
Synthesis of the [11]Cyclacene Framework by Repetitive Diels–Alder Cycloadditions


Abstract
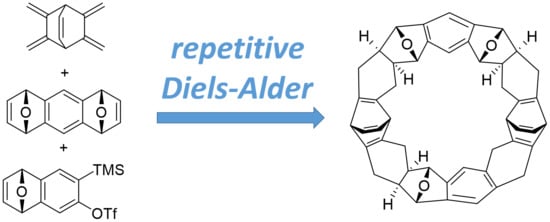
:
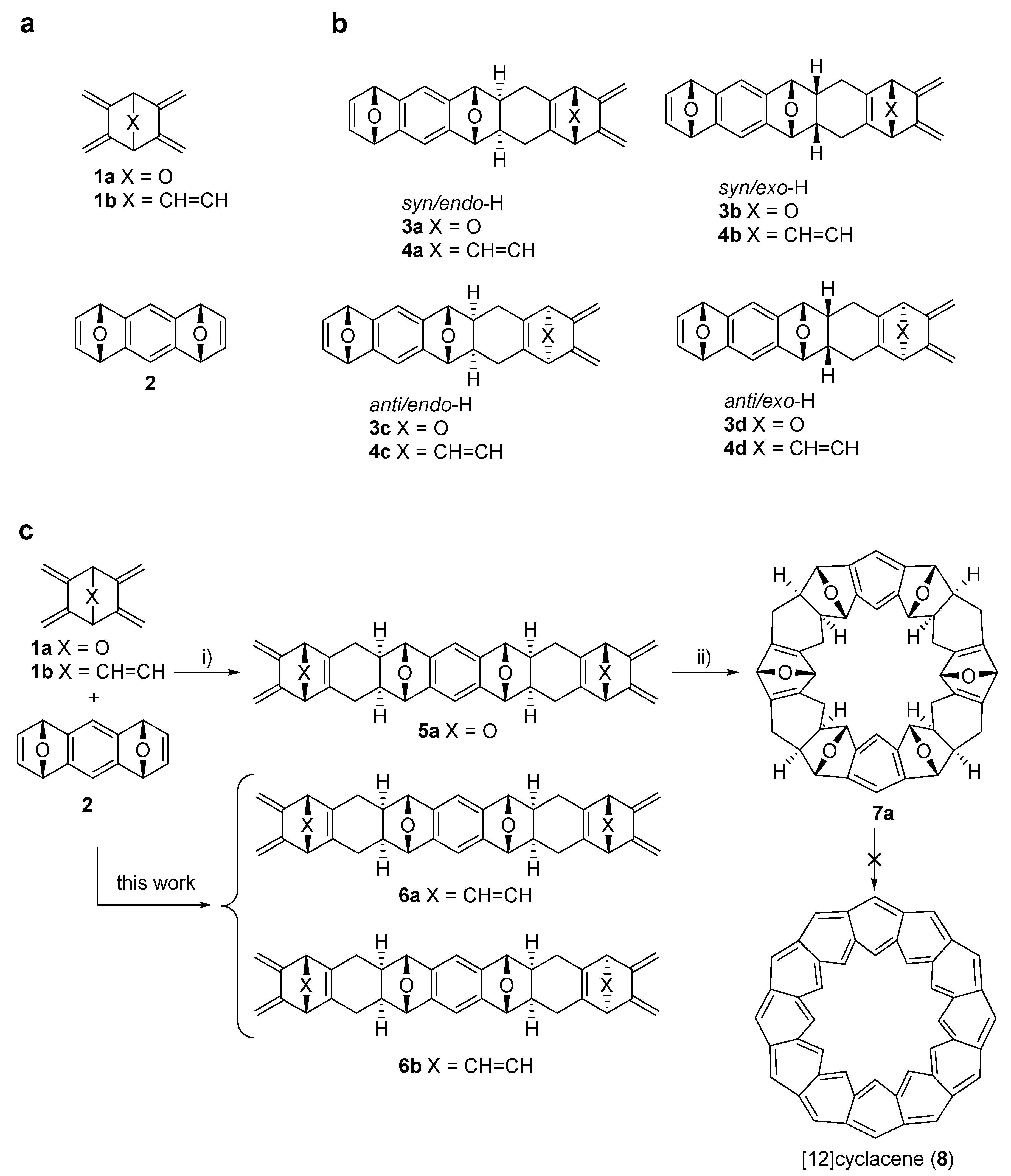
1. Introduction
2. Results and Discussion
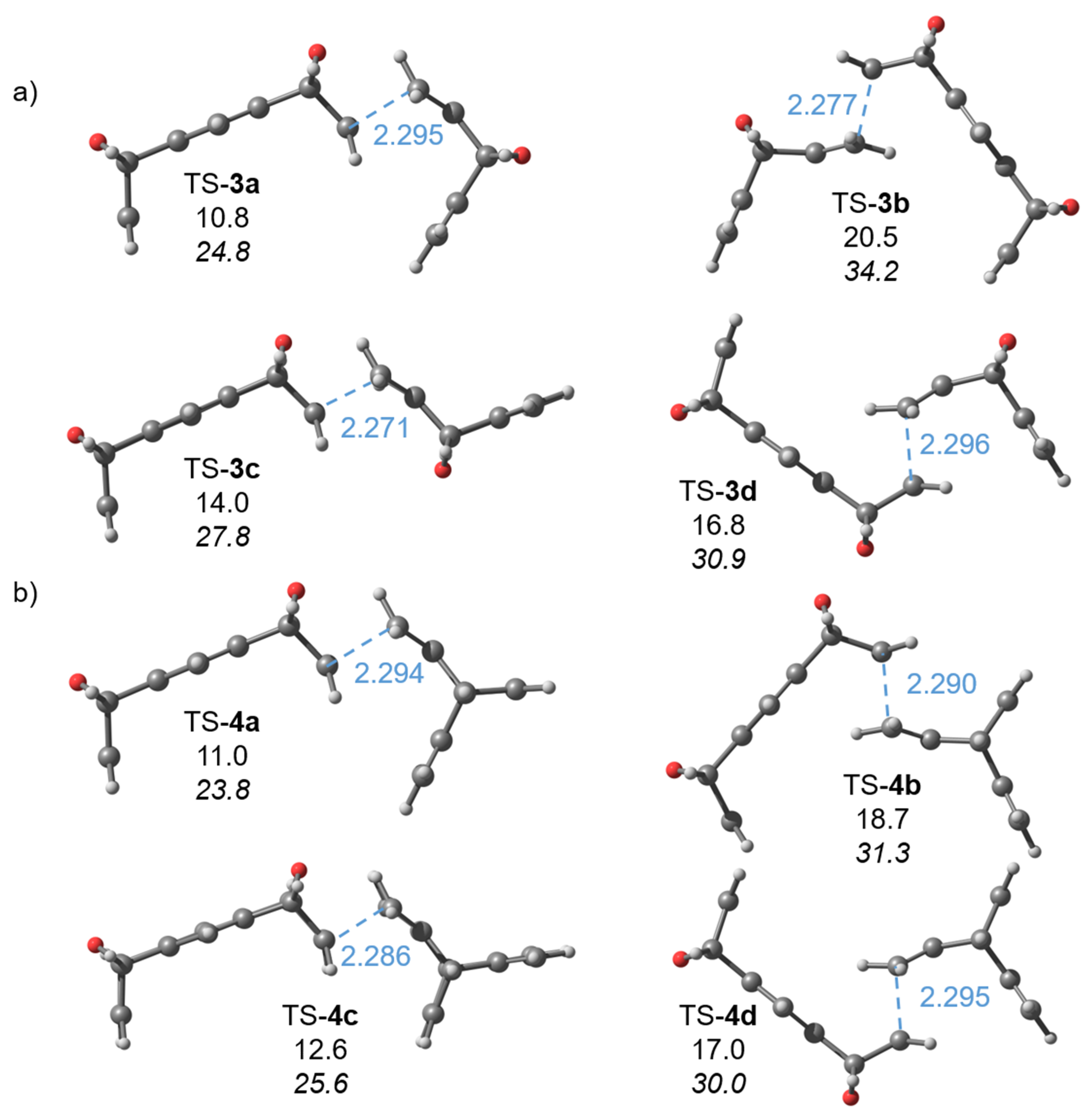
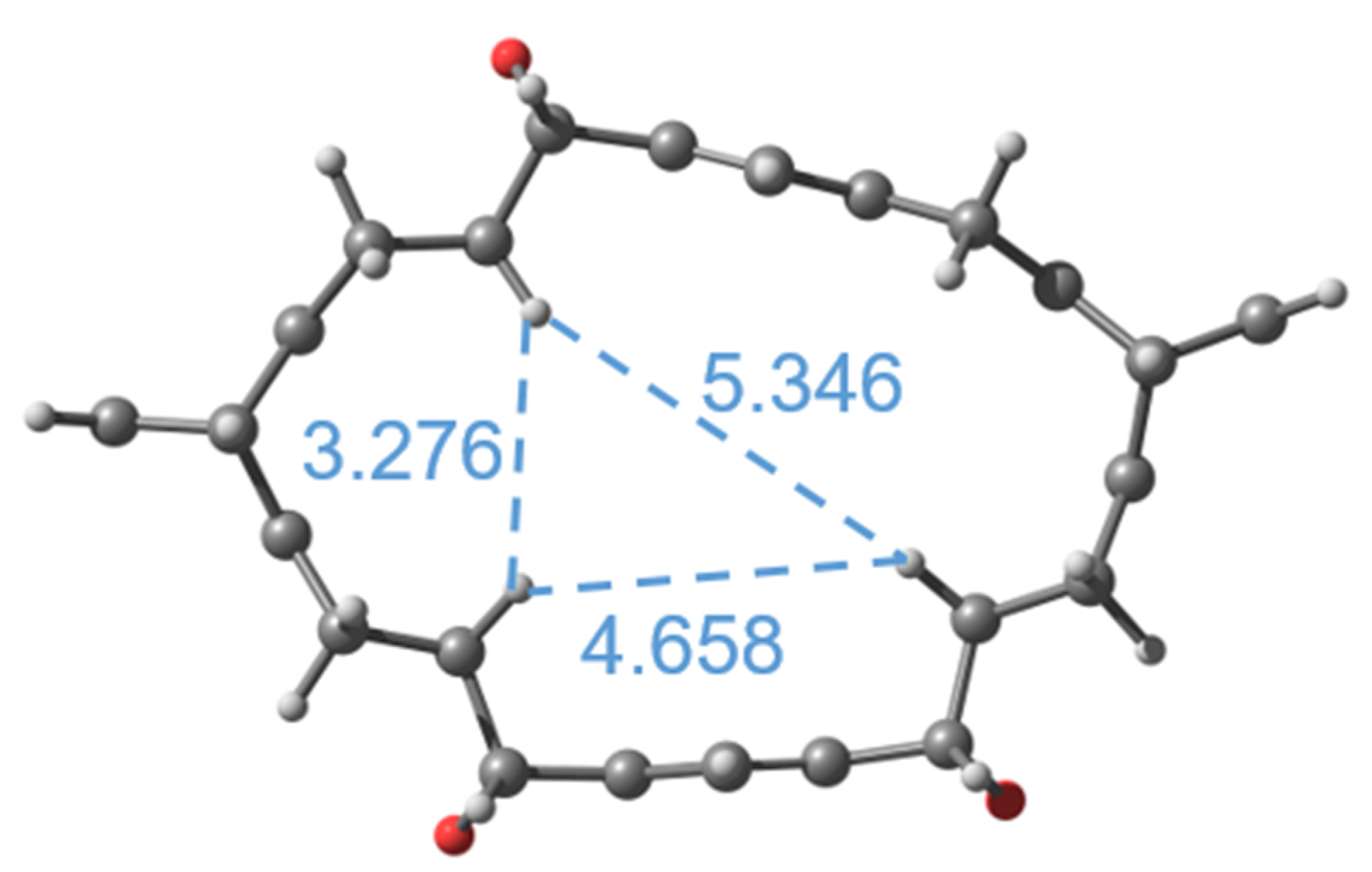
2.1. Computations
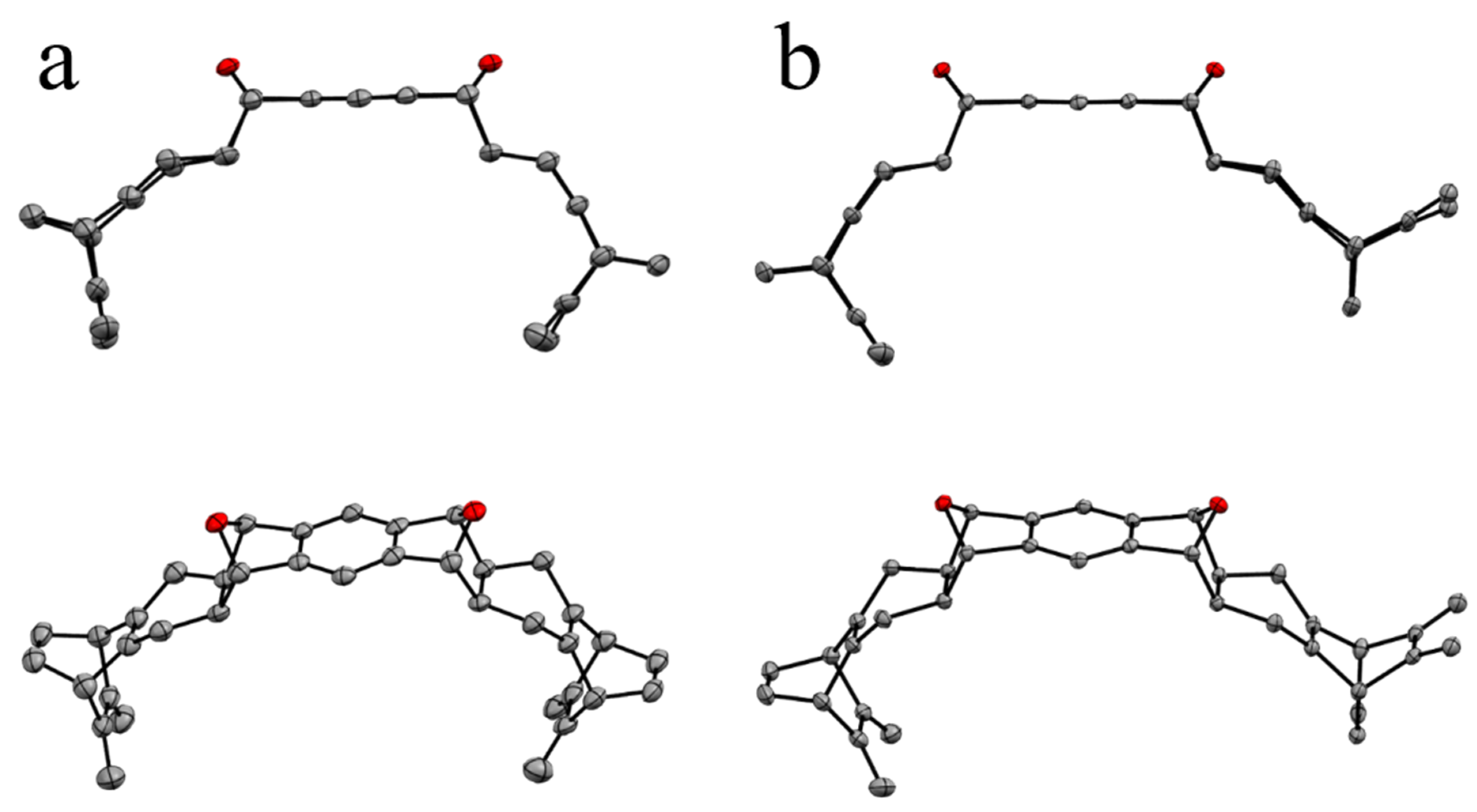
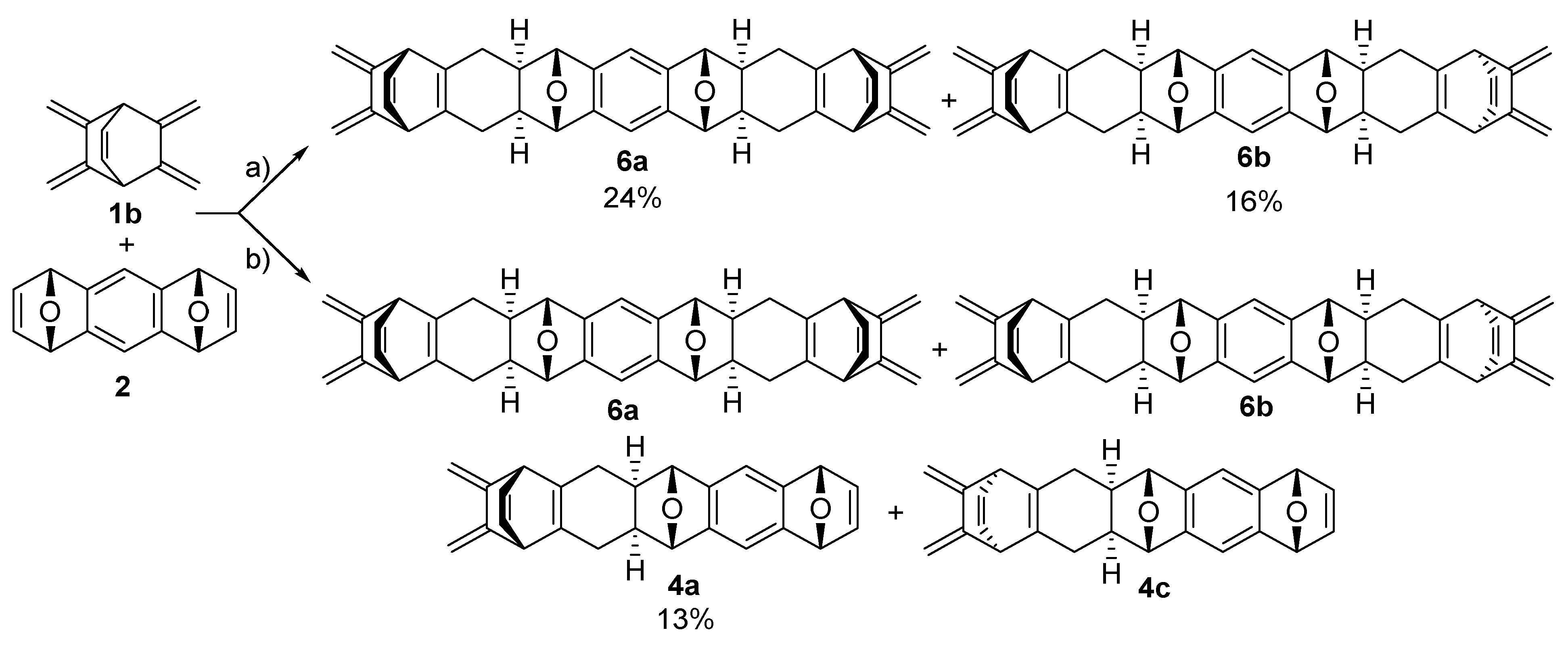
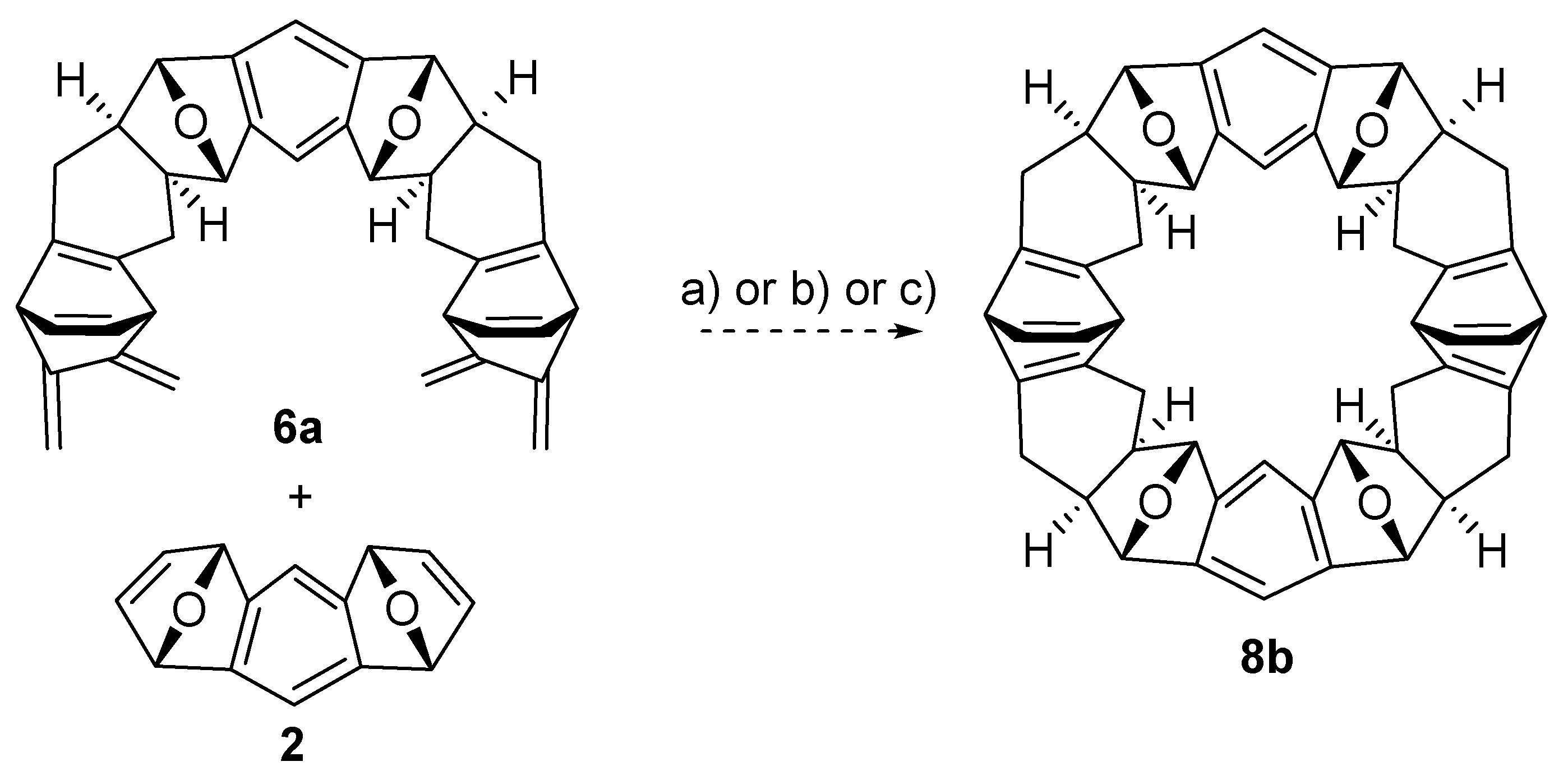
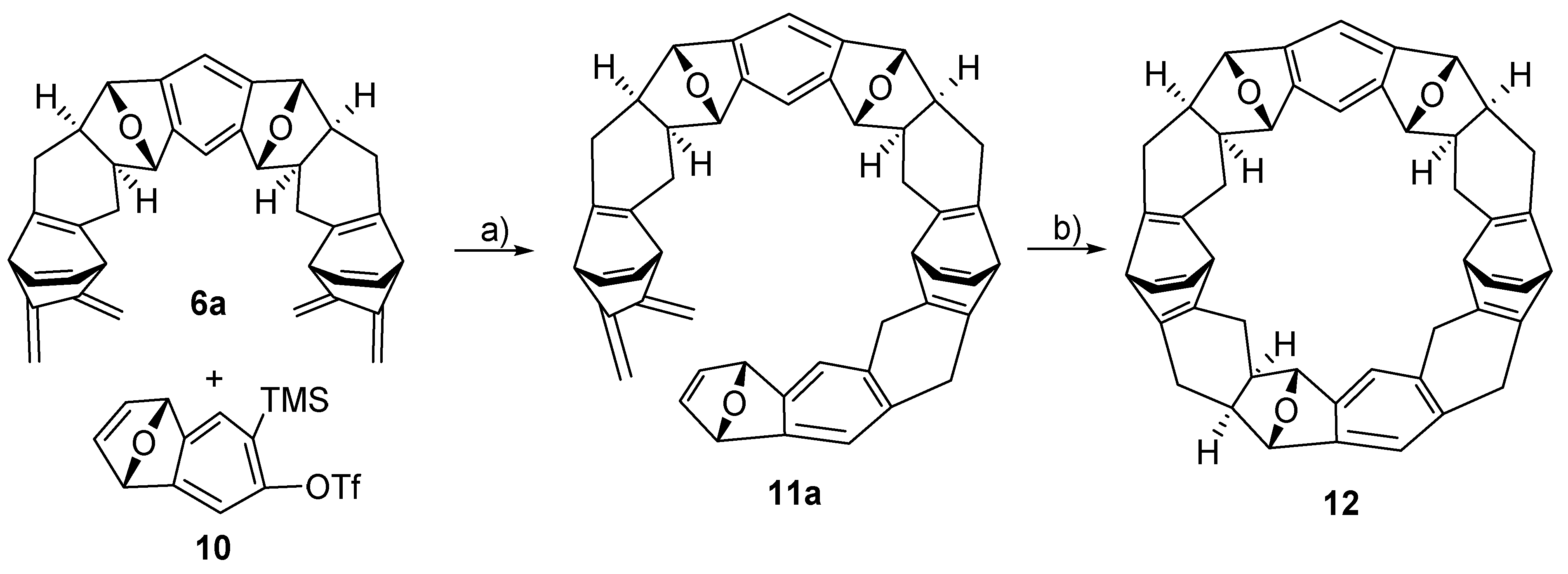
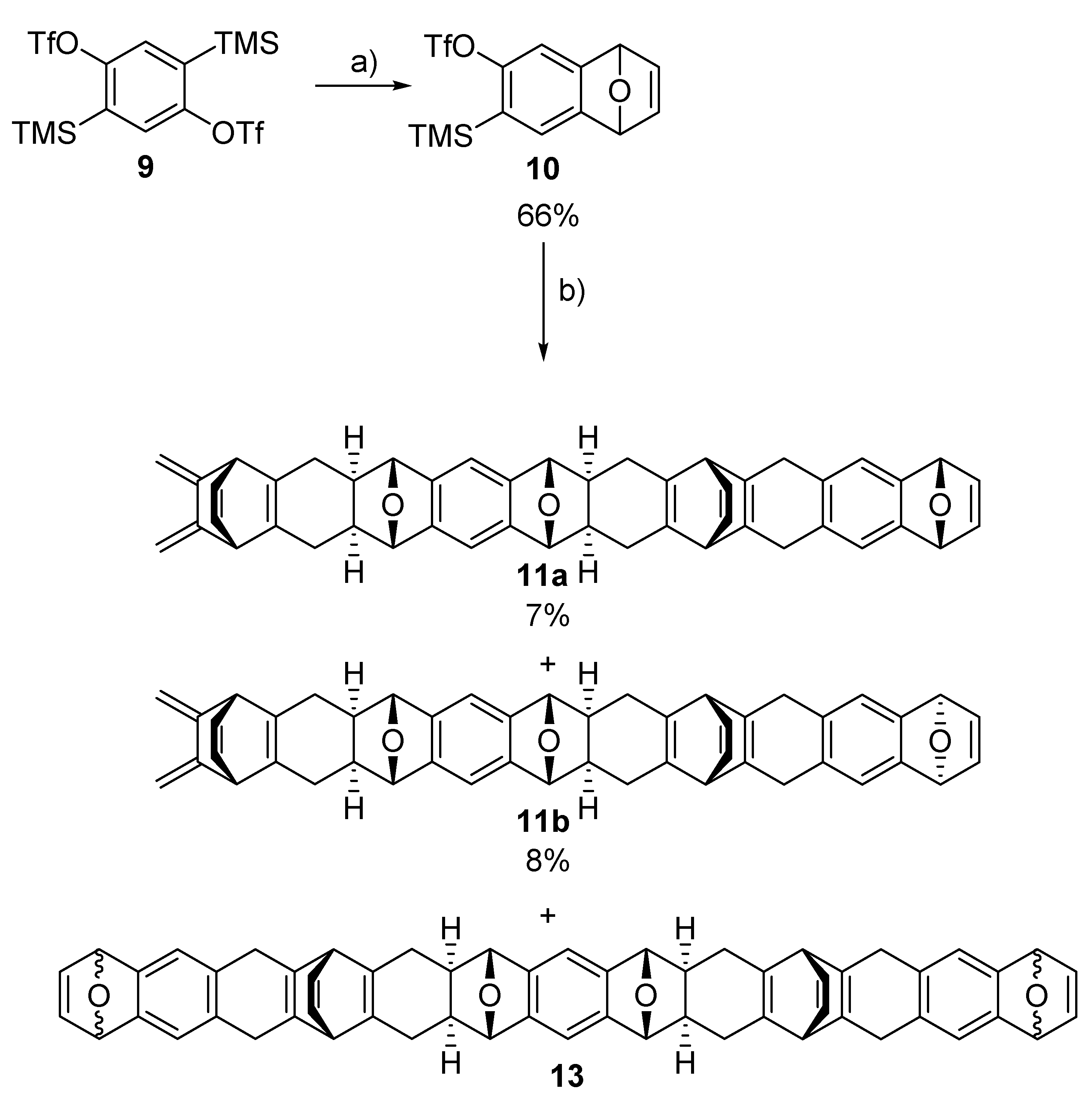
2.2. Synthesis
2.3. Properties of Macrocycle 12
3. Materials and Methods
3.1. General Methods
3.2. Synthetic Procedures
3.3. X-ray Crystallographic Data
3.4. Computational Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe, III. Liebigs Ann. Chem. 1929, 470, 62–103. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe, IV. Ber. Dtsch. Chem. Ges. 1929, 62, 2081–2087. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe, V. Ber. Dtsch. Chem. Ges. 1929, 62, 2087–2090. [Google Scholar] [CrossRef]
- Ichihara, A. Retro-Diels–Alder strategy in natural product synthesis. Synthesis 1987, 1987, 207–222. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder reaction in total synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Ashton, P.R.; Brown, G.R.; Isaacs, N.S.; Giuffrida, D.; Kohnke, F.H.; Mathias, J.P.; Slawin, A.M.Z.; Smith, D.R.; Stoddart, J.F.; Williams, D.J. Molecular LEGO. 1. Substrate-directed synthesis via stereoregular Diels–Alder oligomerizations. J. Am. Chem. Soc. 1992, 114, 6330–6353. [Google Scholar] [CrossRef]
