiotics 2020, 9, 221. [Google Scholar] [CrossRef]
- Tratrat, C.; Haroun, M.; Xenikakis, I.; Liaras, K.; Tsolaki, E.; Eleftheriou, P.; Petrou, A.; Aldhubiab, B.; Attimarad, M.; Venugopala, K.N.; et al. Design, Synthesis, Evaluation of Antimicrobial Activity and Docking Studies of New Thiazole-based Chalcones. Curr. Top. Med. Chem. 2019, 19, 356–375. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Kositzi, K.; Tsolaki, E.; Petrou, A.; Aldhubiab, B.; Attimarad, M.; Harsha, S.; Geronikaki, A.; Venugopala, K.; et al. New Benzothiazole-based Thiazolidinones as Potent Antimicrobial Agents. Design, synthesis and Biological Evaluation. Curr. Top. Med. Chem. 2018, 18, 75–87. [Google Scholar] [CrossRef]
- Pitta, E.; Tsolaki, E.; Geronikaki, A.; Petrović, J.; Glamočlija, J.; Soković, M.; Crespan, E.; Maga, G.; Bhunia, S.S.; Saxena, A.K. 4-Thiazolidinone derivatives as potent antimicrobial agents: Microwave-assisted synthesis, biological evaluation and docking studies. MedChemComm 2014, 6, 319–326. [Google Scholar] [CrossRef]
- Reddy, D.S.; Hosamani, K.M.; Devarajegowda, H.C.; Kurjogi, M.M. A facile synthesis and evaluation of new biomolecule-based coumarin–thiazoline hybrids as potent anti-tubercular agents with cytotoxicity, DNA cleavage and X-ray studies. RSC Adv. 2015, 5, 64566–64581. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, R.; Kachroo, M. Synthesis and biological evaluation of anti-tubercular activity of Schiff bases of 2-Amino thiazoles, Bioorg. Med. Chem. Lett. 2020, 30, 3166. [Google Scholar] [CrossRef]
- Abhale, Y.K.; Sasane, A.V.; Chavan, A.P.; Deshmukh, K.K.; Kotapalli, S.S.; Ummanni, R.; Sayyad, S.F.; Mhaske, P.C. Synthesis and biological screening of 2′-aryl/benzyl-2-aryl-4-methyl-4′,5-bithiazolyls as possible anti-tubercular and antimicrobial agents. Eur. J. Med. Chem. 2015, 94, 340–347. [Google Scholar] [CrossRef]
- Shaikh, M.S.; Palkar, M.B.; Patel, H.M.; Rane, R.A.; Alwan, W.S.; Shaikh, M.M.; Shaikh, I.M.; Hampannavar, G.A.; Karpoormath, R. Design and synthesis of novel carbazolo–thiazoles as potential anti-mycobacterial agents using a molecular hybridization approach. RSC Adv. 2014, 4, 62308–62320. [Google Scholar] [CrossRef]
- Jagadale, S.M.; Abhale, Y.K.; Pawar, H.R.; Shinde, A.; Bobade, V.D.; Chavan, A.P.; Sarkar, D.; Mhaske, P.C. Synthesis of New Thiazole and Pyrazole Clubbed 1,2,3-Triazol Derivatives as Potential Antimycobacterial and Antibacterial Agents. Polycycl. Aromat. Compd. 2020, 1–22. [Google Scholar] [CrossRef]
- Karale, U.B.; Krishna, V.S.; Krishna, E.V.; Choudhari, A.S.; Shukla, M.; Gaikwad, V.R.; Mahizhaveni, B.; Chopra, S.; Misra, S.; Sarkar, D.; et al. Synthesis and biological evaluation of 2,4,5-trisubstituted thiazoles as antituberculosis agents effective against drug-resistant tuberculosis. Eur. J. Med. Chem. 2019, 178, 315–328. [Google Scholar] [CrossRef]
- Kesicki, E.A.; Bailey, M.A.; Ovechkina, Y.; Early, J.V.; Alling, T.; Bowman, J.; Zuniga, E.S.; Dalai, S.; Kumar, N.; Masquelin, T.; et al. Synthesis and Evaluation of the 2-Aminothiazoles as Anti-Tubercular Agents. PLoS ONE 2016, 11, e0155209. [Google Scholar] [CrossRef] [Green Version]
- Abo-Ashour, M.F.; Eldehna, W.M.; George, R.F.; Abdel-Aziz, M.M.; Elaasser, M.M.; Abou-Seri, S.M.; Gawad, N.M.A. Synthesis and biological evaluation of 2-aminothiazole-thiazolidinone conjugates as potential antitubercular agents. Future Med. Chem. 2018, 10, 1405–1419. [Google Scholar] [CrossRef]
- Makam, P.; Kankanala, R.; Prakash, A.; Kannan, T. 2-(2-Hydrazinyl)thiazole derivatives: Design, synthesis and in vitro antimycobacterial studies. Eur. J. Med. Chem. 2013, 69, 564–576. [Google Scholar] [CrossRef]
