
The Glu143 Residue Might Play a Significant Role in T20 Peptide Binding to HIV-1 Receptor gp41: An In Silico Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
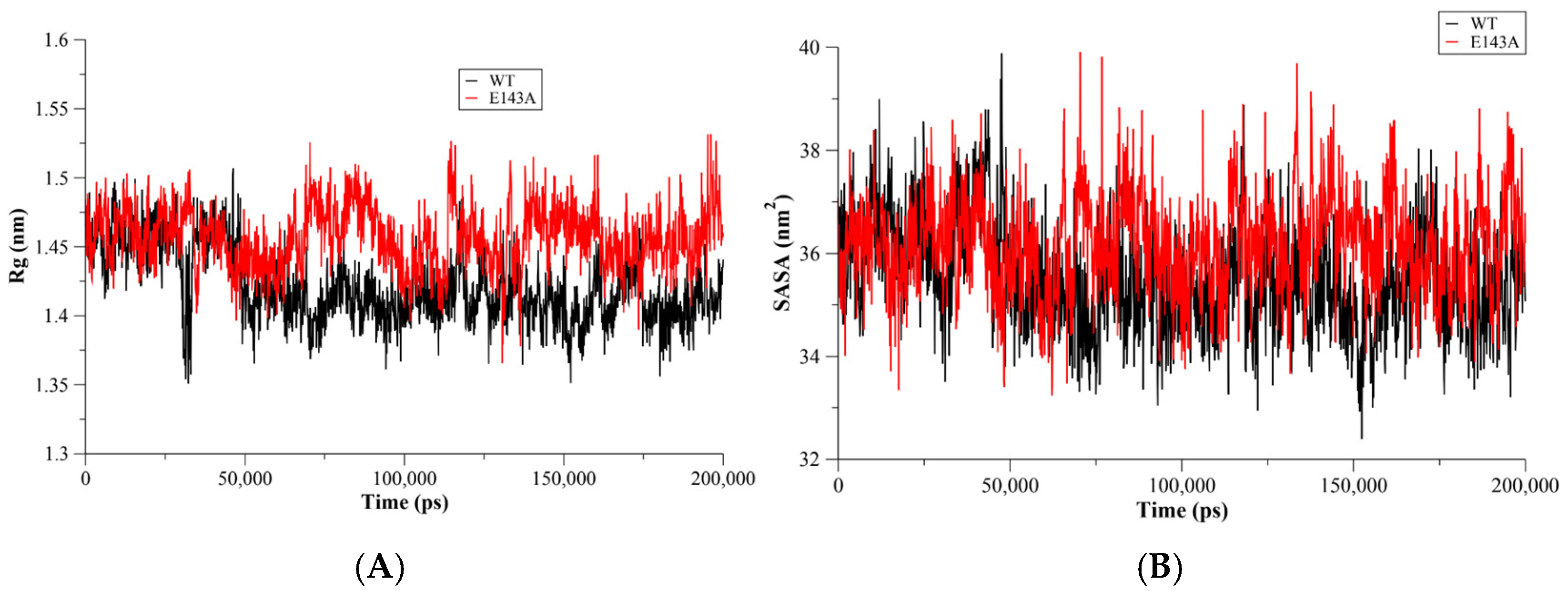
2. Results and Discussion
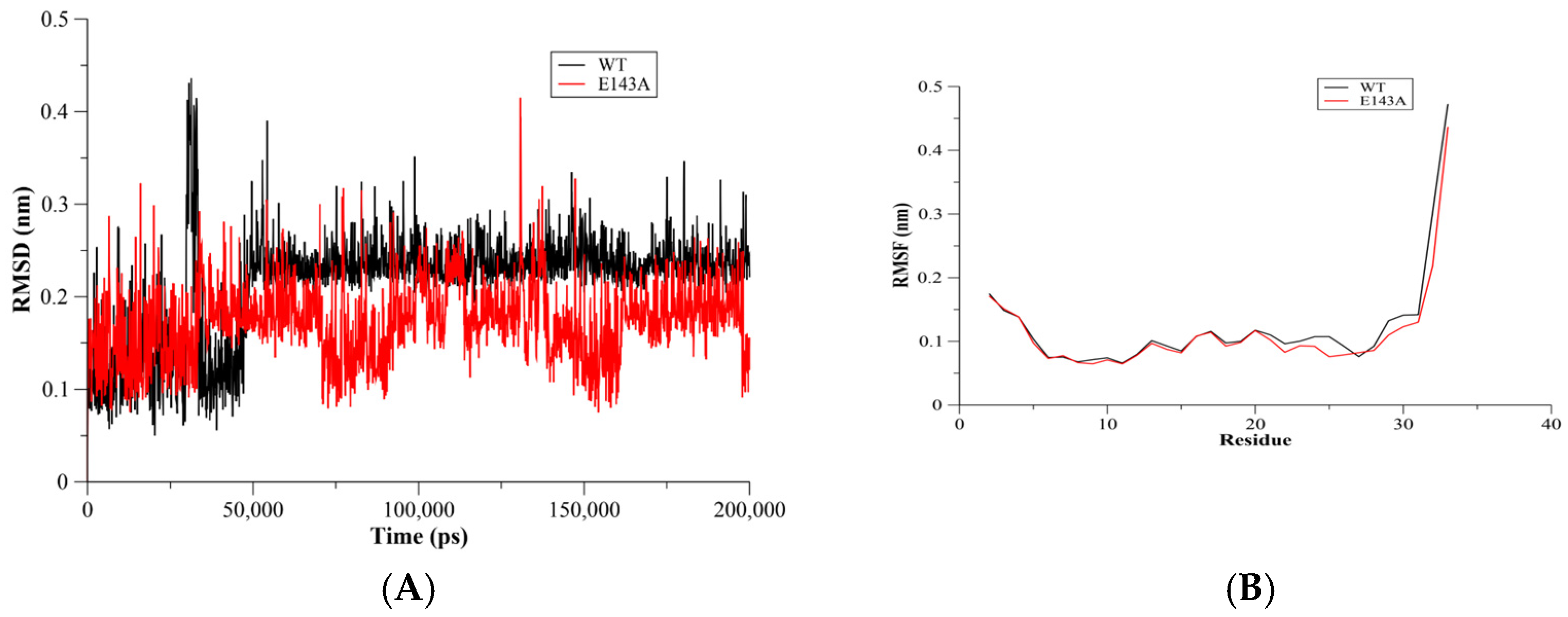
2.1. RMSD and RMSF Assessments
2.2. Conformational Changes
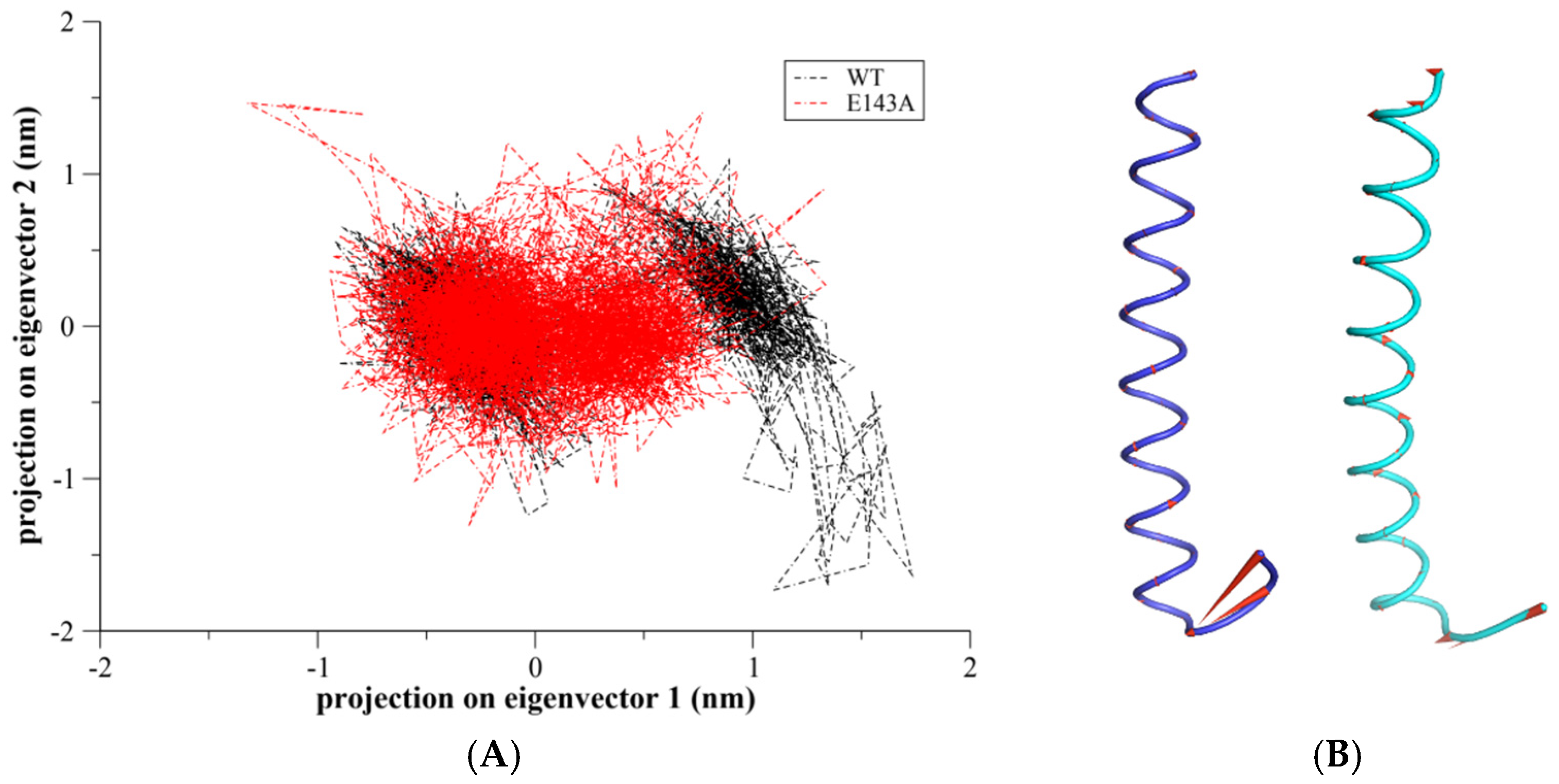
2.3. Principal Component Analysis (PCA)
2.4. The Free Energy Landscape (FEL)
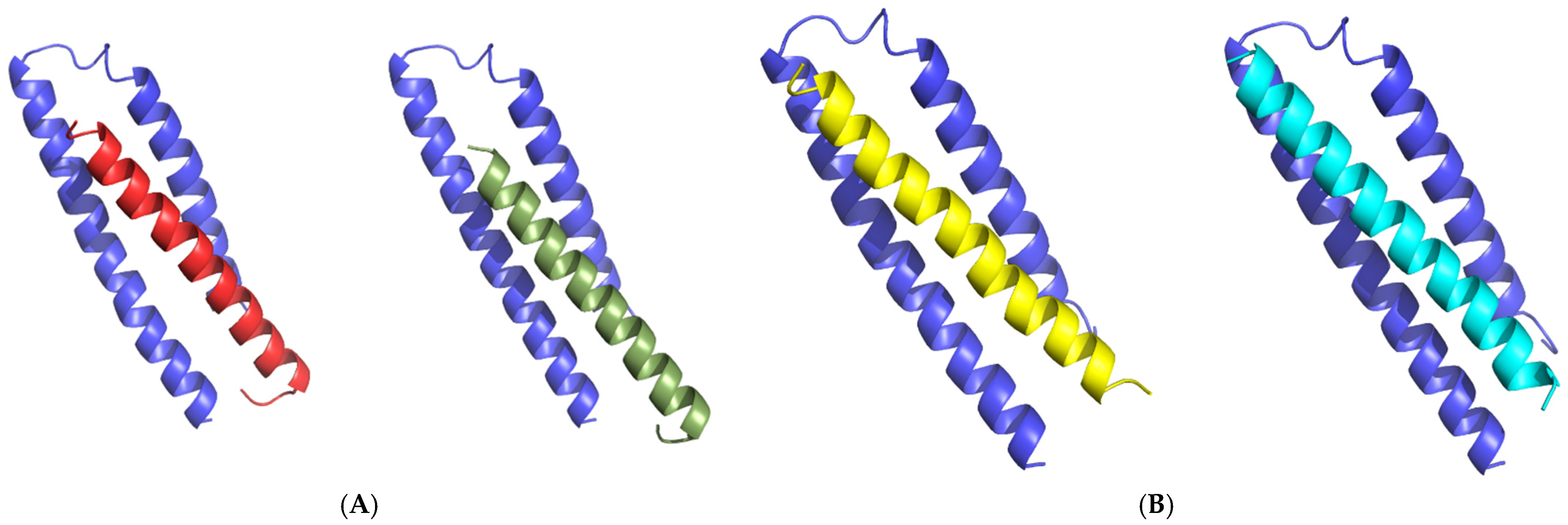
2.5. T20 Docking to the gp41 Receptor
3. Materials and Methods
3.1. Structural Preparation
3.2. MD Simulations
3.3. Docking and Visualization
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Caffrey, M. HIV Envelope: Challenges and Opportunities for Development of Entry Inhibitors. Trends Microbiol. 2011, 19, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The Structural Biology of HIV-1: Mechanistic and Therapeutic Insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, P.M.; Lawrence, M.C. The Structural Biology of Type I Viral Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ding, X.; Zhu, Y.