
Hetero-Diels-Alder Reactions of In Situ-Generated Azoalkenes with Thioketones; Experimental and Theoretical Studies †

 , , , ,
, , , ,
Abstract
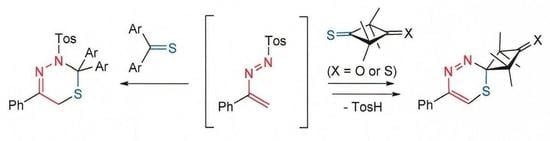
:
1. Introduction
2. Results and Discussion
2.1. Experimental Work
2.2. Mechanistic Investigations by DFT Calculations
3. Materials and Methods
3.1. Materials
3.2. Analytical Methods and Equipment
3.3. Quantum Chemical Calculations
3.4. Synthesis
Reactions of Azoalkenes 7a,b with Thioketones 1a–k and Thiochalcone 1l—General Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mlostoń, G.; Grzelak, P.; Hamera-Faldyga, R.; Jasiński, M.; Pipiak, P.; Urbaniak, K.; Albrecht, Ł.; Hejmanowska, J.; Heimgartner, H. Aryl, hetaryl, and ferrocenyl thioketones as versatile building blocks for exploration in the organic chemistry of sulfur. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 204–211. [Google Scholar] [CrossRef]
- Huisgen, R.; Fisera, L.; Giera, H.; Sustmann, R. Thiones as superdipolarophiles. Rates and equilibria of nitrone cycloadditions to thioketones. J. Am. Chem. Soc. 1995, 117, 9671–9685. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Jasiński, M.; Würthwein, E.-U.; Heimgartner, H.; Zimmer, R.; Reissig, H.-U. The [4+2]-cycloaddition of α-nitrosoalkenes with thiochalcones as a prototype of periselective hetero-Diels-Alder reactions—Experimental and computational studies. Chem. Eur. J. 2020, 26, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. First (3+2)-cycloadditions of thiochalcones as C=S dipolarophiles: Efficient synthesis of 1,3,4-thiadiazoles via reactions with fluorinated nitrile imines. Synthesis 2017, 49, 2129–2137. [Google Scholar]
- Huisgen, R.; Li, X.; Giera, H.; Langhals, E. “Thiobenzophenone S-methylide” (=(diphenylmethylidenesulfonio)methanide), and C,C multiple bonds: Cycloadditions and dipolarophilic reactivities. Helv. Chim. Acta 2001, 84, 981–999. [Google Scholar] [CrossRef]
- Rohr, U.; Schatz, J.; Sauer, J. Thio- and selenocarbonyl compounds as “superdienophiles” in [4+2] cycloadditions. Eur. J. Org. Chem. 1998, 1998, 2875–2883. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Selenophen-2-yl substituted thiocarbonyl ylides—At the borderline of dipolar and diradical reactivity. Helv. Chim. Acta 2015, 98, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Pipiak, P.; Linden, A.; Heimgartner, H. Studies on the reactions of thiocarbonyl S-methanides with hetaryl thioketones. Helv. Chim. Acta 2015, 98, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Pipiak, P.; Heimgartner, H. Diradical reaction mechanisms in [3+2]-cycloadditions of hetaryl thioketones with alkyl- or trimethylsilyl-substituted diazomethanes. Beilstein J. Org. Chem. 2016, 12, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Hamera-Fałdyga, R.; Celeda, M.; Heimgartner, H. Efficient synthesis of ferrocifenes and other ferrocenyl substituted ethylenes via a ‘sulfur approach’. Org. Biomol. Chem. 2018, 16, 4350–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlostoń, G.; Jasiński, R.; Kula, K.; Heimgartner, H. A DFT study on the Barton-Kellogg reaction—The molecular mechanism of the formation of thiiranes in the reaction between diphenyldiazomethane and diaryl thioketones. Eur. J. Org. Chem. 2020, 2020, 176–182. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Utecht, G.; Lentz, D.; Jasiński, M. Trifluoromethylated 2,3-dihydro-1,3,4-thiadiazoles via the regioselective [3+2]-cycloadditions of fluorinated nitrile imines with aryl, hetaryl, and ferrocenyl thioketones. J. Fluor. Chem. 2016, 192, 147–154. [Google Scholar] [CrossRef]