- Kumar, G.V.S.; Prasad, Y.R.; Chandrashekar, S.M. Synthesis and pharmacological evaluation of some novel 4-isopropyl thiazole-based sulfonyl derivatives as potent antimicrobial and antitubercular agents. Med. Chem. Res. 2013, 22, 4239–4252. [Google Scholar] [CrossRef]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo [2, 1-b] thiazole-5-carboxamide anti-tuberculosis compounds. PLoS ONE 2020, 15, e0227224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopala, K.N.; Khedr, M.A.; Pillay, M.; Nayak, S.K.; Chandrashekharappa, S.; Aldhubiab, B.E.; Harsha, S.; Attimard, M.; Odhav, B. Benzothiazole analogs as potential anti-TB agents: Computational input and molecular dynamics. J. Biomol. Struct. Dyn. 2018, 37, 1830–1842. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Pillay, M.; Bhandary, S.; Kandeel, M.; Mahomoodally, F.M.; Morsy, M.A.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; et al. Synthesis and Structural Elucidation of Novel Benzothiazole Derivatives as Anti-tubercular Agents: In-silico Screening for Possible Target Identification. Med. Chem. 2019, 15, 311–326. [Google Scholar] [CrossRef]
- Roy, K.K.; Singh, S.; Sharma, S.K.; Srivastava, R.; Chaturvedi, V.; Saxena, A.K. Synthesis and biological evaluation of substituted 4-arylthiazol-2-amino derivatives as potent growth inhibitors of replicating Mycobacterium tuberculosis H37RV. Bioorganic Med. Chem. Lett. 2011, 21, 5589–5593. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, A.H.; Habash, M.; Maghrabi, I.A.; Taha, M.O. Synthesis and evaluation of novel diphenylthiazole derivatives as potential anti-inflammatory agents. Med. Chem. Res. 2015, 24, 3681–3695. [Google Scholar] [CrossRef]
- Kamble, R.D.; Meshram, R.J.; Hese, S.V.; More, R.A.; Kamble, S.S.; Gacche, R.N.; Dawane, B.S. Synthesis and in silico investigation of thiazoles bearing pyrazoles derivatives as anti-inflammatory agents. Comput. Biol. Chem. 2016, 61, 86–96. [Google Scholar] [CrossRef]
- Khillare, L.D.; Bhosle, M.R.; Deshmukh, A.R.; Mane, R.A. Synthesis and anti-inflammatory evaluation of new pyrazoles bearing biodynamic thiazole and thiazolidinone scaffolds. Med. Chem. Res. 2014, 24, 1380–1386. [Google Scholar] [CrossRef]
- Tiperciuc, B.; Pârvu, A.; Tamaian, R.; Nastasă, C.M.; Ionuţ, I.; Oniga, O. New anti-inflammatory thiazolyl-carbonyl-thiosemicarbazides and thiazolyl-azoles with antioxidant properties as potential iNOS inhibitors. Arch. Pharmacal Res. 2013, 36, 702–714. [Google Scholar] [CrossRef]
- Haiba, M.E.; El-Karim, S.S.A.; Gouhar, R.S.; El-Zahar, M.I.; El-Awdan, S.A. Synthesis and evaluation of anti-inflammatory and analgesic activity of some substituted thiazolyl and thaizolidinonyl tetrahydronapthalene derivatives. Med. Chem. Res. 2014, 23, 3418–3435. [Google Scholar] [CrossRef]
- Chandak, N.; Kumar, P.; Kaushik, P.; Varshney, P.; Sharma, C.; Kaushik, D.; Jain, S.; Aneja, K.R.; Sharma, P. Dual evaluation of some novel 2-amino-substituted coumarinylthiazoles as anti-inflammatory-antimicrobial agents and their docking studies with COX-1/COX-2 active sites. J. Enzym. Inhib. Med. Chem. 2013, 29, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Yum, E.K.; Cheon, H.G.; Cho, Y.S. Synthesis and Biological Evaluation of N-aryl-4-aryl-1,3-Thiazole-2-Amine Derivatives as Direct 5-Lipoxygenase Inhibitors. Chem. Biol. Drug Des. 2012, 80, 89–98. [Google Scholar] [CrossRef]
- Sherif, Y.E.-S.; El-Asmy, A.A.-H.; Lotfy, M. 4-Hydroxy-2-methyl-N-(2-thiazole)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide (EX15) and its Cu(II) Complex as New Oxicam Selective Cyclooxygenase-2 Inhibitors. Croat. Chem. Acta 2012, 85, 19–26. [Google Scholar] [CrossRef]
- Kamat, V.; Santosh, R.; Poojary, B.; Nayak, S.P.; Kumar, B.K.; Sankaranarayanan, M.; Faheem; Khanapure, S.; Barretto, D.A.; Vootla, S.K. Pyridine- and Thiazole-Based Hydrazides with Promising Anti-inflammatory and Antimicrobial Activities along with Their In Silico Studies. ACS Omega 2020, 5, 25228–25239. [Google Scholar] [CrossRef]
