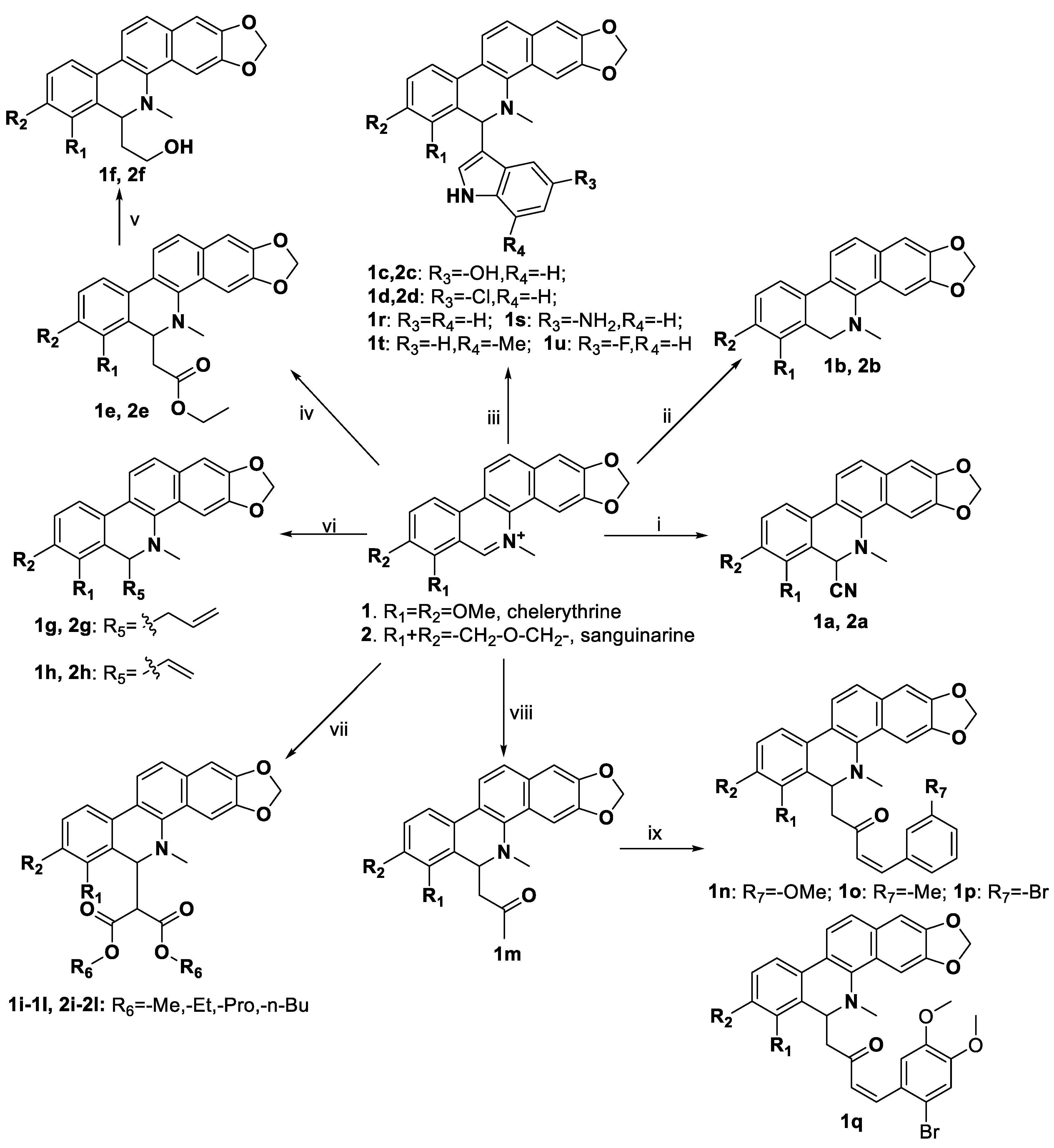
3.3. Procedure for Synthesizing Benzophenanthridine Alkaloid Derivatives
3.3.1. Synthesis of Compounds 1a and 2a
Trimethylsilyl cyanlde (TMSCN) (200 μL, 0.194 mmol) and 4-dimethylaminopyridine (DMAP) (60 mg, 0.492 mmol) were added to a stirred solution of 1 or 2 (0.144 mmol) in dry dichloromethane (DCM) (10 mL) at room temperature, and the reaction mixture was stirred under reflux for 14 h. After the reaction was complete, the mixture was washed with saturated NaHCO3 solution three times and filtered. The filtrate was washed with an aqueous hydrochloric acid solution (0.1 mol/L, 5 × 10 mL), and then the organic layer was collected, dried over anhydrous Na2SO4, and concentrated under vacuum. The crude products were washed with methanol and filtered, and then dried under vacuum to obtain target compounds 1a and 2a.
Compound 1a: light-yellow powder; yield: 65.1%; 1H-NMR (600 MHz, CDCl3) δ 7.74 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.16 (s, 1H), 7.10 (d, J = 8.5 Hz, 1H), 6.09 (s, 2H), 5.67 (s, 1H), 4.03 (s, 3H), 3.98 (s, 3H), 2.65 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 152.37, 148.53, 147.97, 146.08, 138.61, 131.31, 126.66, 125.15, 125.05, 122.94, 120.72, 119.86, 119.37, 118.34, 113.50, 104.48, 101.22, 100.60, 61.29, 56.00, 48.58, 41.56. HR-ESI-MS (m/z) calculated for C22H19O4N2 [M + H]+ 375.1331, found 375.1339.