- Ali, K.A.; Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. [3+2]-Cycloadditions of nitrile imines with hetaryl thioketones. J. Sulfur Chem. 2017, 38, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Kowalski, M.K.; Obijalska, E.; Heimgartner, H. Efficient synthesis of fluoroalkylated 1,4,2-oxathiazoles via regioselective [3+2]-cycloaddition of fluorinated nitrile oxides with thioketones. J. Fluor. Chem. 2017, 199, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Kowalczyk, M.; Augustin, A.U.; Jones, P.G.; Werz, D.B. Ferrocenyl substituted tetrahydrothiophenes via formal [3+2]-cycloaddition reactions of ferrocenyl thioketones with donor-acceptor cyclopropanes. Beilstein J. Org. Chem. 2020, 16, 1288–1295. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Taming of thioketones—The first asymmetric thio-Diels-Alder reaction with thioketones as heterodienophiles. Eur. J. Org. Chem. 2017, 2017, 950–954. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Woodward, R.B.; Hoffmann, R. The conservation of orbital symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- McKee, M.L.; Mlostoń, G.; Urbaniak, K.; Heimgartner, H. Dimerization reactions of aryl selenophen-2-yl-substituted thiocarbonyl S-methanides as diradical processes: A computational study. Beilstein J. Org. Chem. 2017, 13, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiński, R.; Dresler, E. On the question of zwitterionic intermediates in the [3+2] cycloaddition reactions: A critical review. Organics 2020, 1, 49–69. [Google Scholar] [CrossRef]
- Mlostoń, G.; Grzelak, P.; Linden, A.; Heimgartner, H. Thia-Diels–Alder reactions of hetaryl thioketones with nonactivated 1,3-dienes leading to 3,6-dihydro-2H-pyrans: Evidence for a diradical mechanism. Chem. Heterocycl. Compd. 2017, 53, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Urbaniak, K.; Zimmer, R.; Reissig, H.-U.; Heimgartner, H. Hetero-Diels-Alder reactions of conjugated nitrosoalkenes with ferrocenyl, hetaryl and cycloaliphatic thioketones. Chem. Sel. 2018, 3, 11724–11728. [Google Scholar]
- Attanasi, O.A.; De Crescentini, L.; Filippone, P.; Mantellini, F.; Santeusanio, S. 1,2-Diaza-1,3-butadienes; just a nice class of compounds, or powerful tools in organic chemistry? Reviewing an experience. ARKIVOC 2002, xi, 274–292. [Google Scholar] [CrossRef] [Green Version]
- Lemos, A. Addition and cycloaddition reactions of phosphinyl- and phosphonyl-2H-azirines, nitrosoalkenes and azoalkenes. Molecules 2009, 14, 4098–4119. [Google Scholar] [CrossRef] [Green Version]
