
The Structural Rule Distinguishing a Superfold: A Case Study of Ferredoxin Fold and the Reverse Ferredoxin Fold


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
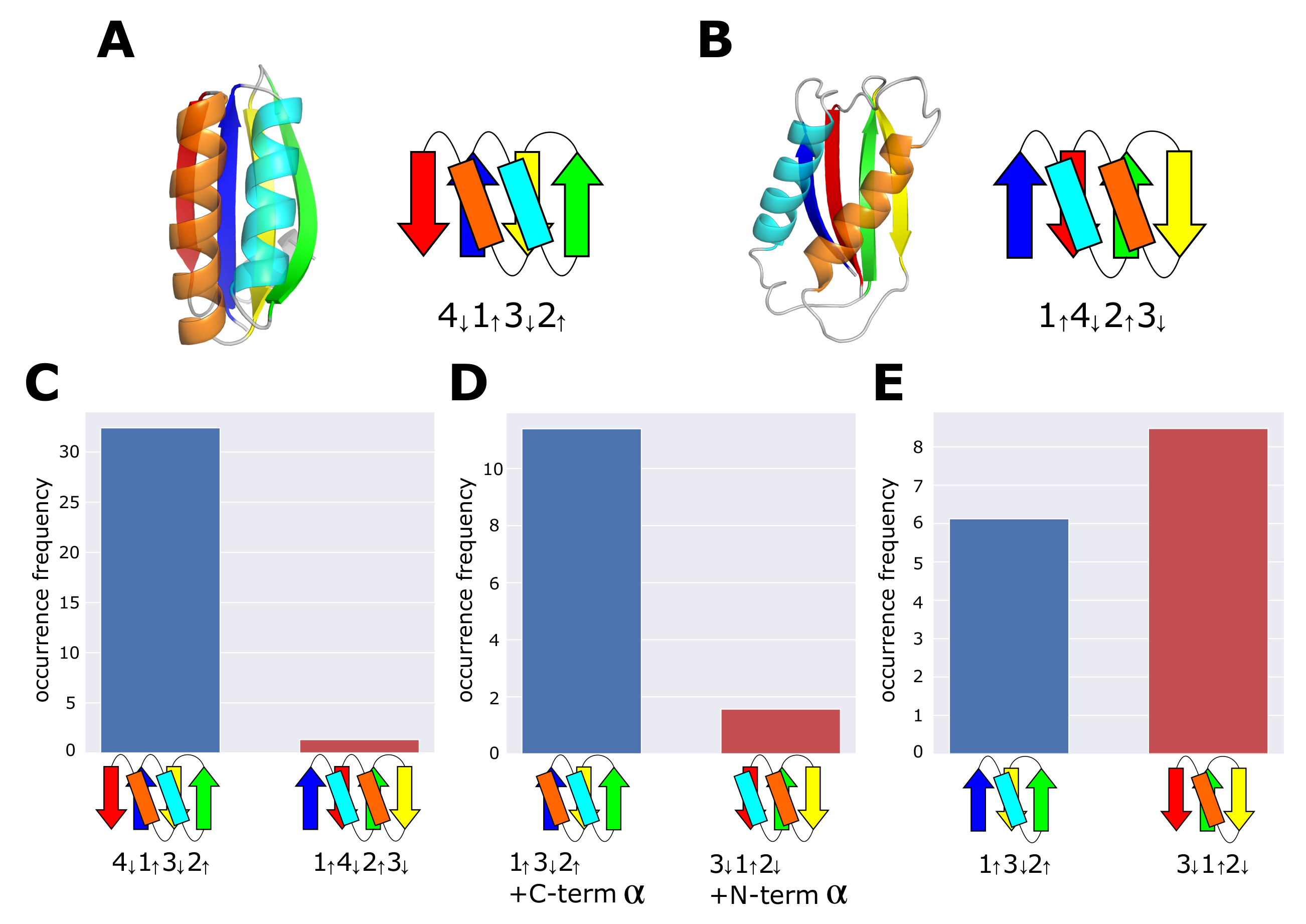
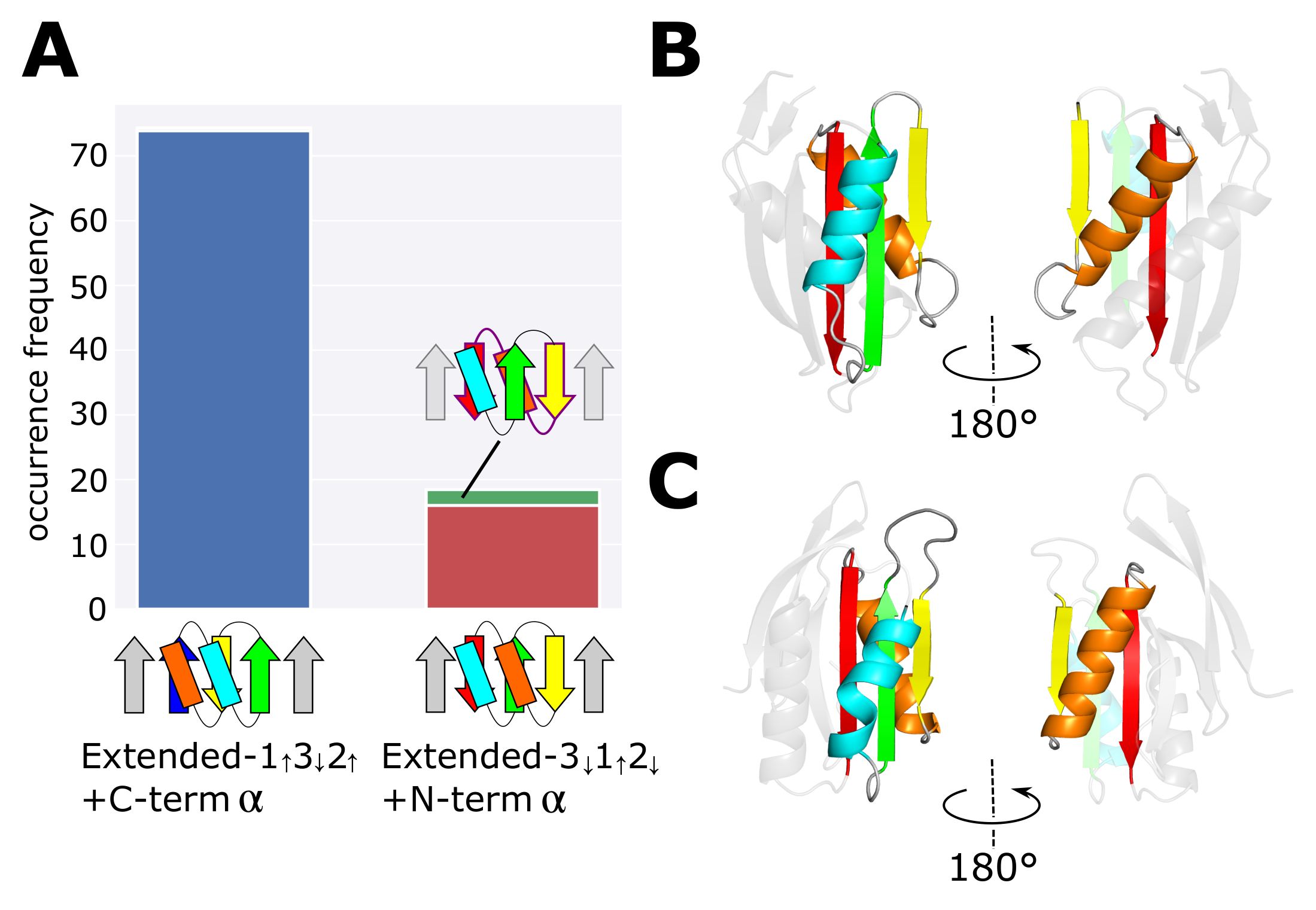
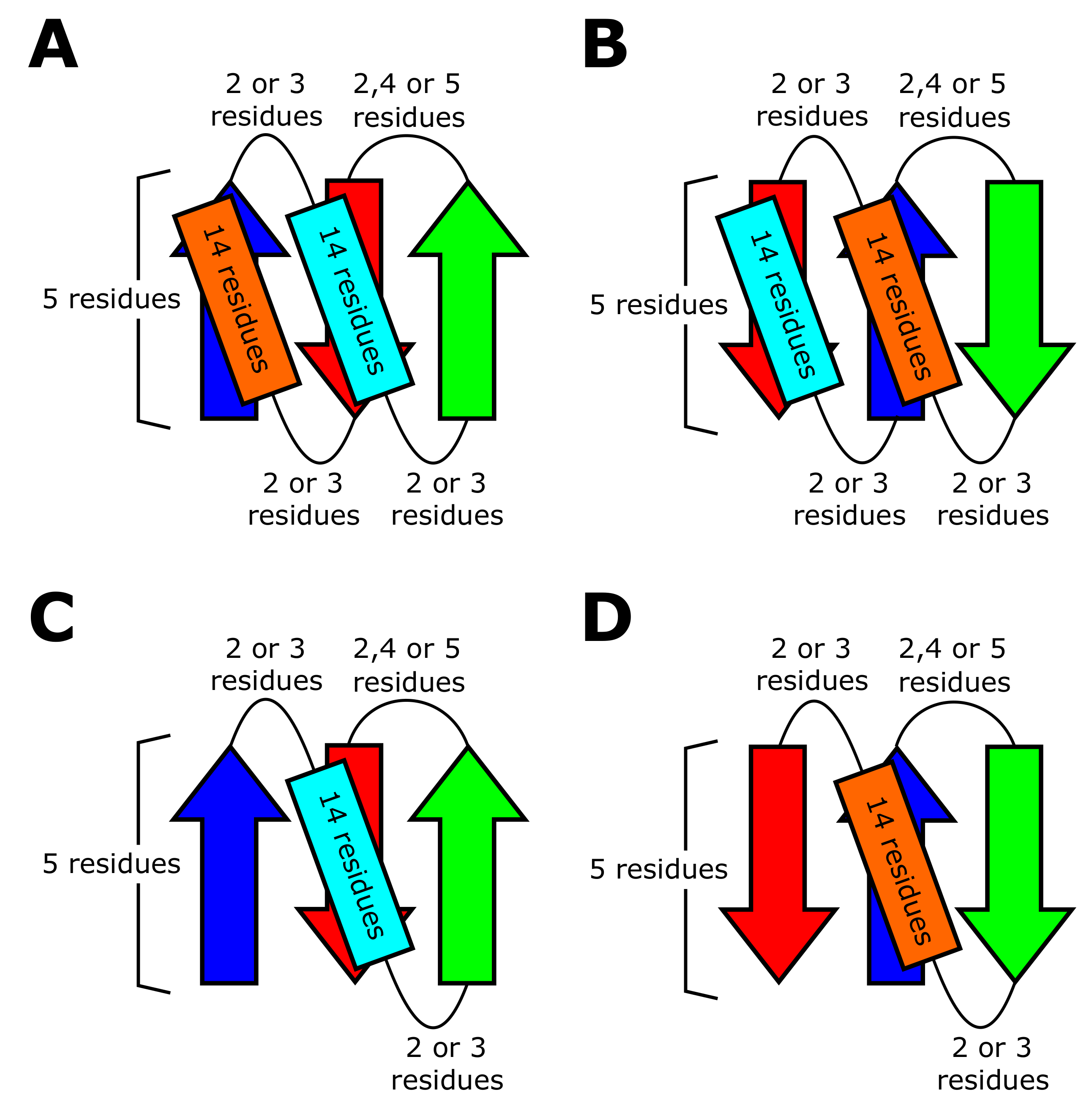
2.1. Occurrence Frequency of Topologies
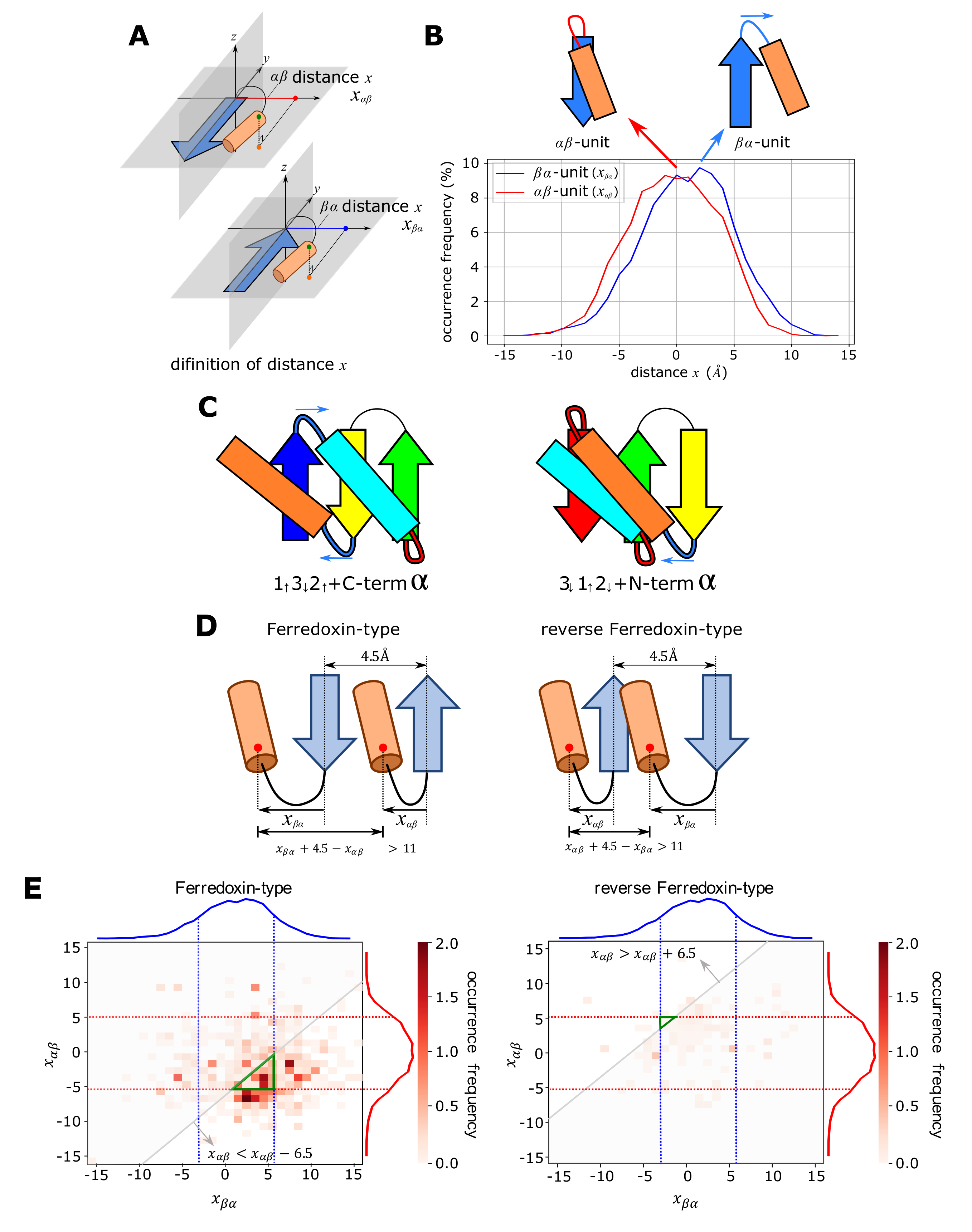
2.2. Conflict between Structural Preferences of - and -Units
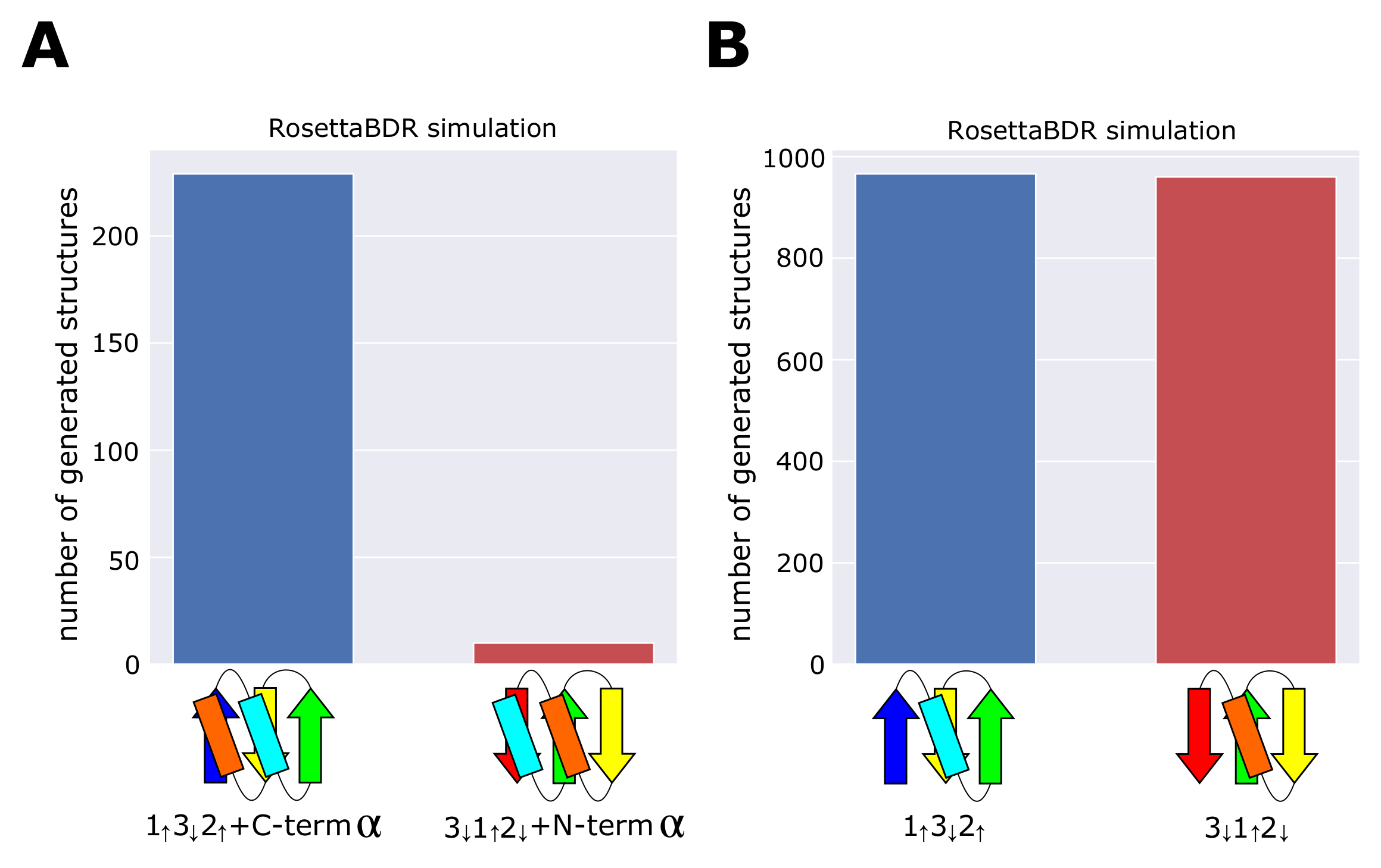
2.3. Minimum Frustration Rule
3. Discussion
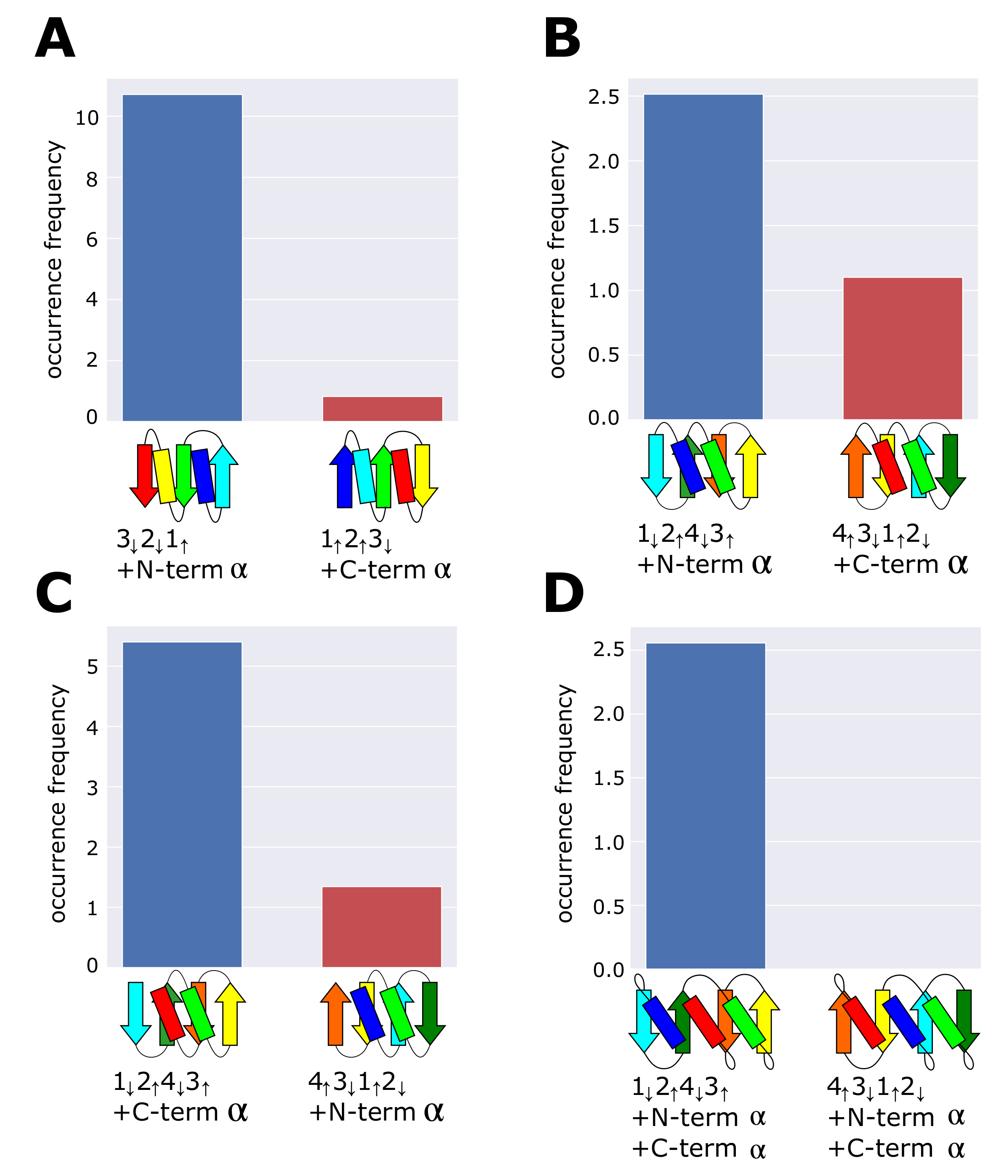
3.1. Occurrence Frequency of Other Structures
3.2. The Left-Handed -Unit Is Selectively Found in the Structures
3.3. Frustration and Function
4. Materials and Methods
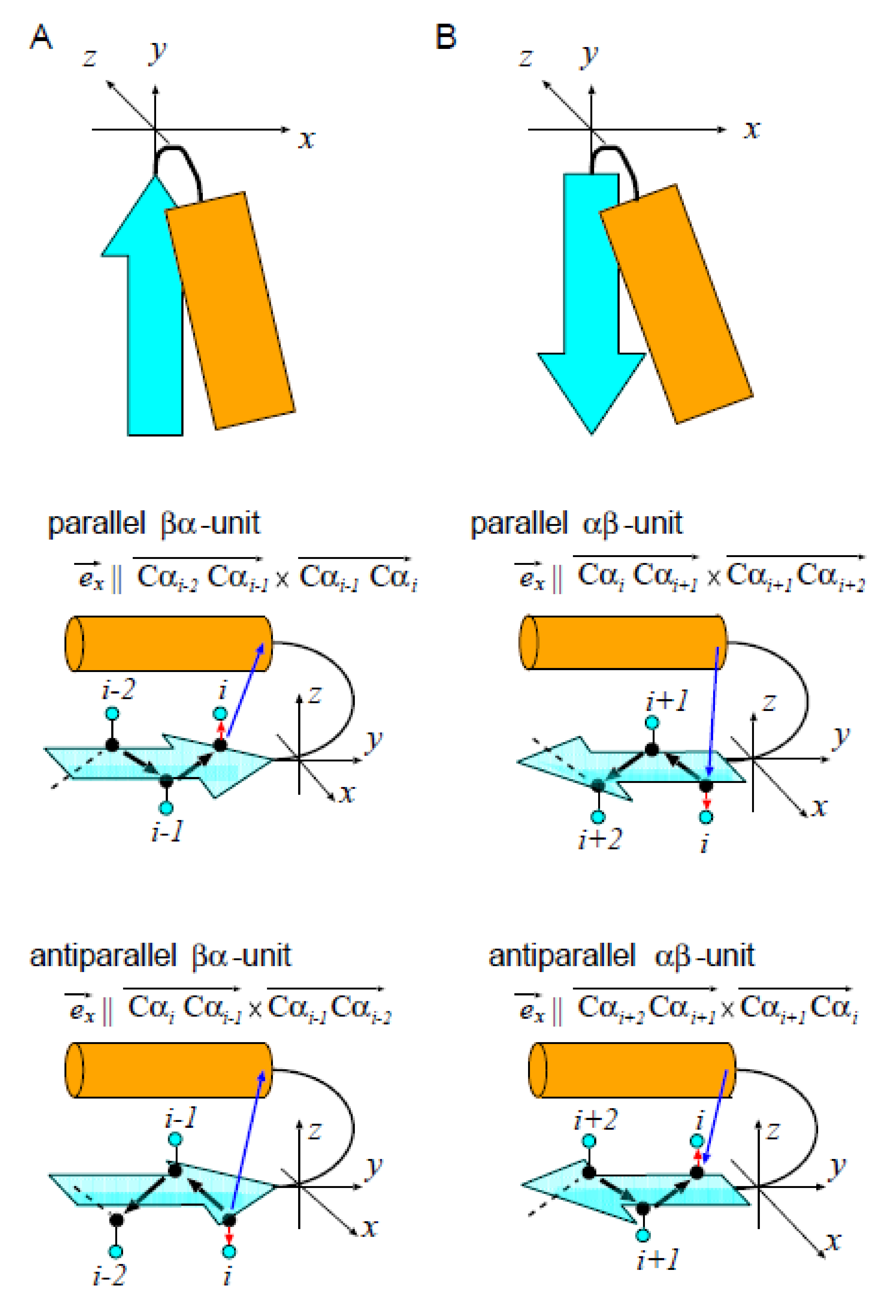
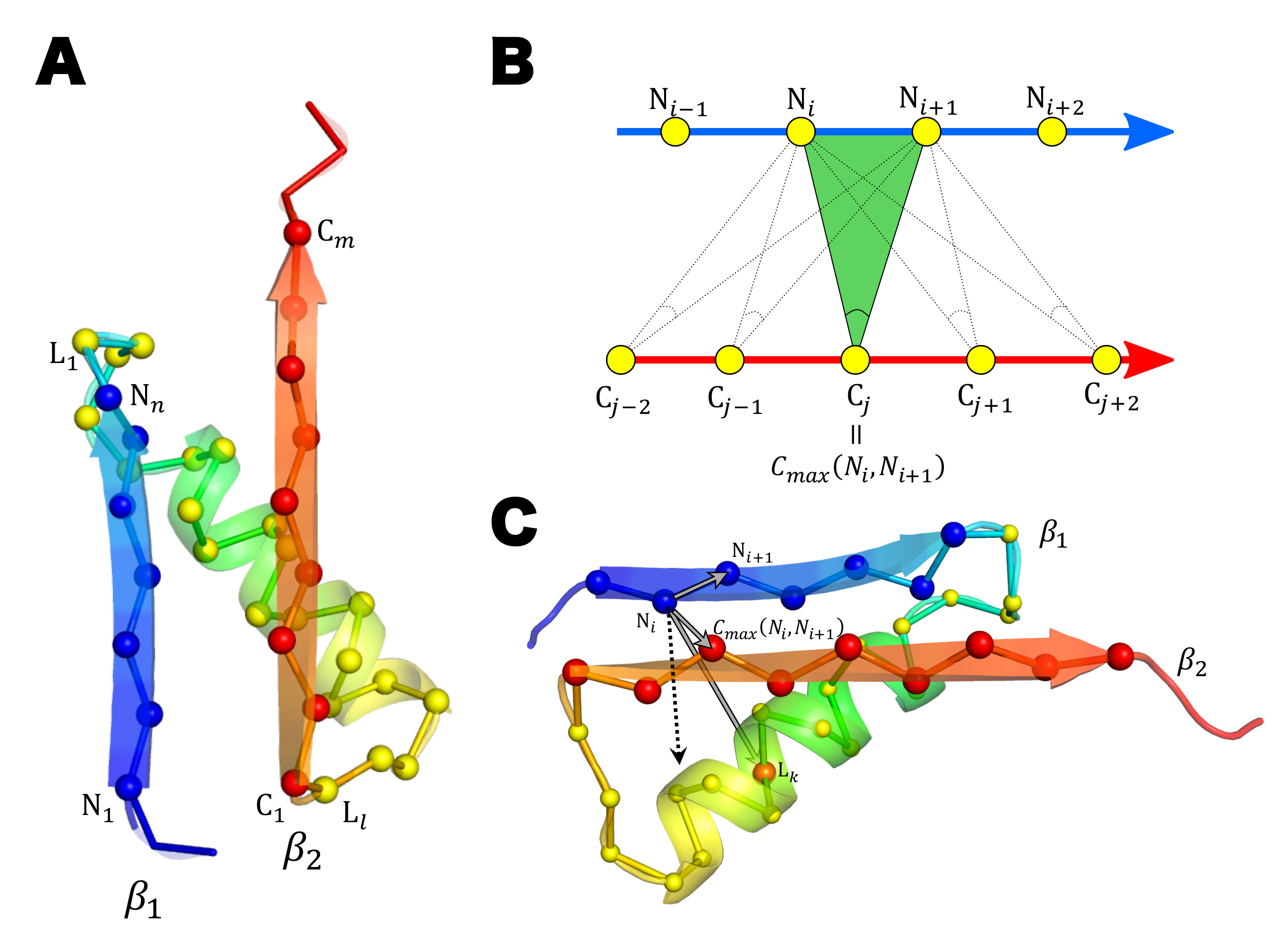
4.1. Detecting the Position of the C/N Terminal -Helix
4.2. Definition of the Distance x between the Plane of -Pleats and the -Helix in the - or -Unit
4.3. Rosetta Folding Simulations
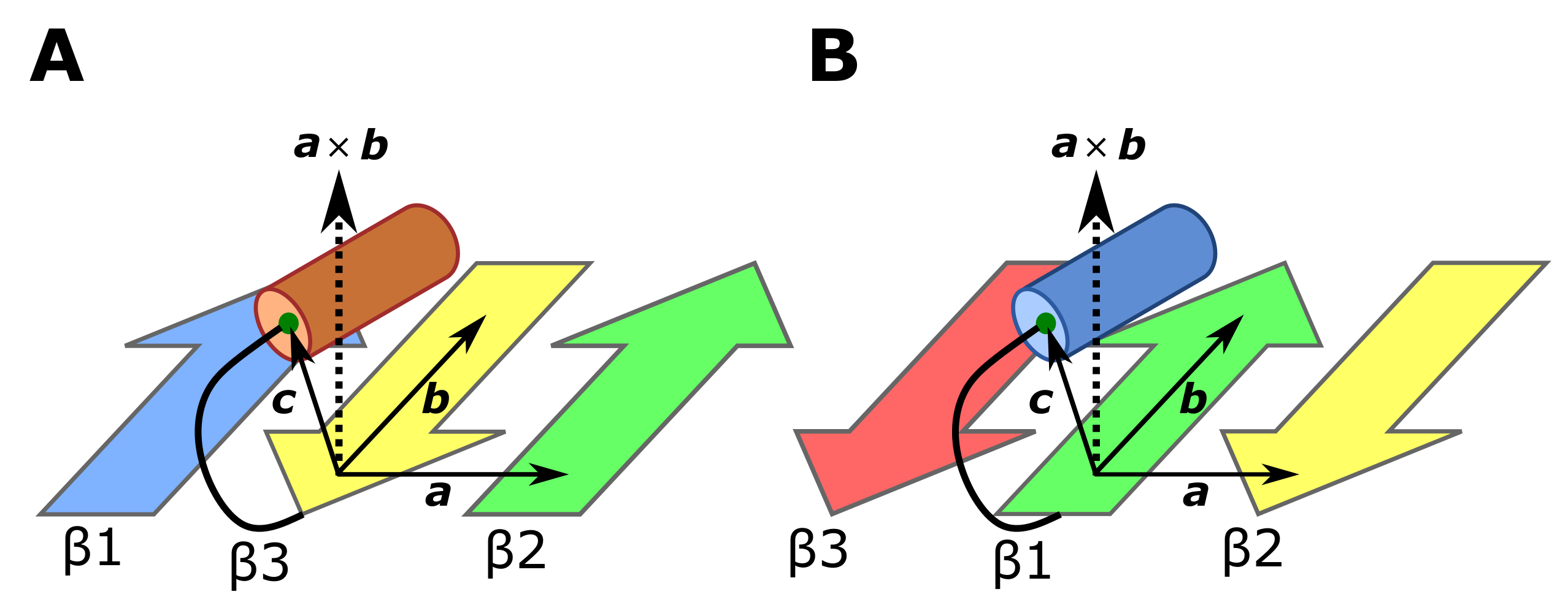
4.4. Score to Detect the Left-Handed -Unit
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCOP | Structural Classification of Proteins |