Compound 2a: khaki powder; yield: 61.2%; 1H-NMR (600 MHz, CDCl3) δ 7.72 (d, J = 8.5 Hz, 1H), 7.68 (s, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.15 (s, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.14 (dd, J = 15.9, 1.6 Hz, 2H), 6.10 (d, J = 1.9 Hz, 2H), 5.35 (s, 1H), 2.69 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 148.61, 148.02, 147.82, 145.03, 138.48, 131.28, 126.70, 125.92, 125.35, 123.15, 120.08, 117.39, 117.11, 109.41, 107.62, 104.47, 102.20, 101.26, 100.57, 48.75, 41.44. HR-ESI-MS (m/z) calculated for C21H15O4N2 [M + H]+ 359.1018, found 359.1026.
3.3.2. Synthesis of Compounds 1b and 2b
NaBH4 (10 mg, 0.264 mmol) was added to a solution of 1 or 2 (0.052 mmol) in MeOH (5 mL) at room temperature. The reaction mixture was stirred for 0.5 h at the same temperature. After the reaction was complete, acetic acid was added to remove the excess NaBH4 and concentrated under vacuum. The residue was dissolved in dry DCM and extracted with saturated aqueous NaCl (3 × 10 mL). The organic layer was collected, dried over anhydrous Na2SO4, and concentrated under vacuum. Finally, the crude products were purified by silica gel column chromatography (petroleum ether (PE)/ethyl acetate (EA), 10:1) to obtain the target compounds.
Compound 1b: light-yellow powder; yield: 89.7%; 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.5 Hz, 1H), 7.70 (s, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.07 (s, 2H), 4.32 (s, 2H), 3.95 (s, 3H), 3.90 (s, 3H), 2.62 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 152.30, 148.09, 147.49, 146.14, 142.75, 130.83, 126.41, 126.30, 126.27, 124.28, 123.80, 120.15, 118.70, 111.00, 104.37, 101.04, 100.75, 61.12, 55.84, 48.76, 41.32. HR-ESI-MS (m/z) calculated for C21H20O4N [M + H]+ 350.1381, found 350.1387.
Compound 2b: white powder; yield: 94.7%; 1H-NMR (600 MHz, CDCl3) δ 7.71 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.5 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.13 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.07 (d, J = 7.7 Hz, 4H), 4.23 (s, 2H), 2.65 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 148.17, 147.54, 147.13, 144.66, 142.54, 130.84, 127.29, 126.55, 124.43, 123.97, 120.38, 116.22, 113.65, 107.21, 104.37, 101.35, 101.07, 100.77, 48.47, 41.59. HR-ESI-MS (m/z) calculated for C20H16O4N [M + H]+ 334.1069, found 334.1074.
3.3.3. Synthesis of Compounds 1c, 1d, 1r–u, and 2c, 2d
The indole compounds (two equivalents) were added to a solution of 1 or 2 (0.052 mmol) in CH3CN (10 mL) at room temperature. Each reaction mixture was stirred at the same temperature until the reaction was complete and then concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography to obtain the target compounds.
Compound 1c: The crude product was purified by silica gel column chromatography (PE/EA, 1.5:1) to obtain the target compound 1c: white powder; yield: 43.5%; 1H-NMR (600 MHz, acetone-d6) δ 9.39 (s, 1H), 7.82–7.75 (m, 2H), 7.71 (d, J = 8.5 Hz, 1H), 7.66 (s, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.17 (d, J = 8.6 Hz, 1H), 7.05 (s, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.63 (dd, J = 8.6, 2.4 Hz, 1H), 6.18 (q, J = 1.2 Hz, 1H), 6.05 (d, J = 1.0 Hz, 1H), 5.99 (d, J = 1.0 Hz, 1H), 5.96 (d, J = 1.1 Hz, 1H), 3.96 (s, 3H), 3.77 (s, 3H), 2.88 (s, 3H); 13C-NMR (151 MHz, Acetone-d6) δ 152.46, 150.59, 147.87, 147.38, 146.41, 141.07, 131.77, 131.00, 128.23, 128.14, 127.44, 125.62, 124.32, 123.92, 123.27, 119.77, 118.74, 115.88, 111.65, 111.41, 111.28, 104.30, 103.85, 101.06, 100.71, 60.13, 55.23, 54.52, 41.61. HR-ESI-MS (m/z) calculated for C29H25O5N2 [M + H]+ 481.1755, found 481.1758.
Compound 1d: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 1d: white powder; yield: 41.9%; 1H-NMR (600 MHz, DMSO-d6) δ 10.69 (d, J = 2.6 Hz, 1H), 8.05 (d, J = 2.1 Hz, 1H), 7.78 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.51 (s, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.19–7.16 (m, 2H), 7.00 (dd, J = 8.6, 2.1 Hz, 1H), 6.22–6.20 (m, 1H), 6.07 (d, J = 6.0 Hz, 2H), 5.86 (d, J = 1.1 Hz, 1H), 3.91 (s, 3H), 3.72 (s, 3H), 2.78 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ 152.47, 148.16, 147.52, 146.15, 140.73, 135.41, 130.91, 128.18, 127.46, 126.98, 125.42, 125.21, 124.20, 123.83, 123.50, 121.33, 120.29, 119.66, 119.57, 116.19, 113.43, 112.48, 104.53, 101.58, 100.26, 61.04, 56.17, 54.27, 42.32. HR-ESI-MS (m/z) calculated for C29H24O3N2Cl [M + H]+ 499.1413, found 499.1419.