- Cory, R.M.; McPhail, C.L.; Dikmans, A.J.; Vittal, J.J. Macrocyclic cyclophane belts via double Diels–Alder cycloadditions: Macroannulation of bisdienes by bisdienophiles. Synthesis of a key precursor to an [8]cyclacene. Tetrahedron Lett. 1996, 37, 1983–1986. [Google Scholar] [CrossRef]
- Neudorff, W.D.; Lentz, D.; Anibarro, M.; Schlüter, A.D. The carbon skeleton of the belt region of fullerene C84 (D2). Chem. Eur. J. 2003, 9, 2745–2757. [Google Scholar] [CrossRef]
- Schulz, F.; García, F.; Kaiser, K.; Pérez, D.; Guitián, E.; Gross, L.; Peña, D. Exploring a route to cyclic acenes by on-surface synthesis. Angew. Chem. Int. Ed. 2019, 58, 9038–9042. [Google Scholar] [CrossRef] [Green Version]
- Ashton, P.R.; Isaacs, N.S.; Kohnke, F.H.; Slawin, A.M.Z.; Spencer, C.M.; Stoddart, J.F.; Williams, D.J. Towards the making of [12]collarene. Angew. Chem. Int. Ed. Engl. 1988, 27, 966–969. [Google Scholar] [CrossRef]
- Girreser, U.; Giuffrida, D.; Kohnke, F.H.; Mathias, J.P.; Philip, D.; Stoddart, J.F. The structure-directed synthesis of cyclacene and polyacene derivatives. Pure Appl. Chem. 1993, 65, 119–125. [Google Scholar] [CrossRef]
- Kohnke, F.H.; Mathias, J.P.; Stoddart, J.F. Structure-directed synthesis of new organic materials. Angew. Chem. Int. Ed. Engl. 1989, 28, 1103–1110. [Google Scholar] [CrossRef]