- Attanasi, O.A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F.R.; Santeusanio, S. Cultivating the passion to build heterocycles from 1,2-diaza-1,3-dienes; the force of imagination. Eur. J. Org. Chem. 2009, 2009, 3109–3127. [Google Scholar] [CrossRef]
- Lopes, S.M.M.; Cardoso, A.L.; Lemos, A.; Pinho e Melo, T.M.V.D. Recent advances in the chemistry of conjugated nitrosoalkenes and azoalkenes. Chem. Rev. 2018, 118, 11324–11352. [Google Scholar] [CrossRef]
- Wei, L.; Shen, C.; Hu, Y.-Z.; Tao, H.-Y.; Wang, C.-J. Enantioselective synthesis of multi-nitrogen containing heterocycles using azoalkenes as key intermediates. Chem. Commun. 2019, 55, 6672–6684. [Google Scholar] [CrossRef]
- Gao, S.; Chen, J.-R.; Hu, X.-Q.; Cheng, H.-G.; Lu, L.-Q.; Xiao, W.-J. Copper-catalyzed enantioselective inverse electron-demand hetero-Diels–Alder reactions of diazadienes with enol ethers: Efficient synthesis of chiral pyridazines. Adv. Synth. Catal. 2013, 355, 3539–3544. [Google Scholar] [CrossRef]
- Zhong, X.; Lv, J.; Luo, S. [4+2] Cycloaddition of in situ generated 1,2-diaza-1,3-dienes with simple olefins: Facile approaches to tetrahydropyridazines. Org. Lett. 2015, 17, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Tao, H.-Y.; Wang, C.-J. PPh3-Mediated [4+2]- and [4+1]-annulations of maleimides with azoalkenes: Access to fused tetrahydropyridazine/pyrrolidinedione and spiro-dihydropyrazole/pyrrolidinedione derivatives. Org. Lett. 2017, 19, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Mei, G.-J.; Zheng, W.; Gonçalves, T.P.; Tang, X.; Huang, K.-W.; Lu, Y. Catalytic Asymmetric formal [3+2] cycloaddition of azoalkenes with 3-vinylindoles: Synthesis of 2,3-dihydropyrroles. iScience 2020, 23, e100873. [Google Scholar] [CrossRef] [Green Version]
- Bonini, B.F.; Maccagnani, G.; Mazzanti, G.; Rosini, G.; Foresti, E. Cycloaddition reactions of sulphines and thiones with azoalkenes. J. Chem. Soc. Perkin Transl. I 1981, 1, 2322–2327. [Google Scholar] [CrossRef]
- Quan, B.-X.; Zhuo, J.-R.; Zhao, J.-Q.; Zhang, M.-L.; Zhou, M.-Q.; Zhang, X.-M.; Yuan, W.-C. [4+1] annulation reaction of cyclic pyridinium ylides with in situ generated azoalkenes for the construction of spirocyclic skeletons. Org. Biomol. Chem. 2020, 18, 1886–1891. [Google Scholar] [CrossRef]
- Johnson, C.K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Mlostoń, G.; Grzelak, P.; Maciej, M.; Linden, A.; Heimgartner, H. Hetero-Diels-Alder reactions of hetaryl and aryl thioketones with acetylenic dienophiles. Beilstein J. Org. Chem. 2015, 11, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Nonhebel, D.C. The chemistry of cyclopropylmethyl and related radicals. Chem. Soc. Rev. 1993, 22, 347–359. [Google Scholar] [CrossRef]
- Firestone, R.A. The low energy of concert in many symmetry-allowed cycloadditions supports a stepwise-diradical mechanism. Int. J. Chem. Kinet. 2013, 45, 415–428. [Google Scholar] [CrossRef]
- Firestone, R.A. The diradical mechanism for 1,3-dipolar cycloadditions and related thermal pericyclic reactions. Tetrahedron 1977, 33, 3009–3039. [Google Scholar] [CrossRef]