Compound 1r: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 1r: white solid; yield: 52.5%; 1H-NMR (600 MHz, CDCl3) δ 8.18 (dd, J = 7.8, 1.1 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.68 (s, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.56 (s, 1H), 7.37 (d, J = 8.5 Hz, 1H), 7.20–7.18 (m, 1H), 7.17–7.15 (m, 1H), 7.11 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.05 (d, J = 8.5 Hz, 1H), 6.98 (s, 1H), 6.26 (dd, J = 2.5, 1.1 Hz, 1H), 6.04 (d, J = 1.2 Hz, 1H), 6.00 (d, J = 1.4 Hz, 1H), 5.94 (d, J = 1.4 Hz, 1H), 3.98 (s, 3H), 3.80 (s, 3H), 2.89 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 152.23, 147.76, 147.26, 146.31, 141.13, 136.51, 130.89, 128.20, 127.46, 127.23, 125.85, 124.01, 123.30, 122.99, 121.65, 120.49, 119.64, 119.22, 118.95, 117.60, 111.35, 110.85, 104.08, 101.21, 100.81, 61.19, 55.83, 54.35, 42.32. HR-ESI-MS (m/z) calculated for C29H23O4N2 [M − H]− 463.1646, found 463.1652.
Compound 1s: The crude product was purified by silica gel column chromatography (PE/EA, 1:1) to obtain the target compound 1s: brown powder; yield: 40.0%; 1H-NMR (600 MHz, DMSO-d6) δ 9.96 (d, J = 2.7 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.53 (s, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.25 (d, J = 2.2 Hz, 1H), 7.18 (s, 1H), 7.15 (d, J = 8.6 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.42 (dd, J = 8.5, 2.2 Hz, 1H), 6.07 (s, 1H), 6.03 (s, 1H), 5.87 (d, J = 2.5 Hz, 1H), 5.74 (s, 1H), 3.90 (s, 3H), 3.67 (s, 3H), 2.77 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ 152.40, 147.98, 147.44, 145.99, 141.23, 141.03, 130.88, 130.68, 128.27, 128.15, 127.20, 125.35, 124.30, 123.58, 123.41, 120.25, 119.33, 114.72, 112.28, 112.18, 111.83, 104.44, 103.52, 101.46, 100.71, 60.89, 56.12, 54.49, 42.54. HR-ESI-MS (m/z) calculated for C29H26O4N3 [M + H]+ 480.1914, found 480.1918.
Compound 1t: The crude product was purified by silica gel column chromatography (PE/EA, 3:1) to obtain the target compound 1t: white powder; yield: 36.4%; 1H-NMR (600 MHz, CDCl3) δ 8.00 (d, J = 7.9 Hz, 1H), 7.73–7.66 (m, 2H), 7.63 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.19 (s, 1H), 7.15–7.04 (m, 2H), 6.95 (s, 1H), 6.87 (d, J = 7.1 Hz, 1H), 6.06 (dd, J = 2.5, 1.2 Hz, 1H), 6.01 (d, J = 1.1 Hz, 1H), 5.99 (s, 1H), 5.92 (d, J = 1.4 Hz, 1H), 3.99 (s, 3H), 3.77 (s, 3H), 2.88 (s, 3H), 2.15 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 152.24, 147.79, 147.26, 146.31, 141.15, 136.01, 130.87, 128.34, 127.45, 126.62, 125.84, 124.04, 123.28, 122.83, 122.12, 119.96, 119.64, 119.35, 118.89, 118.08, 117.78, 111.33, 104.10, 101.19, 100.81, 61.19, 55.86, 54.36, 42.32, 16.22. HR-ESI-MS (m/z) calculated for C30H27O4N2 [M + H]+ 479.1959, found 479.1065.
Compound 1u: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 1u: white powder; yield: 43.3%; 1H-NMR (600 MHz, DMSO-d6) δ 10.59 (s, 1H), 7.78 (d, J = 8.7 Hz, 1H), 7.75–7.68 (m, 2H), 7.54 (s, 1H), 7.42 (d, J = 8.6 Hz, 1H), 7.17 (d, J = 8.4 Hz, 3H), 6.84 (td, J = 9.2, 2.6 Hz, 1H), 6.22 (d, J = 2.5 Hz, 1H), 6.07 (d, J = 11.6 Hz, 2H), 5.84 (s, 1H), 3.91 (s, 3H), 3.72 (s, 3H), 2.78 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ 156.20, 152.46, 148.12, 147.51, 146.13, 140.80, 133.60, 130.91, 127.53, 127.01, 125.69, 125.24, 124.18, 123.77, 120.28, 119.56, 116.47, 112.82, 112.42, 109.61, 109.43, 104.90, 104.53, 101.56, 100.33, 61.02, 56.15, 54.35, 42.30. HR-ESI-MS (m/z) calculated for C29H24O4N2F [M + H]+ 483.1710, found 483.1715.
Compound 2c: The crude product was purified by silica gel column chromatography (PE/EA, 1:1) to obtain the target compound 2c: khaki powder; yield: 35.8%; 1H-NMR (600 MHz, CDCl3) δ 7.69 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 2.5 Hz, 1H), 7.50 (s, 1H), 7.40 (dd, J = 9.7, 8.3 Hz, 2H), 7.02 (d, J = 9.2 Hz, 2H), 6.94 (d, J = 8.1 Hz, 1H), 6.70 (dd, J = 8.6, 2.5 Hz, 1H), 6.35–6.30 (m, 1H), 6.06 (dd, J = 9.4, 1.5 Hz, 2H), 6.01 (d, J = 1.4 Hz, 1H), 5.96 (d, J = 1.3 Hz, 1H), 5.73 (d, J = 1.1 Hz, 1H), 2.88 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 149.22, 147.89, 147.35, 147.07, 144.90, 140.92, 131.86, 130.91, 127.68, 127.53, 126.66, 125.24, 123.97, 123.57, 119.95, 116.58, 111.83, 111.60, 111.54, 107.50, 104.75, 104.16, 102.06, 101.42, 101.14, 100.89, 54.33, 42.70. HR-ESI-MS (m/z) calculated for C28H21O5N2 [M + H]+ 465.1441, found 465.1445.