- Moldovan, C.M.; Oniga, O.; Pârvu, A.; Tiperciuc, B.; Verite, P.; Pirnau, A.; Crişan, O.; Bojiţă, M.; Pop, R. Synthesis and anti-inflammatory evaluation of some new acyl-hydrazones bearing 2-aryl-thiazole. Eur. J. Med. Chem. 2011, 46, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Tratrat, C.; Haroun, M.; Paparisva, A.; Kamoutsis, C.; Petrou, A.; Gavalas, A.; Eleftheriou, P.; Geronikaki, A.; Venugopala, K.; Kochkar, H.; et al. New Substituted 5-Benzylideno-2-Adamantylthiazol[3,2-b][1,2,4]Triazol-6(5H)ones as Possible Anti-Inflammatory Agents. Molecules 2021, 26, 659. [Google Scholar] [CrossRef]
- Gore, V.A.; Tekale, S.U.; Bhale, S.P.; Rajani, D.P.; Domb, A.J.; Pawar, R.P. Synthesis and biological evaluation of novel thiazole hydrazines as antimicrobial and antimalarial agents. Lett. Appl. Nano. Biosci. 2021, 10, 1846–1855. [Google Scholar] [CrossRef]
- Sahu, S.; Ghosh, S.K.; Gahtori, P.; Singh, U.P.; Bhattacharyya, D.R.; Bhat, H.R. In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor. Pharmacol. Rep. 2019, 71, 762–767. [Google Scholar] [CrossRef]
- Makam, P.; Thakur, P.K.; Kannan, T. In vitro and in silico antimalarial activity of 2-(2-hydrazinyl)thiazole derivatives. Eur. J. Pharm. Sci. 2014, 52, 138–145. [Google Scholar] [CrossRef]
- Guimarães, D.S.M.; Luz, L.S.D.S.; Nascimento, S.B.D.; Silva, L.R.; Martins, N.R.D.M.; de Almeida, H.G.; Reis, V.D.S.; Maluf, S.E.C.; Budu, A.; Marinho, J.A.; et al. Improvement of antimalarial activity of a 3-alkylpiridine alkaloid analog by replacing the pyridine ring to a thiazole-containing heterocycle: Mode of action, mutagenicity profile, and Caco-2 cell-based permeability. Eur. J. Pharm. Sci. 2019, 138, 105015. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Hassan, A.M.; El Razik, H.A.A.; El-Miligy, M.M.; El-Agroudy, E.J.; Bekhit, A.E.-D.A. New heterocyclic hybrids of pyrazole and its bioisosteres: Design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015, 94, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.G.; Douelle, F.; Feng, T.-S.; Nchinda, A.T.; Younis, Y.; White, K.L.; Wu, Q.; Ryan, E.; Burrows, J.N.; Waterson, D.; et al. Novel Orally Active Antimalarial Thiazoles. J. Med. Chem. 2011, 54, 7713–7719. [Google Scholar] [CrossRef] [PubMed]
- Sujatha, K.; Ommi, N.B.; Mudiraj, A.; Babu, P.P.; Vedula, R.R. Synthesis of thiazolyl hydrazonothiazolamines and 1,3,4-thiadiazinyl hydrazonothiazolamines as a class of antimalarial agents. Arch. der Pharm. 2019, 352, e1900079. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Heras-Dueña, L.D.L.; Mata-Cantero, L.; Diaz-Hernandez, B.; Vazquez-Muñiz, M.-J.; Ghidelli-Disse, S.; Drewes, G.; Fernandez-Alvaro, E.; Baker, D.A. High-throughput screening of the Plasmodium falciparum cGMP-dependent protein kinase identified a thiazole scaffold which kills erythrocytic and sexual stage parasites. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Kalita, J.M.; Ghosh, S.K.; Sahu, S.; Dutta, M. Rational design and microwave assisted synthesis of some novel phenyl thiazolyl clubbed s-triazine derivatives as antimalarial antifolate. Futur. J. Pharm. Sci. 2017, 3, 11–17. [Google Scholar] [CrossRef]
- Bueno, J.M.; Carda, M.; Crespo, B.; Cuñat, A.C.; de Cozar, C.; León, M.L.; Marco, J.A.; Roda, N.; Sanz-Cervera, J.F. Design, synthesis and antimalarial evaluation of novel thiazole derivatives. Bioorganic Med. Chem. Lett. 2016, 26, 3938–3944. [Google Scholar] [CrossRef]
- Sharma, I.; Sullivan, M.; McCutchan, T.F. In Vitro Antimalarial Activity of Novel Semisynthetic Nocathiacin I Antibiotics. Antimicrob. Agents Chemother. 2015, 59, 3174–3179. [Google Scholar] [CrossRef] [Green Version]
- Osman, H.; Yusufzai, S.K.; Khan, M.S.; Abd Razik, B.M.; Sulaiman, O.; Mohamad, S.; Gansau, J.A.; Ezzat, M.O.; Parumasivam, T.; Hassan, M.Z. New thiazolyl-coumarin hybrids: Design, synthesis, characterization, X-ray crystal structure, antibacterial and antiviral evaluation. J. Mol. Struct. 2018, 1166, 147–154. [Google Scholar] [CrossRef]