- Kohnke, F.H.; Slawin, A.M.Z.; Stoddart, J.F.; Williams, D.J. Molecular belts and collars in the making: A hexaepoxyoctacosahydro [12]cyclacene derivative. Angew. Chem. Int. Ed. Engl. 1987, 26, 892–894. [Google Scholar] [CrossRef]
- Kohnke, F.H.; Stoddart, J.F. The evolution of molecular belts and collars. Pure Appl. Chem. 1989, 61, 1581–1586. [Google Scholar] [CrossRef]
- Heilbronner, E. Molecular Orbitals in homologen Reihen mehrkerniger aromatischer Kohlenwasserstoffe: I. Die Eigenwerte von LCAO-MO’s in homologen Reihen. Helv. Chim. Acta 1954, 37, 921–935. [Google Scholar] [CrossRef]
- Gleiter, R.; Hellbach, B.; Gath, S.; Schaller, R.J. From superphanes to beltenes. Pure Appl. Chem. 2006, 78, 699–706. [Google Scholar] [CrossRef]
- Hellbach, B.; Rominger, F.; Gleiter, R. Synthesis of beltenes by reactions of 5,6,11,12-tetradehydrodibenzo[a,e]cyclooctene with [CpCo(CO)2] derivatives. Angew. Chem. Int. Ed. 2004, 43, 5846–5849. [Google Scholar] [CrossRef]
- Takaba, H.; Omachi, H.; Yamamoto, Y.; Bouffard, J.; Itami, K. Selective synthesis of [12]cycloparaphenylene. Angew. Chem. Int. Ed. 2009, 48, 6112–6116. [Google Scholar] [CrossRef]
- Stuparu, M.; Lentz, D.; Rüegger, H.; Schlüter, A.D. Exploring the chemistry of a double-stranded cycle with the carbon skeleton of the belt region of the C84 fullerene. Eur. J. Org. Chem. 2007, 2007, 88–100. [Google Scholar] [CrossRef]