Compound 2d: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 2d: white powder; yield: 27.5%; 1H-NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), 7.70–7.66 (m, 2H), 7.64 (s, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 7.06 (d, J = 1.5 Hz, 2H), 7.02 (s, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.38 (dd, J = 2.6, 1.2 Hz, 1H), 6.09 (q, J = 1.5 Hz, 2H), 6.01 (dd, J = 18.7, 1.3 Hz, 2H), 5.75 (d, J = 1.2 Hz, 1H), 2.89 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 148.09, 147.46, 147.14, 144.85, 140.72, 134.82, 130.90, 127.84, 127.40, 126.51, 125.08, 124.11, 123.95, 123.68, 122.11, 119.91, 119.63, 116.71, 116.37, 115.47, 111.92, 107.62, 104.15, 101.46, 100.97, 54.18, 42.70, 14.21. HR-ESI-MS (m/z) calculated for C28H20O4N2Cl [M + H]+ 483.1102, found 483.1106.
3.3.4. Synthesis of Compounds 1e and 2e
Ethyl trimethylsilyl acetate (24 mg, 0.164 mmol) and CsF (23 mg, 0.151 mmol) were added to a stirred solution of 1 or 2 (0.052 mmol) in CH3CN (15 mL) at room temperature. Each reaction mixture was stirred for 4–5 h at the same temperature until the reaction was complete and then concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography to obtain the target products.
Compound 1e: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 1e: white solid; yield: 52.0%; 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.6 Hz, 1H), 7.59–7.55 (m, 2H), 7.50 (d, J = 8.4 Hz, 1H), 7.12 (s, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.06 (s, 2H), 5.03 (dd, J = 11.1, 4.4 Hz, 1H), 4.22–4.13 (m, 2H), 3.99 (s, 3H), 3.95 (s, 3H), 2.67 (s, 3H), 2.42–2.29 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 171.70, 152.11, 147.96, 147.51, 145.76, 139.40, 131.07, 127.96, 127.56, 124.92, 123.80, 123.09, 119.76, 118.81, 111.61, 104.30, 100.99, 100.94, 61.04, 60.27, 55.83, 55.10, 42.88, 39.19, 14.23. HR-ESI-MS (m/z) calculated for C25H25O6N Na [M + Na]+ 458.1569, found 458.1574.
Compound 2e: The crude product was purified by silica gel column chromatography (PE/EA, 6:1) to obtain the target compound 2e: white powder; yield: 47.5%; 1H-NMR (600 MHz, CDCl3) δ 7.71 (d, J = 8.6 Hz, 1H), 7.57 (s, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.12 (s, 1H), 6.89 (d, J = 8.1 Hz, 1H), 6.13–6.01 (m, 4H), 4.85 (dd, J = 8.5, 6.9 Hz, 1H), 4.18 (m, J = 10.8, 7.2 Hz, 2H), 2.68 (s, 3H), 2.41 (d, J = 7.8 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 171.40, 148.07, 147.57, 147.11, 144.48, 139.27, 131.06, 127.67, 125.81, 124.01, 123.24, 119.99, 116.46, 115.73, 107.68, 104.30, 101.56, 101.02, 100.91, 60.41, 54.81, 43.14, 39.00, 14.25. HR-ESI-MS (m/z) calculated for C24H21O6N Na [M + Na]+ 442.1257, found 442.1261.
3.3.5. Synthesis of Compounds 1f and 2f
Compound 1e or 2e (0.043 mmol) in dry tetrahydrofuran (THF, 5 mL) was cooled to 5 °C. After 5 min, LiAlH4 (0.75 mmol, 300 μL) was slowly added to the mixture under an argon atmosphere, followed by stirring for 0.5 h at 5 °C. After the reaction was complete, it was quenched with a 15% NaOH aqueous solution and extracted with DCM (3 × 5 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography to obtain the target compounds.
Compound 1f: The crude product was purified by silica gel column chromatography (PE/EA, 2:1) to obtain the target compound 1f: white solid; yield: 73.8%; 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.6 Hz, 1H), 7.58 (s, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 8.5 Hz, 1H), 7.14 (s, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.06 (dd, J = 12.0, 1.4 Hz, 2H), 4.68 (dd, J = 9.4, 5.4 Hz, 1H), 3.97 (s, 3H), 3.96 (s, 3H), 3.79 (ddd, J = 11.2, 9.3, 3.5 Hz, 1H), 3.71 (dt, J = 10.9, 4.6 Hz, 1H), 2.70 (s, 3H), 1.81 (dtd, J = 14.0, 9.4, 4.5 Hz, 1H), 1.51 (dtd, J = 14.1, 5.1, 3.4 Hz, 1H); 13C-NMR (151 MHz, CDCl3) δ 152.13, 148.47, 147.59, 145.69, 139.20, 131.11, 128.74, 127.06, 124.62, 124.23, 123.80, 119.89, 119.26, 111.33, 104.63, 101.14, 99.95, 61.76, 61.14, 57.14, 55.81, 42.63, 35.47. HR-ESI-MS (m/z) calculated for C23H24O5N [M + H]+ 394.1643, found 394.1649.