| ECOD | Evolutionary Classification of protein Domains |
| PDB | Protein Data Bank |
References
- Tramontano, A.; Cozzetto, D. Supramolecular Structure and Function 8; Springer: Berlin/Heidelberg, Germany, 2004; pp. 15–29. [Google Scholar]
- Sadowski, M.I.; Jones, D.T. The sequence-structure relationship and protein function prediction. Curr. Opin. Str. Biol. 2019, 19, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Tatsumi-Koga, R.; Liu, G.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Baker, D. Principles for designing ideal protein structures. Nature 2012, 491, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos, E.; Chidyausiku, T.K.; McShan, A.C.; Evangelidis, T.; Nerli, S.; Carter, L.; Nivón, L.G.G.; Davis, A.; Oberdorfer, G.; Tripsianes, K.; et al. De novo design of a non-local β-sheet protein with high stability and accuracy. Nat. Struct. Mol. Biol. 2018, 25, 1028–1034. [Google Scholar] [CrossRef]
- Murata, H.; Imakawa, H.; Koga, N.; Chikenji, G. The register shift rules for βαβ-motifs for de novo protein design. PLoS ONE 2021, 16, e0256895. [Google Scholar] [CrossRef]
- Minami, S.; Kobayashi, N.; Sugiki, T.; Nagashima, T.; Fujiwara, T.; Koga, R.; Chikenji, G.; Koga, N. Exploration of novel αβ-protein folds through de novo design. bioRxiv 2021. [Google Scholar] [CrossRef]
- Huang, P.S.; Feldmeier, K.; Parmeggiani, F.; Velasco, D.A.F.; Höcker, B.; Baker, D. De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat. Chem. Biol. 2016, 12, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Dou, J.; Vorobieva, A.A.; Sheffler, W.; Doyle, L.A.; Park, H.; Bick, M.J.; Mao, B.; Foight, G.W.; Lee, M.Y.; Gagnon, L.A.; et al. De novo design of a fluorescence-activating β-barrel. Nature 2018, 561, 485–491. [Google Scholar] [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.; Hallinan, J.; Bolduc, J.; Parmeggiani, F.; Baker, D.; Stoddard, B.L.; Bradley, P. Rational design of α-helical tandem repeat proteins with closed architectures. Nature 2015, 528, 585–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, F.; Zhang, Y.; Liu, X.; Zhang, J. Estimating the designability of protein structures. bioRxiv 2021. [Google Scholar] [CrossRef]
- Richardson, J.S. beta-Sheet topology and the relatedness of proteins. Nature 1977, 268, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar]