- Abu-Melha, S.; Edrees, M.M.; Riyadh, S.M.; Abdelaziz, M.R.; ElFiky, A.A.; Gomha, S.M. Clean grinding technique: A facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules 2020, 25, 4565. [Google Scholar] [CrossRef]
- Pacca, C.C.; Marques, R.E.; Espindola, J.W.P.; Filho, G.B.; Leite, A.C.L.; Teixeira, M.M.; Nogueira, M.L. Thiosemicarbazones and Phthalyl-Thiazoles compounds exert antiviral activity against yellow fever virus and Saint Louis encephalitis virus. Biomed. Pharmacother. 2017, 87, 381–387. [Google Scholar] [CrossRef]
- Madni, M.; Hameed, S.; Ahmed, M.N.; Tahir, M.N.; Al-Masoudi, N.A.; Pannecouque, C. Synthesis, crystal structure, anti-HIV, and antiproliferative activity of new pyrazolylthiazole derivatives. Med. Chem. Res. 2017, 26, 2653–2665. [Google Scholar] [CrossRef]
- Ke, S.; Li, N.; Ke, T.; Shi, L.; Zhang, Z.; Fang, W.; Zhang, Y.-N.; Wang, K.; Zhou, R.; Wan, Z.; et al. Synthesis and evaluation of steroidal thiazoline conjugates as potential antiviral agents. Future Med. Chem. 2018, 10, 2589–2605. [Google Scholar] [CrossRef] [PubMed]
- Mayhoub, A.S.; Khaliq, M.; Kuhn, R.J.; Cushman, M. Design, Synthesis, and Biological Evaluation of Thiazoles Targeting Flavivirus Envelope Proteins. J. Med. Chem. 2011, 54, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Mayhoub, A.; Khaliq, M.; Botting, C.; Li, Z.; Kuhn, R.J.; Cushman, M. An investigation of phenylthiazole antiflaviviral agents. Bioorganic Med. Chem. 2011, 19, 3845–3854. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-M.; Wang, J.-C.; Hu, H.-S.; Wu, P.-S.; Yang, C.-C.; Wu, C.-P.; Pu, S.-Y.; Hsu, T.-A.; Jiaang, W.-T.; Chao, Y.-S.; et al. Resistance Analysis and Characterization of a Thiazole Analogue, BP008, as a Potent Hepatitis C Virus NS5A Inhibitor. Antimicrob. Agents Chemother. 2012, 56, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Dawood, K.M.; Eldebss, T.M.; El-Zahabi, H.S.; Yousef, M.H. Synthesis and antiviral activity of some new bis-1,3-thiazole derivatives. Eur. J. Med. Chem. 2015, 102, 266–276. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Salakhutdinov, N.F. Synthesis and antiviral activity of camphor-based 1, 3-thiazolidin-4-one and thiazole derivatives as Orthopoxvirus-reproduction inhibitors. Med. Chem. Comm. 2018, 9, 1746–1753. [Google Scholar] [CrossRef]
- Duan, L.-M.; Yu, H.-Y.; Li, Y.-L.; Jia, C.-J. Design and discovery of 2-(4-(1H-tetrazol-5-yl)-1H-pyrazol-1-yl)-4-(4-phenyl) thiazole derivatives as cardiotonic agents via inhibition of PDE3. Bioorg. Med. Chem. 2015, 23, 6111–6117. [Google Scholar] [CrossRef]
- Shi, D.-H.; Ma, X.-D.; Liu, Y.-W.; Min, W.; Yin, F.-J.; Tang, Z.-M.; Song, M.-Q.; Lu, C.; Song, X.-K.; Liu, W.-W.; et al. Synthesis, Crystal Structure and Biological Evaluation of Novel 2-Phenylthiazole Derivatives as Butyrylcholinesterase Inhibitors. J. Chem. Res. 2018, 42, 366–370. [Google Scholar] [CrossRef]
- Sağlık, B.N.; Levent, S.; Osmaniye, D.; Çevik, U.A.; Çavuşoğlu, B.K.; Özkay, Y.; Koparal, A.S.; Kaplancıklı, Z.A. Design, Synthesis, and Biological Activity Evaluation of New Donepezil-like Compounds Bearing Thiazole Ring for the Treatment of Alzheimer’s Disease. Crystals 2020, 10, 637. [Google Scholar] [CrossRef]