Compound 2f: The crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound 2f: white solid; yield: 70.5%; 1H-NMR (600 MHz, CDCl3) δ 7.71 (d, J = 8.5 Hz, 1H), 7.57 (s, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.13 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.07 (dd, J = 3.1, 1.5 Hz, 2H), 6.05 (dd, J = 3.3, 1.5 Hz, 2H), 4.50 (dd, J = 10.0, 4.9 Hz, 1H), 3.87–3.78 (m, 3H), 2.71 (s, 3H), 1.85–1.76 (m, 1H), 1.58 (dq, J = 14.5, 4.8 Hz, 1H); 13C-NMR (151 MHz, CDCl3) δ 148.52, 147.61, 147.10, 144.34, 139.01, 131.09, 127.18, 125.55, 124.38, 123.90, 120.13, 116.91, 116.71, 107.50, 104.62, 101.47, 101.16, 99.93, 61.58, 56.81, 43.08, 35.20. HR-ESI-MS (m/z) calculated for C22H20O5N [M + H]+ 378.1331, found 378.1336.
3.3.6. Synthesis of Compounds 1g, 1h, 2g and 2h
RMgBr (1.0 mol/L, 1.5 equivalents) was added to a solution of 1 or 2 (0.052 mmol) in dry THF (10 mL) under an argon atmosphere at room temperature. The reaction mixture was stirred at the same temperature until the reaction was complete and then concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography to obtain the target compounds.
Compound 1g: white solid; yield: 80.5%; the crude product was purified by silica gel column chromatography (PE/EA, 49:1) to obtain the pure compound 1g. 1H-NMR (600 MHz, CDCl3) δ 7.77–7.70 (m, 2H), 7.57 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.14 (s, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.08–6.07 (m, 2H), 6.06–5.99 (m, 1H), 4.99 (ddd, J = 10.2, 2.2, 1.1 Hz, 1H), 4.88 (dq, J = 17.2, 1.4 Hz, 1H), 4.52 (dd, J = 9.7, 5.3 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 2.66 (s, 3H), 2.28–2.05 (m, 2H); 13C-NMR (151 MHz, CDCl3) δ 152.14, 147.95, 147.45, 145.83, 140.06, 136.37, 130.99, 129.74, 127.54, 124.78, 123.68, 123.53, 119.87, 118.84, 115.78, 111.09, 104.32, 100.99, 100.73, 61.02, 58.23, 55.78, 42.71, 38.47. HR-ESI-MS (m/z) calculated for C24H24O4N [M + H]+ 390.1694, found 390.1700.
Compound 1h: white solid; yield: 78.9%; the crude product was purified by silica gel column chromatography (PE/EA, 47:1) to obtain the target compound 1h. 1H-NMR (600 MHz, CDCl3) δ 7.78 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.12 (s, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.07 (s, 2H), 5.83 (ddd, J = 17.2, 10.4, 4.5 Hz, 1H), 5.13 (dt, J = 4.2, 2.0 Hz, 1H), 4.94–4.80 (m, 2H), 3.96 (s, 3H), 3.95 (s, 3H), 2.69 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 152.16, 148.08, 147.45, 146.31, 140.61, 138.12, 130.89, 127.79, 127.23, 125.18, 123.78, 123.65, 119.87, 118.93, 115.15, 111.38, 104.41, 101.02, 100.78, 60.97, 59.32, 55.80, 42.38. HR-ESI-MS (m/z) calculated for C23H22O4N [M + H]+ 376.1539, found 376.1543.
Compound 2g: white solid; yield: 80.1%; the crude product was purified by silica gel column chromatography (PE/EA, 49:1) to obtain the target compound 2g. 1H-NMR (600 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 2H), 7.50 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.13 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.10–6.03 (m, 4H), 5.98 (ddt, J = 17.2, 10.2, 7.0 Hz, 1H), 5.05–4.98 (m, 1H), 4.92 (dq, J = 17.2, 1.6 Hz, 1H), 4.30 (dd, J = 9.1, 6.0 Hz, 1H), 2.67 (s, 3H), 2.20 (ddt, J = 50.5, 14.3, 7.4 Hz, 2H); 13C-NMR (151 MHz, CDCl3) δ 148.04, 147.50, 146.95, 144.61, 139.99, 135.72, 130.96, 127.65, 125.74, 123.73, 123.72, 120.10, 117.46, 116.44, 116.27, 107.24, 104.31, 101.32, 101.01, 100.98, 58.13, 43.03, 38.32. HR-ESI-MS (m/z) calculated for C23H20O4N [M + H]+ 374.1384, found 374.1387.
Compound 2h: white solid; yield: 78.6%; the crude product was purified by silica gel column chromatography (PE/EA, 49:1) to obtain the target compound 2h. 1H-NMR (600 MHz, CDCl3) δ 7.78 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.13 (s, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.16–6.03 (m, 4H), 5.91–5.76 (m, 1H), 5.02–4.91 (m, 1H), 4.93–4.86 (m, 2H), 2.72 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 148.18, 147.51, 147.02, 145.08, 140.41, 137.12, 130.90, 127.38, 126.07, 123.89, 123.86, 120.10, 116.52, 115.57, 115.29, 107.52, 104.40, 101.47, 101.05, 100.79, 59.34, 42.77. HR-ESI-MS (m/z) calculated for C22H18O4N [M + H]+ 360.1225, found 360.1230.