- Ruczinski, I.; Kooperberg, C.; Bonneau, R.; Baker, D. Distributions of beta sheets in proteins with application to structure prediction. Proteins 2002, 48, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Chitturi, B.; Shi, S.; Kinch, L.N.; Grishin, N.V. Compact Structure Patterns in Proteins. J. Mol. Biol. 2016, 428, 4392–4412. [Google Scholar] [CrossRef]
- Minami, S.; Chikenji, G.; Ota, M. Rules for connectivity of secondary structure elements in protein: Two-layer αβ sandwiches. Protein Sci. 2017, 26, 2257–2267. [Google Scholar] [CrossRef]
- Orengo, C.A.; Jones, D.T.; Thornton, J.M. Protein superfamilles and domain superfolds. Nature 1994, 372, 631–634. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Salem, G.M.; Hutchinson, E.G.; Orengo, C.A.; Thornton, J.M. Correlation of observed fold frequency with the occurrence of local structural motifs. J. Mol. Biol. 1999, 287, 969–981. [Google Scholar] [CrossRef]
- Kinoshita, K.; Kidera, A.; Go, N. Diversity of functions of proteins with internal symmetry in spatial arrangement of secondary structural elements. Protein Sci. 1999, 8, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef] [PubMed]
- Chandonia, J.M.; Fox, N.K.; Brenner, S.E. SCOPe: Classification of large macromolecular structures in the structural classification of proteins-extended database. Nucleic Acids Res. 2019, 47, D475–D481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Kim, S.H. The anatomy of protein beta-sheet topology. J. Mol. Biol. 2000, 299, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Schaeffer, R.D.; Liao, Y.; Kinch, L.N.; Pei, J.; Shi, S.; Kim, B.H.; Grishin, N.V. ECOD: An evolutionary classification of protein domains. PLoS Comput. Biol. 2014, 10, e1003926. [Google Scholar] [CrossRef]
- Andreeva, A.; Howorth, D.; Chandonia, J.M.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. Data growth and its impact on the SCOP database: New developments. Nucleic Acids Res. 2008, 36, D419–D425. [Google Scholar] [CrossRef] [Green Version]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef] [Green Version]
- Street, T.O.; Fitzkee, N.C.; Perskie, L.L.; Rose, G.D. Physical-chemical determinants of turn conformations in globular proteins. Protein Sci. 2007, 16, 1720–1727. [Google Scholar] [CrossRef] [Green Version]