- Cory, R.M.; McPhail, C.L. Transformations of a macrocyclic cyclophane belt into advanced [8]cyclacene and [8]cyclacene triquinone precursors. Tetrahedron Lett. 1996, 37, 1987–1990. [Google Scholar] [CrossRef]
- Chen, H.; Miao, Q. Recent advances and attempts in synthesis of conjugated nanobelts. Phys. Org. Chem. 2020, 33, 1–20. [Google Scholar] [CrossRef]
- Cheung, K.Y.; Segawa, Y.; Itami, K. Synthetic strategies of carbon nanobelts and related belt-shaped polycyclic aromatic hydrocarbons. Chem. Eur. J. 2020, 26, 14791–14801. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.-H.; Wang, M.-X. Zigzag hydrocarbon belts. CCS Chem. 2020, 2, 916–931. [Google Scholar] [CrossRef]
- Gleiter, R.; Esser, B.; Kornmayer, S.C. Cyclacenes: Hoop-shaped systems composed of conjugated rings. Acc. Chem. Res. 2009, 42, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Tobe, Y. Molecular loops and belts. Chem. Rev. 2006, 106, 5274–5290. [Google Scholar] [CrossRef]
- Gupta, D.; Omont, A.; Bettinger, H.F. Energetics of formation of cyclacenes from 2,3-didehydroacenes and implications for astrochemistry. Chem. Eur. J. 2021, 27, 4605–4616. [Google Scholar] [CrossRef]
- Sadowsky, D.; McNeill, K.; Cramer, C.J. Electronic structures of [n]-cyclacenes (n = 6–12) and short, hydrogen-capped, carbon nanotubes. Faraday Discuss. 2010, 145, 507–521. [Google Scholar] [CrossRef]
- Segawa, Y.; Yagi, A.; Ito, H.; Itami, K. A theoretical study on the strain energy of carbon nanobelts. Org. Lett. 2016, 18, 1430–1433. [Google Scholar] [CrossRef]