3.3.7. Synthesis of Compounds 1i–l and 2i–l
Malonate diester compounds (1.5 equivalents) were added to a solution of 1 or 2 (0.115 mmol) in CH3CN (20 mL) at room temperature. The reaction mixture was stirred for 5–14 h at the same temperature until the reaction was complete and then concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target products.
Compound 1i: white solid; yield: 36.3%; 1H-NMR (600 MHz, CDCl3) δ 7.76 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.44 (s, 1H), 7.13 (s, 1H), 7.02 (d, J = 8.5 Hz, 1H), 6.06 (dd, J = 8.6, 1.4 Hz, 2H), 5.24 (d, J = 10.8 Hz, 1H), 3.94 (s, 3H), 3.94 (s, 3H), 3.66 (s, 3H), 3.59 (s, 3H), 3.40 (d, J = 10.9 Hz, 1H), 2.71 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 168.39, 167.22, 151.93, 148.10, 147.56, 146.81, 138.45, 131.07, 127.11, 125.08, 124.31, 124.02, 123.26, 119.81, 118.97, 112.45, 104.45, 101.06, 100.60, 61.01, 57.56, 55.91, 55.28, 52.43, 52.10, 42.22. HR-ESI-MS (m/z) calculated for C26H26O8N [M + H]+ 480.1643, found 480.1653.
Compound 1j: white solid; yield: 30.9%; 1H-NMR (600 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.12 (d, J = 2.2 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.05 (s, 2H), 5.25 (d, J = 10.8 Hz, 1H), 4.25–4.11 (m, 2H), 4.05–3.96 (m, 2H), 3.94 (s, 3H), 3.93 (s, 3H), 3.37 (d, J = 10.8 Hz, 1H), 2.72 (s, 3H), 1.12 (t, J = 7.1 Hz, 3H), 1.04 (t, J = 7.2 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 168.03, 167.03, 151.92, 148.01, 147.50, 146.87, 138.62, 131.05, 127.17, 125.14, 124.53, 123.92, 123.37, 119.81, 118.85, 112.15, 104.41, 101.04, 100.76, 61.25, 61.01, 60.96, 57.35, 55.82, 55.33, 42.11, 13.96, 13.64. HR-ESI-MS (m/z) calculated for C28H29O8N Na [M + Na]+ 530.1776, found 530.1785.
Compound 1k: white solid; yield: 30.9%; 1H-NMR (600 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.52–7.45 (m, 2H), 7.12 (s, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.04 (s, 2H), 5.27 (d, J = 10.9 Hz, 1H), 5.11 (h, J = 6.3 Hz, 1H), 4.84 (p, J = 6.3 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.32 (d, J = 10.9 Hz, 1H), 2.71 (s, 3H), 1.24 (d, J = 6.2 Hz, 3H), 1.11 (dd, J = 9.0, 6.3 Hz, 6H), 0.95 (d, J = 6.4 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 167.65, 166.42, 151.96, 147.98, 147.48, 146.92, 138.71, 131.04, 127.22, 125.24, 124.50, 123.82, 123.52, 119.82, 118.76, 112.00, 104.38, 101.00, 100.90, 68.59, 68.40, 60.85, 56.91, 55.76, 55.48, 41.95, 21.68, 21.62, 21.57, 21.08. HR-ESI-MS (m/z) calculated for C30H33O8N Na [M + Na]+ 558.2089, found 558.2098.
Compound 1l: white solid; yield: 38.6%; 1H-NMR (600 MHz, CDCl3) δ 7.76 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.12 (s, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.05 (dd, J = 8.1, 1.4 Hz, 2H), 5.25 (d, J = 10.8 Hz, 1H), 4.17 (t, J = 6.7 Hz, 2H), 4.09 (qt, J = 10.7, 6.8 Hz, 2H), 3.94 (s, 3H), 3.92 (s, 3H), 3.39 (d, J = 1.9 Hz, 1H), 2.71 (s, 3H), 1.47–1.35 (m, 4H), 1.27–1.11 (m, 4H), 0.85 (dt, J = 12.9, 7.4 Hz, 6H); 13C-NMR (151 MHz, CDCl3) δ 168.09, 167.05, 151.95, 148.02, 147.50, 146.82, 138.65, 131.03, 127.19, 125.14, 124.54, 123.90, 123.41, 119.80, 118.84, 112.06, 104.38, 101.02, 100.79, 65.40, 64.97, 60.95, 57.28, 55.74, 55.31, 42.06, 30.45, 30.21, 19.03, 18.90, 13.70, 13.67. HR-ESI-MS (m/z) calculated for C32H37O8N Na [M + Na]+ 586.2406, found 586.2411.
Compound 2i: light-orange solid; yield: 35.9%; 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.6 Hz, 1H), 7.52 (d, J = 8.5 Hz, 1H), 7.42 (s, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.13 (s, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.09–6.01 (m, 4H), 5.12 (d, J = 11.1 Hz, 1H), 3.70 (s, 3H), 3.61 (s, 3H), 3.47 (d, J = 11.1 Hz, 1H), 2.69 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 167.98, 167.01, 148.16, 147.62, 147.13, 145.35, 138.40, 131.08, 124.48, 124.27, 123.99, 123.34, 120.00, 117.03, 112.39, 108.33, 104.43, 101.51, 101.08, 100.57, 57.33, 55.16, 52.40, 52.31, 42.38. HR-ESI-MS (m/z) calculated for C25H21O8N Na [M + Na]+ 486.1150, found 486.1159.