- Lesk, A.M.; Brändén, C.I.; Chothia, C. Structural principles of α/β barrel proteins: The packing of the interior of the sheet. Proteins Str. Funct. Bioinform. 1989, 5, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Finkelstein, A.V. General architecture of the α-helical globule. J. Mol. Biol. 1988, 204, 749–769. [Google Scholar] [CrossRef]
- Nagi, A.D.; Regan, L. An inverse correlation between loop length and stability in a four-helix-bundle protein. Fold. Des. 1997, 2, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Linse, S.; Thulin, E.; Nilsson, H.; Stigler, J. Benefits and constrains of covalency: The role of loop length in protein stability and ligand binding. Sci. Rep. 2020, 10, 20108. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S. Handedness of crossover connections in beta sheets. Proc. Natl. Acad. Sci. USA 1976, 73, 2619–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, M.J.E.; Thornton, J.M. On the conformation of proteins: The handedness of the connection between parallel β-strands. J. Mol. Biol. 1976, 110, 269–283. [Google Scholar] [CrossRef]
- Cole, B.J.; Bystroff, C. Alpha helical crossovers favor right-handed supersecondary structures by kinetic trapping: The phone cord effect in protein folding. Protein Sci. 2009, 18, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, D.U.; Komives, E.A.; Wolynes, P.G. Frustration, function and folding. Curr. Opin. Struct. Biol. 2018, 48, 68–73. [Google Scholar] [CrossRef]
- Parra, R.G.; Schafer, N.P.; Radusky, L.G.; Tsai, M.Y.; Guzovsky, A.B.; Wolynes, P.G.; Ferreiro, D.U. Protein frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 2016, 44, W356–W360. [Google Scholar] [CrossRef]
- Ferreiro, D.U.; Hegler, J.A.; Komives, E.A.; Wolynes, P.G. On the role of frustration in the energy landscapes of allosteric proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 3499–3503. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, D.U.; Hegler, J.A.; Komives, E.A.; Wolynes, P.G. Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. USA 2007, 104, 19819–19824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleishman, S.; Leaver-Fay, A.; Corn, J.E.; Strauch, E.M.; Khare, S.D.; Koga, N.; Ashworth, J.; Murphy, P.; Richter, F.; Lemmon, G.; et al. RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 2011, 6, e20161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.R.; Koga, N.; Tatsumi-Koga, R.; Liu, G.; Clouser, A.F.; Montelione, G.T.; Baker, D. Control over overall shape and size in de novo designed proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E5478–E5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wintjens, R.T.; Rooman, M.J.; Wodak, S.J. Automatic classification and analysis of alpha alpha-turn motifs in proteins. J. Mol. Biol. 1996, 255, 235–253. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishina, T.; Nakajima, M.; Sasai, M.; Chikenji, G. The Structural Rule Distinguishing a Superfold: A Case Study of Ferredoxin Fold and the Reverse Ferredoxin Fold. Molecules 2022, 27, 3547. https://doi.org/10.3390/molecules27113547
Nishina T, Nakajima M, Sasai M, Chikenji G. The Structural Rule Distinguishing a Superfold: A Case Study of Ferredoxin Fold and the Reverse Ferredoxin Fold. Molecules. 2022; 27(11):3547. https://doi.org/10.3390/molecules27113547
Chicago/Turabian StyleNishina, Takumi, Megumi Nakajima, Masaki Sasai, and George Chikenji. 2022. "The Structural Rule Distinguishing a Superfold: A Case Study of Ferredoxin Fold and the Reverse Ferredoxin Fold" Molecules 27, no. 11: 3547. https://doi.org/10.3390/molecules27113547