- Zuzak, R.; Dorel, R.; Kolmer, M.; Szymonski, M.; Godlewski, S.; Echavarren, A.M. Higher acenes by on-surface dehydrogenation: From heptacene to undecacene. Angew. Chem. Int. Ed. 2018, 57, 10500–10505. [Google Scholar] [CrossRef] [Green Version]
- Krüger, J.; García, F.; Eisenhut, F.; Skidin, D.; Alonso, J.M.; Guitián, E.; Pérez, D.; Cuniberti, G.; Moresco, F.; Peña, D. Decacene: On-surface generation. Angew. Chem. Int. Ed. 2017, 56, 11945–11948. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Eisenhut, F.; Skidin, D.; Lehmann, T.; Ryndyk, D.A.; Cuniberti, G.; García, F.; Alonso, J.M.; Guitián, E.; Pérez, D.; et al. Electronic Resonances and Gap Stabilization of Higher Acenes on a Gold Surface. ACS Nano 2018, 12, 8506–8511. [Google Scholar] [CrossRef] [PubMed]
- Eisenhut, F.; Kühne, T.; García, F.; Fernández, S.; Guitián, E.; Pérez, D.; Trinquier, G.; Cuniberti, G.; Joachim, C.; Peña, D.; et al. Dodecacene generated on surface: Reopening of the energy gap. ACS Nano 2020, 14, 1011–1017. [Google Scholar] [CrossRef]
- Urgel, J.I.; Mishra, S.; Hayashi, H.; Wilhelm, J.; Pignedoli, C.A.; Di Giovannantonio, M.; Widmer, R.; Yamashita, M.; Hieda, N.; Ruffieux, P.; et al. On-surface light-induced generation of higher acenes and elucidation of their open-shell character. Nat. Commun. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, C.; Bettinger, H.F. Pushing the limits of acene chemistry: The recent surge of large acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef]