Compound 2j: white solid; yield: 33.8%; 1H-NMR (600 MHz, CDCl3) δ 7.74 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.45 (s, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.12 (s, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.09–5.97 (m, 4H), 5.13 (d, J = 11.1 Hz, 1H), 4.21 (ddq, J = 40.9, 10.7, 7.1 Hz, 2H), 4.11–3.90 (m, 2H), 3.43 (d, J = 11.1 Hz, 1H), 2.69 (s, 3H), 1.16 (t, J = 7.1 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 167.58, 166.80, 148.08, 147.57, 147.12, 145.40, 138.57, 131.07, 127.26, 126.10, 124.20, 123.44, 120.01, 116.94, 112.57, 108.18, 104.41, 101.44, 101.07, 100.72, 61.34, 61.29, 57.14, 55.33, 42.28, 13.99, 13.77. HR-ESI-MS (m/z) calculated for C27H25O8N Na [M + Na]+ 514.1461, found 514.1472.
Compound 2k: white solid; yield: 25.6%; 1H-NMR (600 MHz, CDCl3) δ 7.74 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.12 (s, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.12–5.76 (m, 4H), 5.17–5.06 (m, 2H), 4.91–4.76 (m, 1H), 3.38 (d, J = 11.1 Hz, 1H), 2.69 (s, 3H), 1.29 (d, J = 6.3 Hz, 3H), 1.16 (d, J = 6.3 Hz, 3H), 1.13 (d, J = 6.2 Hz, 3H), 1.00 (d, J = 6.2 Hz, 3H); 13C-NMR (151 MHz, CDCl3) δ 167.03, 166.35, 148.04, 147.55, 147.14, 145.47, 138.65, 131.06, 127.26, 126.18, 124.11, 123.53, 119.99, 116.87, 112.58, 108.04, 104.37, 101.36, 101.03, 100.87, 68.80, 68.70, 56.71, 55.59, 42.14, 21.76, 21.61, 21.47, 21.23. HR-ESI-MS (m/z) calculated for C29H29O8N Na [M + Na]+ 542.1781, found 542.1785.
Compound 2l: white solid; yield: 28.3%; 1H-NMR (600 MHz, CDCl3) δ 7.74 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.45 (s, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.12 (s, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.11–5.99 (m, 4H), 5.12 (d, J = 11.1 Hz, 1H), 4.18 (dt, J = 10.9, 6.7 Hz, 1H), 4.10 (dt, J = 10.7, 6.8 Hz, 1H), 4.03–3.90 (m, 2H), 3.43 (d, J = 3.2 Hz, 1H), 2.69 (s, 3H), 1.55–1.38 (m, 4H), 1.30–1.23 (m, 2H), 1.22–1.14 (m, 2H), 0.89–0.84 (m, 6H); 13C-NMR (151 MHz, CDCl3) δ 167.59, 166.85, 148.08, 147.56, 147.16, 145.39, 138.59, 131.05, 127.26, 126.09, 124.18, 123.44, 119.98, 116.94, 112.58, 108.15, 104.37, 101.45, 101.05, 100.74, 65.23, 65.19, 57.07, 55.34, 42.24, 30.47, 30.26, 18.98, 18.87, 13.66, 13.63. HR-ESI-MS (m/z) calculated for C31H33O8N Na [M + Na]+ 570.2095, found 570.2098.
3.3.8. Synthesis of Compound 1m
To a stirred solution of 1 (100 mg, 0.287 mmol) in acetone (100 mL) was added a 20% solution of Na2CO3 in water at room temperature. The reaction mixture was stirred under reflux for 24 h. After the reaction was complete, the mixture was concentrated under vacuum. The residue was dissolved in DCM and extracted with saturated aqueous NaCl (3 × 10 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated under vacuum. After that, the crude product was purified by silica gel column chromatography (PE/EA, 4:1) to obtain the target compound.
Compound 1m: white solid; yield: 90.2%; 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.54 (s, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.13 (s, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.11–6.04 (m, 2H), 5.07 (dd, J = 11.2, 3.7 Hz, 1H), 3.98 (s, 3H), 3.95 (s, 3H), 2.66 (s, 3H), 2.60 (dd, J = 15.0, 11.2 Hz, 1H), 2.28 (dd, J = 15.0, 3.7 Hz, 1H), 2.09 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 194.64, 145.84, 142.33, 141.82, 140.00, 134.55, 127.31, 124.79, 124.05, 121.81, 120.98, 120.48, 117.40, 116.55, 110.13, 103.85, 100.94, 100.57, 65.75, 61.19, 60.40, 53.31, 49.78, 39.52. HR-ESI-MS (m/z) calculated for C24H23O5N Na [M + Na]+ 428.1465, found 428.1468.
3.3.9. Synthesis of Compounds 1n–q
Aromatic aldehydes (3 equivalents), benzoic acid (30 mg, 0.246 mmol), and piperidine (300 μL) were added to a stirred solution of 1m (30 mg, 0.074 mmol) in toluene (10 mL) at room temperature. The reaction mixture was stirred under reflux until the reaction was complete and then concentrated under vacuum. After that, the crude products were purified by silica gel column chromatography to obtain the target compounds.