- Matczak, P.; Mlostoń, G.; Hamera-Fałdyga, R.; Görls, H.; Weigand, W. Structure of diferrocenyl thioketone: From molecule to crystal. Molecules 2019, 24, e3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Errata: Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Weigend, R.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Scheibye, S.; Nilsson, N.H.; Lawesson, S.O. Studies on organophosphorus compounds. 10. Syntheses of thioketones. Bull. Soc. Chim. Belg. 1978, 87, 223–228. [Google Scholar] [CrossRef]
- Campaigne, E.; Reid, W.B. Thiocarbonyls. 2. Thiofluorenone. J. Amer Chem. Soc. 1946, 68, 769–770. [Google Scholar] [CrossRef]
- Elam, E.U.; Davis, H.E. Chemistry of dimethylketene dimer. 7. Dimers of dimethylthioketene. J. Org. Chem. 1967, 32, 1562–1565. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Coltart, D.M. Copper(I)-catalyzed addition of Grignard reagents to in situ derived N-sulfonyl azoalkenes: An umpolung alkylation procedure applicable to the formation of up to three contiguous quaternary centers. J. Am. Chem. Soc. 2010, 132, 4546–4547. [Google Scholar] [CrossRef]
- Lai, E.C.K.; Mackay, D.; Taylor, N.J.; Kenneth, N.; Watson, K.N. Competitive [4+2] and [3+2] cycloadditions of nitrosoalkenes to the lmino bond of bicyclic 1,2-oxazines. J. Chem. Soc. Perkin Trans. 1 1990, 1990, 1497–1506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chen, Z.; Meng, L.; Ding, Z.; Hu, J. Construction of Versatile N-heterocycles from in situ generated 1,2-diaza-1,3-dienes. Curr. Org. Chem. 2019, 23, 164–187. [Google Scholar] [CrossRef]
- Grosso, C.; Lieber, M.; Brigas, A.F.; Pinho e Melo, T.M.V.D.; Lemos, A. Regioselectivity in hetero Diels−Alder reactions. J. Chem. Educ. 2019, 96, 148–152. [Google Scholar] [CrossRef]
- Zhao, H.-W.; Pang, H.-L.; Zhao, Y.-D.; Liu, Y.-Y.; Zhao, L.-J.; Chen, X.-Q.; Song, X.-Q.; Feng, N.-N.; Du, J. Construction of 2,3,4,5-tetrahydro-1,2,4-triazines via [4+2] cycloaddition of α-halogeno hydrazones to imines. RSC Adv. 2017, 7, 9264–92760. [Google Scholar] [CrossRef] [Green Version]
- Wie, L.; Zhu, Q.; Song, Z.-M.; Liu, K.; Wang, C.-J. Catalytic asymmetric inverse electron demand Diels–Alder reaction of fulvenes with azoalkenes. Chem. Commun. 2018, 54, 2506–2509. [Google Scholar]
- Emamian, S.; Soleymani, M.; Moosavi, S.S. Copper(I)-catalyzed asymmetric aza Diels–Alder reactions of azoalkenes toward fulvenes: A molecular electron density theory study. New J. Chem. 2019, 43, 4765–4776. [Google Scholar] [CrossRef]
- Ciccolini, C.; Giacomo Mari, G.; Francesco, G.; Gatti, F.G.; Giuseppe Gatti, G.; Gianluca Giorg, G.; Fabio Mantellini, F.; Gianfranco Favi, G. Synthesis of polycyclic fused indoline scaffolds through a substrate-guided reactivity switch. J. Org. Chem. 2020, 85, 11409–11425. [Google Scholar] [CrossRef] [PubMed]
- Mei, G.-J.; Tang, X.; Tasdan, Y.; Lu, Y. Enantioselective dearomatization of indoles by an azoalkene-enabled (3+2) reaction: Access to pyrroloindolines. Angew. Chem. Int. Ed. 2020, 59, 648–652. [Google Scholar] [CrossRef]
- Novikova, A.P.; Perova, N.M.; Chupakhin, O.N. Synthesis and properties of functional derivatives of 1,3,4-thiadiazines and condensed systems based on these systems (Review). Chem. Heterocycl. Comp. 1991, 27, 1159–1172. [Google Scholar] [CrossRef]