- Geiger, T.; Schundelmeier, S.; Hummel, T.; Ströbele, M.; Leis, W.; Seitz, M.; Zeiser, C.; Moretti, L.; Maiuri, M.; Cerullo, G.; et al. Modulating the electronic and solid-state structure of organic semiconductors by site-specific substitution: The case of tetrafluoropentacenes. Chem. Eur. J. 2020, 26, 3420–3434. [Google Scholar] [CrossRef]
- Wegener, S.; Müllen, K. 5,6,7,8-Tetramethylenebicyclo [2.2.2]oct-2-ene as “Bis(diene)” in repetitive Diels–Alder reactions. Chem. Ber. 1991, 124, 2101–2103. [Google Scholar] [CrossRef]
- Wegener, S.; Müllen, K. New ladder polymers via repetitive Diels–Alder reaction under high pressure. Macromolecules 1993, 26, 3037–3040. [Google Scholar] [CrossRef]
- Gabioud, R.; Vogel, P. Synthesis and Diels–Alder reactivity of 5,6,7,8-tetramethylidene-2-bicyclo [2.2.2]octanol and -octanone. Selective oxidations of the corresponding bis(irontricarbonyl) complexes. Helv. Chim. Acta 1983, 66, 1134–1147. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Krüger, J.; Eisenhut, F.; Alonso, J.M.; Lehmann, T.; Guitian, E.; Perez, D.; Skidin, D.; Gamaleja, F.; Ryndyk, D.A.; Joachim, C.; et al. Imaging the electronic structure of on-surface generated hexacene. Chem. Commun. 