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Structural and Functional Characterization of HIV-1 Cell Fusion Inhibitor T20. AIDS 2019, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.G.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J.; et al. Enfuvirtide, an HIV-1 Fusion Inhibitor, for Drug-Resistant HIV Infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The First Therapy to Inhibit the Entry of HIV-1 into Host CD4 Lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Klepeis, J.L.; Rendleman, C.A.; Dror, R.O.; Shaw, D.E. 9.6 New Technologies for Molecular Dynamics Simulations. In Comprehensive Biophysics; Egelman, E.H., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 86–104. ISBN 978-0-08-095718-0. [Google Scholar]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. In Docking Screens for Drug Discovery; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 2053, pp. 13–34. [Google Scholar] [CrossRef]
- Akbal-Delibas, B.; Jagodzinski, F.; Haspel, N. A Conservation and Rigidity Based Method for Detecting Critical Protein Residues. BMC Struct. Biol. 2013, 13 (Suppl. S1), S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Alaofi, A.L.; Shahid, M. Mutations of SARS-CoV-2 RBD May Alter Its Molecular Structure to Improve Its Infection Efficiency. Biomolecules 2021, 11, 1273. [Google Scholar] [CrossRef] [PubMed]
- Alaofi, A.L. Exploring Structural Dynamics of the MERS-CoV Receptor DPP4 and Mutant DPP4 Receptors. J. Biomol. Struct. Dyn. 2020, 40, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Relative Solvent Accessible Surface Area Predicts Protein Conformational Changes upon Binding. Structure 2011, 19, 859–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. In Protein Dynamics; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1084, pp. 193–226. [Google Scholar] [CrossRef] [Green Version]
- Nair, M.S.; Shukla, A. Molecular Modeling, Simulation and Principal Component Analysis of Binding of Resveratrol and Its Analogues with DNA. J. Biomol. Struct. Dyn. 2020, 38, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Alaofi, A.L. Probing the Flexibility of Zika Virus Envelope Protein DIII Epitopes Using Molecular Dynamics Simulations. Mol. Simul. 2020, 46, 541–547. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A Web Server to Predict and Analyze the Protein–Protein Complex Based on Computational Docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.5; Schrödinger, LLC: New York, NY, USA, 2015.





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaofi, A.L. The Glu143 Residue Might Play a Significant Role in T20 Peptide Binding to HIV-1 Receptor gp41: An In Silico Study. Molecules 2022, 27, 3936. https://doi.org/10.3390/molecules27123936
Alaofi AL. The Glu143 Residue Might Play a Significant Role in T20 Peptide Binding to HIV-1 Receptor gp41: An In Silico Study. Molecules. 2022; 27(12):3936. https://doi.org/10.3390/molecules27123936
Chicago/Turabian StyleAlaofi, Ahmed L. 2022. "The Glu143 Residue Might Play a Significant Role in T20 Peptide Binding to HIV-1 Receptor gp41: An In Silico Study" Molecules 27, no. 12: 3936. https://doi.org/10.3390/molecules27123936