- Yurttaş, L.; Kaplancıklı, Z.A.; Özkay, Y. Design, synthesis and evaluation of new thiazole-piperazines as acetylcholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 28, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Ghotbi, G.; Mahdavi, M.; Najafi, Z.; Moghadam, F.H.; Hamzeh-Mivehroud, M.; Davaran, S.; Dastmalchi, S. Design, synthesis, biological evaluation, and docking study of novel dual-acting thiazole-pyridiniums inhibiting acetylcholinesterase and β-amyloid aggregation for Alzheimer’s disease. Bioorganic Chem. 2020, 103, 104186. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, A.; Ali, M.; Sarwar, R.; Mehsud, S.; Farooq, U.; Halimi, S.M.A.; Khan, I.; Al-Harrasi, A. Combined in Vitro and in Silico Studies for the Anticholinesterase Activity and Pharmacokinetics of Coumarinyl Thiazoles and Oxadiazoles. Front. Chem. 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmaniye, D.; Sağlık, B.N.; Çevik, U.A.; Levent, S.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A.; Turan, G. Synthesis and AChE Inhibitory Activity of Novel Thiazolylhydrazone Derivatives. Molecules 2019, 24, 2392. [Google Scholar] [CrossRef] [Green Version]
- Mumtaz, A.; Shoaib, M.; Zaib, S.; Shah, M.S.; Bhatti, H.A.; Saeed, A.; Hussain, I.; Iqbal, J. Synthesis, molecular modelling and biological evaluation of tetrasubstituted thiazoles towards cholinesterase enzymes and cytotoxicity studies. Bioorganic Chem. 2018, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Javid, M.T.; Ullah, H.; Wadood, A.; Taha, M.; Ashraf, M.; Ain, Q.U.; Khan, M.A.; Khan, F.; Mirza, S.; et al. Synthesis, molecular docking, acetylcholinesterase and butyrylcholinesterase inhibitory potential of thiazole analogs as new inhibitors for Alzheimer disease. Bioorganic Chem. 2015, 62, 106–116. [Google Scholar] [CrossRef]
- Sagar, S.R.; Singh, D.P.; Das, R.D.; Panchal, N.B.; Sudarsanam, V.; Nivsarkar, M.; Vasu, K.K. Pharmacological investigation of quinoxaline-bisthiazoles as multitarget-directed ligands for the treatment of Alzheimer’s disease. Bioorganic Chem. 2019, 89, 102992. [Google Scholar] [CrossRef]
- Sever, B.; Türkeş, C.; Altıntop, M.D.; Demir, Y.; Beydemir, Ş. Thiazolyl-pyrazoline derivatives: In-vitro and in silico evaluation as potential acetylcholinesterase and carbonic anhydrase inhibitors. Int. J. Biol. Macromol. 2020, 163, 1970–1988. [Google Scholar] [CrossRef]
- Shidore, M.; Machhi, J.; Shingala, K.; Murumkar, P.; Sharma, M.K.; Agrawal, N.; Tripathi, A.; Parikh, Z.; Pillai, P.; Yadav, M.R. Benzylpiperidine-linked diarylthiazoles as potential anti-Alzheimer’s agents: Synthesis and biological evaluation. J. Med. Chem. 2016, 59, 5823–5846. [Google Scholar] [CrossRef]
- Sun, Z.-Q.; Tu, L.-X.; Zhuo, F.-J.; Liu, S.-X. Design and discovery of Novel Thiazole acetamide derivatives as anticholinesterase agent for possible role in the management of Alzheimer’s. Bioorganic Med. Chem. Lett. 2016, 26, 747–750. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Özdemir, A.; Kaplancikli, Z.A.; Altıntop, M.D.; Temel, H.E.; Çiftçi, G.A. Synthesis and biological evaluation of some thiazole derivatives as new cholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 28, 509–514. [Google Scholar] [CrossRef] [Green Version]
- D’Ascenzio, M.; Chimenti, P.; Gidaro, M.C.; De Monte, C.; De Vita, D.; Granese, A.; Scipione, L.; Di Santo, R.; Costa, G.; Alcaro, S.; et al. (Thiazol-2-yl)hydrazone derivatives from acetylpyridines as dual inhibitors of MAO and AChE: Synthesis, biological evaluation and molecular modeling studies. J. Enzym. Inhib. Med. Chem. 2015, 30, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Sağlık, B.N.; Osmaniye, D.; Çevik, U.A.; Levent, S.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease. Molecules 2020, 25, 4312. [Google Scholar] [CrossRef] [PubMed]
- Hemaida, A.Y.; Hassan, G.S.; Maarouf, A.R.; Joubert, J.; El-Emam, A.A. Synthesis and Biological Evaluation of Thiazole-Based Derivatives as Potential Acetylcholinesterase Inhibitors. ACS Omega 2021, 6, 19202–19211. [Google Scholar] [CrossRef]