2017, 53, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Bruker AXS Inc. COSMO v. 1.61; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Bruker AXS Inc. APEX 2 v. 2012.10_0; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Bruker AXS Inc. SAINT v. 8.34A; Bruker AXS Inc.: Madison: WI, USA, 2013. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruker AXS Inc. APEX 3 V. 2017.3-0; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- Bruker AXS Inc. SAINT v. 8.38A; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- Sheldrick, G. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]









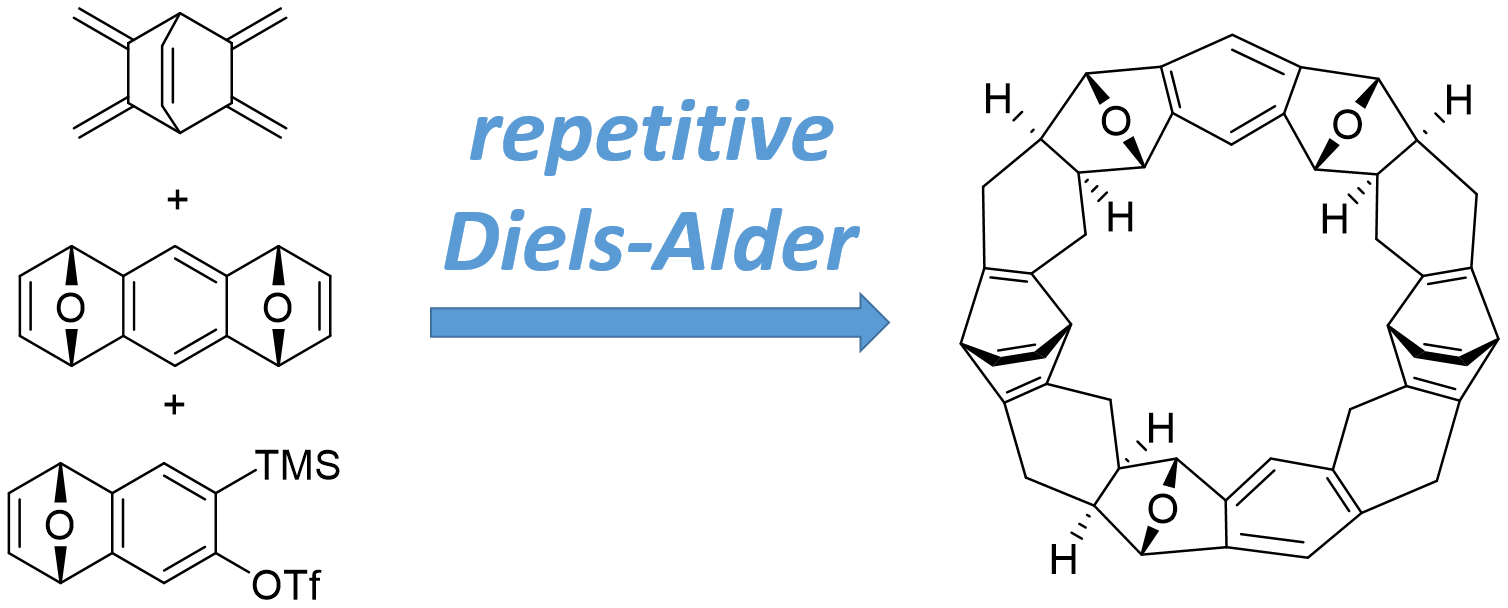{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 11a | 12 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1 | 101.2, CH2 | Ha: 4.70, Hb: 4.86 | 31.16a, CH2 or 31.19a, CH2 | Ha/Hb: 2.57–2.48/2.20–2.04 |
| 2 | 144.4, C | - | 147.5, C or 147.4, C | - |
| 3 | 53.6, CH | 3.88–3.86 | 57.0, CH | 4.14–4.12 |
| 4 | 133.8, CH | 6.39–6.37 | 139.8, CH | 6.75–6.73 |
| 5 | 137.5, C | - | 147.5, C or 147.4, C | - |
| 6 | 30.7, CH2 | Ha/Hb: 2.60–2.55/2.18–2.12 | 31.16a, CH2 or 31.19a, CH2 | Ha/Hb: 2.57–2.48/2.20–2.04 |
| 7 | 43.1, CH | 1.36–1.31 or 1.31–1.25 or 1.25–1.20 | 46.3, CH or 45.8, CH | 1.36–1.31 or 1.31–1.25 or 1.25–1.20 |
| 8 | 85.19 a, CH or 85.24 a, CH | 4.87 | 84.3, CH or 84.0, CH | 4.79 |
| 9 | 144.8, C or 144.7, C | - | 145.2, C or 145.1, C | - |
| 10 | 110.2, CH | 6.95 | 110.0, CH | 6.85 |
| 11 | 144.8, C or 144.7, C | - | 145.2, C or 145.1, C | - |
| 12 | 85.19 a, CH or 85.24 a, CH | 4.85 | 84.3, CH or 84.0, CH | 4.82 |