- Blond, G.; Gulea, M.; Mamane, V. Recent contributions to hetero Diels-Alder reactions. Curr. Org. Chem. 2016, 20, 2161–2210. [Google Scholar] [CrossRef]
- Sachse, F.; Gebauer, K.; Schneider, C. Continuous flow synthesis of 2H-thiopyrans via thia-Diels–Alder reactions of photochemically generated thioaldehydes. Eur. J. Org. Chem. 2021, 2021, 64–71. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatische Reihe. I. Mitteilung: Anlagerungen von ‘Di-en‘-kohlenwasserstoffen. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Norton, J.A. The Diels-Alder chemistry was reviewed in 1942 for the first time, see in the Diels-Alder diene synthesis. Chem. Rev. 1942, 31, 319–523. [Google Scholar] [CrossRef]
- Von Euler, H.; Josephson, K.O. Historically, first report on a (4+2)-cycloaddition reaction originates from a Swedish laboratory: Über Kondensationen an Doppelbindungen. I.: Über die Kondensation von Isopren mit Benzochinon. Ber. Deutsch. Chem. Gesell. 1920, 53, 822–826. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Materials | Van der Waals Complexes | Transition State A | 1,3,4-Thiadiazine 9 | Transition State B | 1,2,3-Thiadiazine 10 |
|---|---|---|---|---|---|---|
| 1 | 7a + 1a [0.0] | 4.3 | 8.2 | −20.8 | 19.3 | −19.3 |
| 2 | 7a + 1b [0.0] | 3.0 | 10.0 | −24.0 | 17.6 | −22.7 |
| 3 | 7a + 1m [0.0] | 3.3 | 13.1 | −15.6 | 33.6 | −18.7 |
| 4 | 7a + 1n [0.0] | 3.9 | 11.9 | −19.4 | 33.5 | −15.3 |
| 5 | 7a + 1i [0.0] | 4.3 | 13.2 [a] 12.0 | −19.1 | 25.9 | −22.8 |
| 6 [b] | 7a + 1i [0.0] | 4.3 | 32.1 | −4.1 [c] | 38.8 | 13.8 [d] |
| Entry | Starting Materials | Transition State A to 1,3,4-Thiadiazine 9 TS-Distances (Å) | Transition State B to 1,2,3-Thiadiazine 10 TS-Distances (Å) | ||
|---|---|---|---|---|---|
| C..S | N..C | C..C | S..N | ||
| 1 | 1a + 7a [0.0] | 2.578 | 3.199 | 2.493 | 2.151 |
| 2 | 1b + 7a [0.0] | 2.393 | 3.097 | 2.370 | 2.226 |
| 3 | 1m + 7a [0.0] | 2.319 | 3.029 | 2.471 | 2.342 |
| 4 | 1n + 7a [0.0] | 2.373 | 3.044 | 2.546 | 2.190 |
| 5 | 1i + 7a | 2.276 | 3.573 | 2.427 | 2.366 |
| 1.833 | 2.529 | ||||
| 6 | 1i + 7a [a] | 1.695 [b] | 3.214 [b] | 2.770 [c] | 1.939 [c] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlostoń, G.; Urbaniak, K.; Sobiecka, M.; Heimgartner, H.; Würthwein, E.-U.; Zimmer, R.; Lentz, D.; Reissig, H.-U. Hetero-Diels-Alder Reactions of In Situ-Generated Azoalkenes with Thioketones; Experimental and Theoretical Studies. Molecules 2021, 26, 2544. https://doi.org/10.3390/molecules26092544
Mlostoń G, Urbaniak K, Sobiecka M, Heimgartner H, Würthwein E-U, Zimmer R, Lentz D, Reissig H-U. Hetero-Diels-Alder Reactions of In Situ-Generated Azoalkenes with Thioketones; Experimental and Theoretical Studies. Molecules. 2021; 26(9):2544. https://doi.org/10.3390/molecules26092544
Chicago/Turabian StyleMlostoń, Grzegorz, Katarzyna Urbaniak, Malwina Sobiecka, Heinz Heimgartner, Ernst-Ulrich Würthwein, Reinhold Zimmer, Dieter Lentz, and Hans-Ulrich Reissig. 2021. "Hetero-Diels-Alder Reactions of In Situ-Generated Azoalkenes with Thioketones; Experimental and Theoretical Studies" Molecules 26, no. 9: 2544. https://doi.org/10.3390/molecules26092544