- Tegginamath, G.; Kamble, R.R.; Kattimani, P.P.; Margankop, S.B. Synthesis of 3-aryl-4-({2-[4-(6-substituted-coumarin-3-yl)-1,3-thiazol-2-yl]hydrazinylidene}methyl/ethyl)-sydnones using silica sulfuric acid and their antidiabetic, DNA cleavage activity. Arab. J. Chem. 2016, 9, S306–S312. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Vázquez, G.; Morales-Vilchis, M.G.; Estrada-Soto, S.; Ramírez-Espinosa, J.J.; Hidalgo-Figueroa, S.; Nava-Zuazo, C.; Tlahuext, H.; Leon-Rivera, I.; Medina-Franco, J.L.; López-Vallejo, F.; et al. Synthesis of 2-{2-[(α/β-naphthalen-1-ylsulfonyl)amino]-1,3-thiazol-4-yl} acetamides with 11β-hydroxysteroid dehydrogenase inhibition and in combo antidiabetic activities. Eur. J. Med. Chem. 2014, 74, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Tang, C.-L.; Ma, W.-P.; Gao, L.-X.; Wei, Y.; Zhang, W.; Li, J.-Y.; Li, J.; Nan, F.-J. Design, synthesis, and biological evaluation of novel 2-ethyl-5-phenylthiazole-4-carboxamide derivatives as protein tyrosine phosphatase 1B inhibitors with improved cellular efficacy. Eur. J. Med. Chem. 2013, 69, 399–412. [Google Scholar] [CrossRef]
- Li, F.; Zhu, Q.; Zhang, Y.; Feng, Y.; Leng, Y.; Zhang, A. Design, synthesis, and pharmacological evaluation of N-(4-mono and 4,5-disubstituted thiazol-2-yl)-2-aryl-3-(tetrahydro-2H-pyran-4-yl)propanamides as glucokinase activators. Bioorganic Med. Chem. 2010, 18, 3875–3884. [Google Scholar] [CrossRef]
- Song, K.-S.; Lee, S.H.; Kim, M.J.; Seo, H.J.; Lee, J.; Lee, S.-H.; Jung, M.E.; Son, E.-J.; Lee, M.; Kim, J.; et al. Synthesis and SAR of Thiazolylmethylphenyl Glucoside as Novel C-Aryl Glucoside SGLT2 Inhibitors. ACS Med. Chem. Lett. 2010, 2, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Ezer, M.; Yildirim, L.T.; Bayro, O.; Verspohl, E.J.; Dundar, O.B. Synthesis and antidiabetic activity of morpholinothiazolyl-2,4-thiazolidindione derivatives. J. Enzym. Inhib. Med. Chem. 2011, 27, 419–427. [Google Scholar] [CrossRef]
- Tang, H.; Yan, Y.; Feng, Z.; de Jesus, R.K.; Yang, L.; Levorse, D.A.; Owens, K.A.; Akiyama, T.E.; Bergeron, R.; Castriota, G.A.; et al. Design and synthesis of a new class of malonyl-CoA decarboxylase inhibitors with anti-obesity and anti-diabetic activities. Bioorganic Med. Chem. Lett. 2010, 20, 6088–6092. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-J.; Lee, W.J.; Kim, E.-A.; Nam, K.D.; Hahn, H.-G.; Choi, S.Y.; Cho, S.-W. Effects of N-adamantyl-4-methylthiazol-2-amine on hyperglycemia, hyperlipidemia and oxidative stress in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2014, 736, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Sravanthi, T.V.; Lulu, S.S.; Vino, S.; Jayasri, M.A.; MohanaPriya, A.; Manju, S.L. Synthesis, docking, and evaluation of novel thiazoles for potent antidiabetic activity. Med. Chem. Res. 2017, 26, 1306–1315. [Google Scholar] [CrossRef]
- Gao, H.-D.; Liu, P.; Yang, Y.; Gao, F. Sulfonamide-1,3,5-triazine–thiazoles: Discovery of a novel class of antidiabetic agents via inhibition of DPP-4. RSC Adv. 2016, 6, 83438–83447. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Ali, R.; Shah, S.; Adil, M.; Akhtar, M.; Wadhwa, R.; Bawa, S.; Sharma, M. 2-[(4-Chlorobenzyl) amino]-4-methyl-1,3-thiazole-5-carboxylic acid exhibits antidiabetic potential and raises insulin sensitivity via amelioration of oxidative enzymes and inflammatory cytokines in streptozotocin˗induced diabetic rats. Biomed. Pharmacother. 2017, 89, 651–659. [Google Scholar] [CrossRef]
- Pandya, D.H.; Sharma, J.A.; Jalani, H.B.; Pandya, A.N.; Sudarsanam, V.; Kachler, S.; Klotz, K.N.; Vasu, K.K. Novel thiazole–thiophene conjugates as adenosine receptor antagonists: Synthesis, biological evaluation and docking studies. Bioorganic Med. Chem. Lett. 2015, 25, 1306–1309. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Yerande, S.G.; Namasivayam, V.; Klapschinski, T.A.; Alnouri, M.W.; El-Tayeb, A.; Müller, C.E. Substituted 4-phenylthiazoles: Development of potent and selective A1, A3 and dual A1/A3 adenosine receptor antagonists. Eur. J. Med. Chem. 2020, 186, 111879. [Google Scholar] [CrossRef]