| 13 | 42.9, CH | 1.77–1.71 | 47.2, CH | 1.36–1.31 or 1.31–1.25 or 1.25–1.20 |
| 14 | 31.4, CH2 | Ha/Hb: 2.70–2.65/2.12–2.06 | 31.7, CH2 | Ha/Hb: 2.57–2.48/2.23–2.17 |
| 15 | 143.8, C | - | 147.8, C | - |
| 16 | 55.3, CH | 4.15–4.13 | 56.3, CH | 4.23–4.21 |
| 17 | 139.5, CH | 6.77–6.75 | 139.0, CH | 6.82–6.80 |
| 18 | 140.8, C | - | 143.3, C | - |
| 19 | 33.7, CH2 | Ha, Hb: 3.49–3.33 | 34.3, CH2 | Ha/Hb: 3.53–3.46/3.26–3.19 |
| 20 | 130.7, C | - | 133.5, C | - |
| 21 | 120.9, CH | 6.93 | 118.8, CH | 6.77 |
| 22 | 146.6, C | - | 143.7, C | - |
| 23 | 82.2, CH | 5.61 | 84.1, CH | 4.81 |
| 24 | 143.0, CH | 6.91 | 46.3, CH or 45.8, CH | 1.36–1.31 or 1.31–1.25 or 1.25–1.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, J.B.; Diab, F.; Maichle-Mössmer, C.; Schubert, H.; Bettinger, H.F. Synthesis of the [11]Cyclacene Framework by Repetitive Diels–Alder Cycloadditions. Molecules 2021, 26, 3047. https://doi.org/10.3390/molecules26103047
Bauer JB, Diab F, Maichle-Mössmer C, Schubert H, Bettinger HF. Synthesis of the [11]Cyclacene Framework by Repetitive Diels–Alder Cycloadditions. Molecules. 2021; 26(10):3047. https://doi.org/10.3390/molecules26103047
Chicago/Turabian StyleBauer, John B., Fatima Diab, Cäcilia Maichle-Mössmer, Hartmut Schubert, and Holger F. Bettinger. 2021. "Synthesis of the [11]Cyclacene Framework by Repetitive Diels–Alder Cycloadditions" Molecules 26, no. 10: 3047. https://doi.org/10.3390/molecules26103047