Compound 1n: The crude product was purified by silica gel column chromatography (PE/EA, 7:1) to obtain the target compound 1n: yellow solid; yield: 62.0%; 1H-NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.36 (s, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.01 (d, J = 7.8 Hz, 2H), 6.92–6.86 (m, 2H), 6.70–6.65 (m, 1H), 6.63 (t, J = 2.0 Hz, 1H), 6.48 (d, J = 16.2 Hz, 1H), 5.90 (d, J = 1.5 Hz, 1H), 5.63 (d, J = 1.6 Hz, 1H), 5.14 (dd, J = 11.4, 4.1 Hz, 1H), 4.02 (s, 3H), 3.96 (s, 3H), 3.80 (s, 3H), 2.98 (dd, J = 13.4, 11.4 Hz, 1H), 2.63 (s, 3H), 2.41 (dd, J = 13.5, 4.1 Hz, 1H); 13C-NMR (151 MHz, CDCl3) δ 199.99, 159.58, 152.24, 148.01, 147.41, 145.64, 142.97, 139.42, 135.85, 131.05, 129.42, 128.51, 127.68, 127.15, 124.86, 123.84, 123.37, 121.06, 119.79, 118.87, 116.16, 112.57, 111.60, 103.97, 101.50, 100.93, 61.13, 56.81, 55.85, 55.23, 42.94, 42.74. HR-ESI-MS (m/z) calculated for C32H29O6N Na [M + Na]+ 546.1876, found 546.1887.
Compound 1o: The crude product was purified by silica gel column chromatography (PE/EA, 8:1) to obtain the target compound 1o: yellow liquid; yield: 58.6%; 1H-NMR (600 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.36 (s, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.05 (s, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.90 (d, J = 16.3 Hz, 1H), 6.77 (s, 1H), 6.47 (d, J = 16.3 Hz, 1H), 5.88 (d, J = 1.6 Hz, 1H), 5.54 (d, J = 1.6 Hz, 1H), 5.14 (dd, J = 11.4, 4.0 Hz, 1H), 4.02 (s,3H), 3.97 (s, 3H), 2.97 (dd, J = 13.4, 11.4 Hz, 1H), 2.63 (s, 3H), 2.39 (dd, J = 13.4, 4.1 Hz, 1H), 2.32 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 200.01, 152.25, 148.00, 147.40, 145.65, 143.28, 139.47, 138.08, 134.43, 131.05, 130.76, 128.96, 128.59, 128.37, 127.35, 127.17, 125.25, 124.87, 123.85, 123.40, 119.82, 118.88, 111.59, 103.87, 101.56, 100.88, 61.14, 56.83, 55.85, 42.97, 42.72, 21.16. HR-ESI-MS (m/z) calculated for C32H29O5N Na [M + Na]+ 530.1925, found 530.1938.
Compound 1p: The crude product was purified by silica gel column chromatography (PE/EA, 8:1) to obtain the target compound 1p: yellow liquid; yield: 47.2%; 1H-NMR (600 MHz, CDCl3) δ 7.80 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.55 (d, J = 8.6 Hz, 1H), 7.44 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.32 (s, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.08 (d, J = 1.8 Hz, 1H), 7.06 (s, 1H), 7.02 (dd, J = 8.8, 2.0 Hz, 2H), 6.76 (d, J = 16.3 Hz, 1H), 6.43 (d, J = 16.3 Hz, 1H), 5.91 (d, J = 1.6 Hz, 1H), 5.68 (d, J = 1.6 Hz, 1H), 5.12 (dd, J = 11.4, 4.3 Hz, 1H), 4.02 (s, 3H), 3.97 (s, 3H), 2.98 (dd, J = 13.2, 11.4 Hz, 1H), 2.62 (s, 3H), 2.39 (dd, J = 13.3, 4.2 Hz, 1H); 13C-NMR (151 MHz, CDCl3) δ 199.84, 152.27, 148.02, 147.35, 145.63, 141.19, 139.30, 136.59, 132.61, 131.09, 130.80, 129.91, 128.62, 128.37, 127.00, 126.62, 124.78, 123.96, 123.42, 122.61, 119.86, 118.94, 111.65, 104.11, 101.35, 101.00, 61.15, 57.03, 55.85, 42.90, 42.71. HR-ESI-MS (m/z) calculated for C31H27O5NBr [M + Na]+ 572.1057, found 572.1067.
Compound 1q: The crude product was purified by silica gel column chromatography (PE/EA, 6:1) to obtain the target compound 1q: yellow solid; yield: 29.9%; 1H-NMR (600 MHz, CDCl3) δ 7.77 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.33 (s, 1H), 7.17–7.12 (m, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.95 (s, 1H), 6.93 (s, 1H), 6.85 (s, 1H), 6.38 (d, J = 16.2 Hz, 1H), 5.89 (d, J = 1.6 Hz, 1H), 5.72 (d, J = 1.6 Hz, 1H), 5.15 (dd, J = 11.3, 4.2 Hz, 1H), 4.03 (s, 3H), 3.97 (s, 3H), 3.91 (s, 3H), 3.87 (s, 3H), 3.06 (dd, J = 13.3, 11.3 Hz, 1H), 2.63 (s, 3H), 2.42 (dd, J = 13.4, 4.2 Hz, 1H); 13C-NMR (151 MHz, CDCl3) δ 200.08, 152.23, 151.09, 148.21, 147.70, 147.10, 145.69, 141.30, 139.29, 131.23, 128.44, 127.80, 127.03, 126.26, 124.93, 124.18, 123.42, 119.77, 118.88, 117.79, 115.28, 111.63, 109.18, 104.10, 101.47, 100.96, 61.15, 56.96, 56.32, 55.87, 42.76, 31.45, 30.21; HR-ESI-MS (m/z) calculated for C33H30O7NBr Na [M + Na]+ 654.1085, found 654.1098.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





