- Inamdar, G.S.; Pandya, A.N.; Thakar, H.M.; Sudarsanam, V.; Kachler, S.; Sabbadin, D.; Moro, S.; Klotz, K.-N.; Vasu, K.K. New insight into adenosine receptors selectivity derived from a novel series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] benzamides and furamides. Eur. J. Med. Chem. 2013, 63, 924–934. [Google Scholar] [CrossRef]
- Sams, A.G.; Mikkelsen, G.K.; Larsen, M.; Langgård, M.; Howells, M.E.; Schrøder, T.J.; Brennum, L.T.; Torup, L.; Jørgensen, E.B.; Bundgaard, C.; et al. Discovery of Phosphoric Acid Mono-{2-[(E/Z)-4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-benzoylimino]-thiazol-3-ylmethyl} Ester (Lu AA47070): A Phosphonooxymethylene Prodrug of a Potent and Selective hA2A Receptor Antagonist. J. Med. Chem. 2011, 54, 751–764. [Google Scholar] [CrossRef]
- Mishra, C.B.; Sharma, D.; Prakash, A.; Kumari, N.; Kumar, N.; Luthra, P.M. Design and synthesis of (4E)-4-(4-substitutedbenzylideneamino)-3-substituted-2,3-dihydro-2-thioxothiazole-5-carbonitrile as novel A2A receptor antagonists. Bioorganic Med. Chem. 2013, 21, 6077–6083. [Google Scholar] [CrossRef]
- Scheiff, A.B.; Yerande, S.G.; El-Tayeb, A.; Li, W.; Inamdar, G.S.; Vasu, K.K.; Sudarsanam, V.; Müller, C.E. 2-Amino-5-benzoyl-4-phenylthiazoles: Development of potent and selective adenosine A1 receptor antagonists. Bioorganic Med. Chem. 2010, 18, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Pandya, A.N.; Baraiya, A.B.; Jalani, H.B.; Pandya, D.; Kaila, J.C.; Kachler, S.; Salmaso, V.; Moro, S.; Klotz, K.-N.; Vasu, K.K. Discovery of 2-aminoimidazole and 2-amino imidazolyl-thiazoles as non-xanthine human adenosine A3receptor antagonists: SAR and molecular modeling studies. MedChemComm 2018, 9, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Kaddouri, Y.; Abrigach, F.; Yousfi, E.B.; El Kodadi, M.; Touzani, R. New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: Synthesis, DFT calculations and molecular docking study. Heliyon 2020, 6, e03185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.V.B.; Srinivasulu, D.; Peddanna, K.; Apparao, C.; Ramesh, P. Synthesis and Antioxidant Activity of New Thiazole Analogues Possessing Urea, Thiourea, and Selenourea Functionality. Synth. Commun. 2015, 45, 2592–2600. [Google Scholar] [CrossRef]
- Mohana, K.N.; Kumar, C.P. Synthesis and Antioxidant Activity of 2-Amino-5-methylthiazol Derivatives Containing 1,3,4-Oxadiazole-2-thiol Moiety. ISRN Org. Chem. 2013, 2013, 620718. [Google Scholar] [CrossRef]
- Tran, N.M.A.; Kumar, M.A.; Chang, S.H.; Kim, M.Y.; Kim, J.-A.; Lee, K.D. Synthesis, Anticancer and Antioxidant Activity of Novel 2,4-Disubstituted Thiazoles. Bull. Korean Chem. Soc. 2014, 35, 1619–1624. [Google Scholar] [CrossRef] [Green Version]
- Drapak, I.; Perekhoda, L.; Demchenko, N.; Suleiman, M.; Rakhimova, M.; Demchuk, I.; Taran, S.; Seredynska, N.; Gerashchenko, I. Cardioprotective Activity of Some 2-Arylimino-1,3-Thiazole Derivatives. Sci. Pharm. 2019, 87, 7. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Liu, A.; Liu, W.; Liu, X.; Ren, Y.; Pei, H.; Huang, L.; Zheng, X.; Huang, M.; Wu, D. Synthesis and Insecticidal Activity of Novel Thiazole Acrylonitrile Derivatives. J. Heterocycl. Chem. 2017, 54, 3395–3402. [Google Scholar] [CrossRef]
- Dai, H.; Xiao, Y.-S.; Li, Z.; Xu, X.-Y.; Qian, X. The thiazoylmethoxy modification on pyrazole oximes: Synthesis and insecticidal biological evaluation beyond acaricidal activity. Chin. Chem. Lett. 2014, 25, 1014–1016. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Krishnappa, M.; Nayak, S.K.; Subrahmanya, B.K.; Vaderapura, J.P.; Chalannavar, R.K.; Gleiser, R.M.; Odhav, B. Synthesis and antimosquito properties of 2,6-substituted benzo[d]thiazole and 2,4-substituted benzo[d]thiazole analogues against Anopheles arabiensis. Eur. J. Med. Chem. 2013, 65, 295–303. [Google Scholar] [CrossRef]
- Nishizawa, E.E.; Mendoza, A.R.; Honohan, T.; Annis, K.A. A Thiazole Compound with Potential Antithrombotic Activity. Thromb. Haemost. 1982, 47, 173–176. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioactive Compounds | Remarks/Conclusions |
|---|---|
 | Fatostatin (125B11): A specific inhibitor of sterol response element-binding proteins (SREBPs) is a newly customized thiazole analogue combined with two aryl groups. Fatostatin inhibits proliferation and increases cellular deaths in cancer cells. The drug may possibly be prescribed in the therapy of uterine carcinoma [44]. |
 | Mirabegron (YM 178): The medication is a selective β3-adrenoceptor stimulant that was recently licensed to alleviate overactive bladder (OAB) [45]. |
 | Ziritaxestat (GLPG1690): An autotaxin inhibitor is being optimized as a potential therapeutic candidate. The medication is still in development because phase 2 clinical studies for the clinical use of Systemic Sclerosis were unsuccessful [46,47]. |
 | WEHI-539 HCl (WEHI-539 hydrochloride): The therapeutic candidate binds to BCL-XL with great affinity and specificity, killing cells effectively by inhibiting its pro-survival function. The molecule is in the developmental stage and is currently being optimized to achieve better physicochemical parameters. If further developed, the medicine could be approved for the management of a host of drug-resistant tumors [48]. |
 | O4I2: The drug candidate is a powerful Oct3/4 inducer that possibly be prescribed to make iPSCs. This molecule is in a preclinical developmental stage and may possibly be used in the treatment of a variety of cancer [49]. |
 | TP0427736: The compound is an effective inhibitor of ALK5 kinase activity. It also suppresses TGF-β1-induced phosphorylation of Smad2/3 in A549 cells. The drug is being optimized for its potential use in androgenic alopecia [50]. |
 | SRT2104 (GSK2245840): The compound is a selective Sirtuin 1(SIRT1) activator engaged in energy homeostasis management. The drug candidate is being investigated for clinical use for Atrophy, Sepsis, Psoriasis, Type 2 Diabetes Mellitus, and Muscular Dystrophy [51,52]. |
 | IRAK inhibitor 6: The therapeutic candidate is a selective Renal Carcinoma Antigen NY REN 64 blocker that can possibly be used to manage bone degradation and rheumatoid arthritis-induced joint inflammation [53]. |
 | SC75741: The therapeutic candidate is a highly effective NF-B inhibitor. Due to its efficacy at inhibiting influenza virus replication, SC75741 is being developed to treat avian influenza-A virus infections [54]. |
 | SRT3025: The active agent is a small molecule activator of the SIRT1 enzyme. The therapy could be used to manage Fanconi anaemia [55]. |
 | UM-164: The active substance is a highly powerful dual c-Src/p38 inhibitor that inhibits both p38 and p38 and has a binding constant Kd of 2.7 nM for c-Src. The drug is the subject of a great deal of research since it claims to be able to cure triple-negative breast cancer [56]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arshad, M.F.; Alam, A.; Alshammari, A.A.; Alhazza, M.B.; Alzimam, I.M.; Alam, M.A.; Mustafa, G.; Ansari, M.S.; Alotaibi, A.M.; Alotaibi, A.A.; et al. Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents. Molecules 2022, 27, 3994. https://doi.org/10.3390/molecules27133994
Arshad MF, Alam A, Alshammari AA, Alhazza MB, Alzimam IM, Alam MA, Mustafa G, Ansari MS, Alotaibi AM, Alotaibi AA, et al. Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents. Molecules. 2022; 27(13):3994. https://doi.org/10.3390/molecules27133994
Chicago/Turabian StyleArshad, Mohammed F., Aftab Alam, Abdullah Ayed Alshammari, Mohammed Bader Alhazza, Ibrahim Mohammed Alzimam, Md Anish Alam, Gulam Mustafa, Md Salahuddin Ansari, Abdulelah M. Alotaibi, Abdullah A. Alotaibi, and et al. 2022. "Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents" Molecules 27, no. 13: 3994. https://doi.org/10.3390/molecules27133994