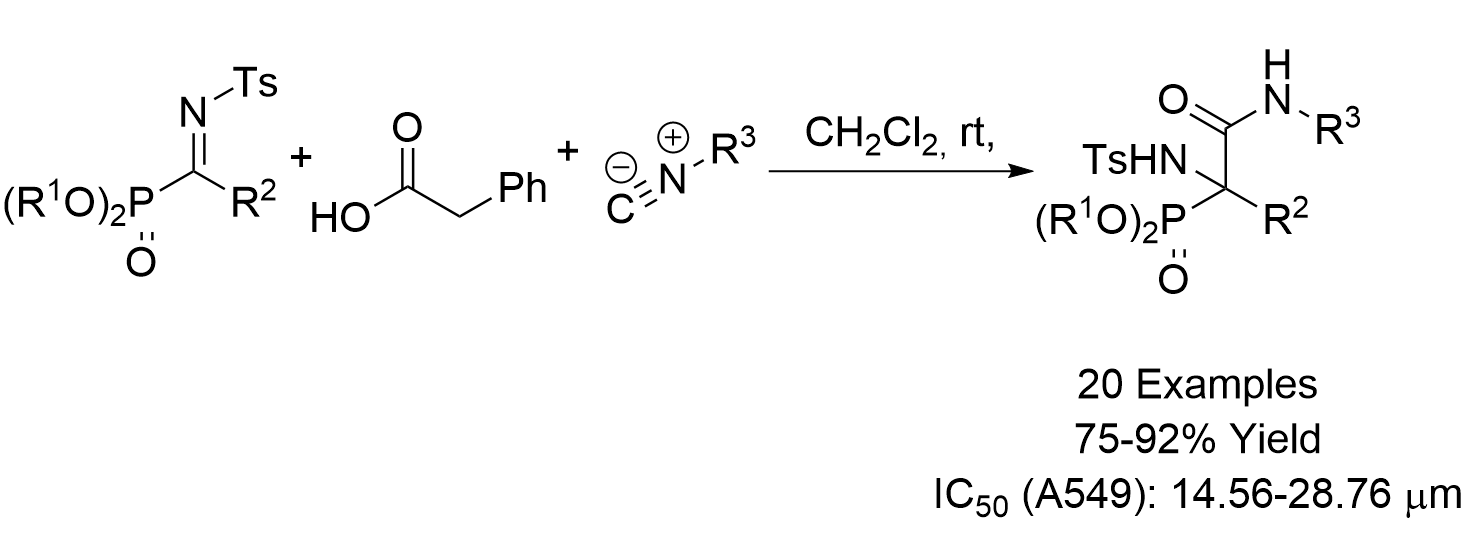
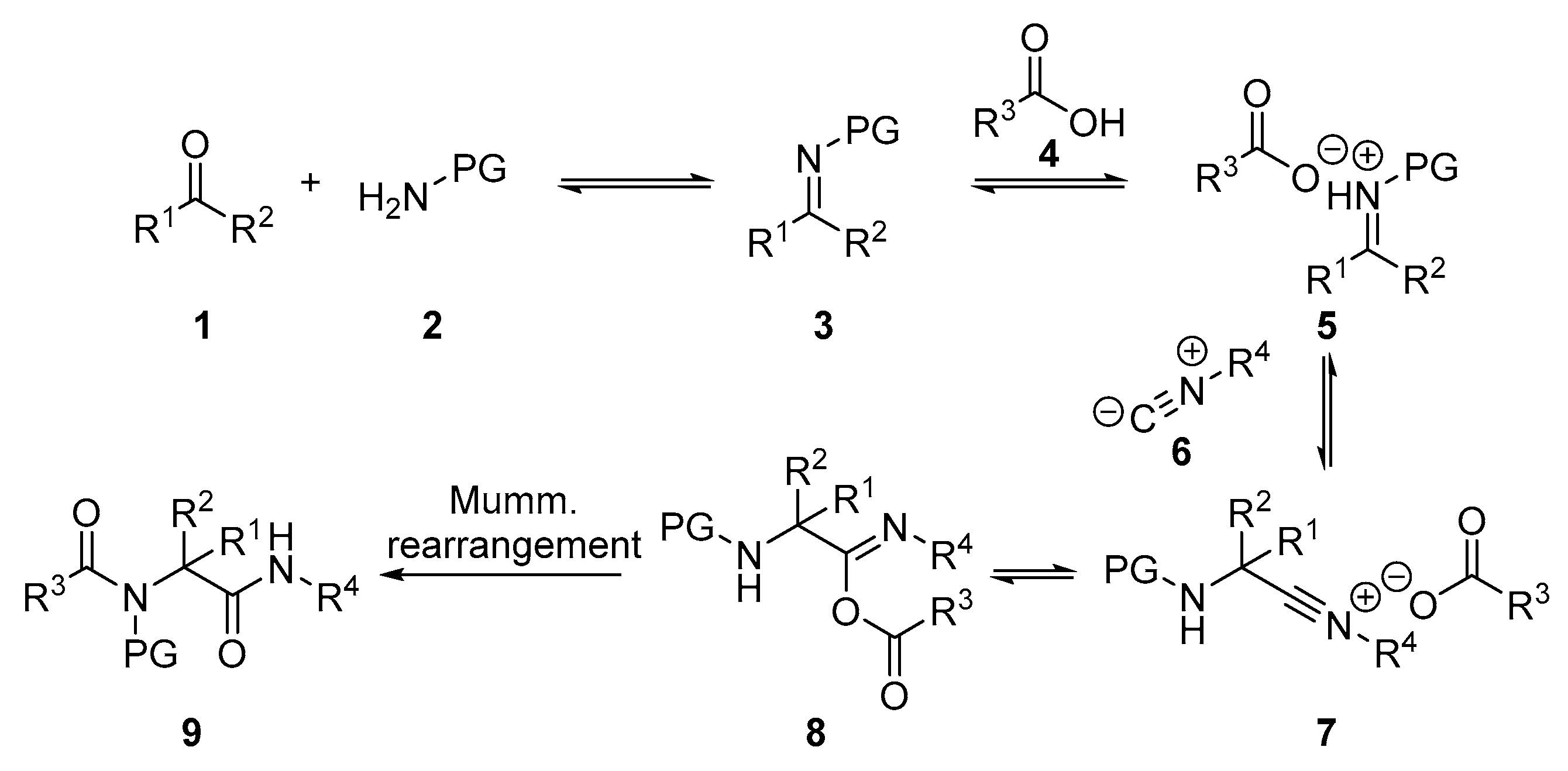
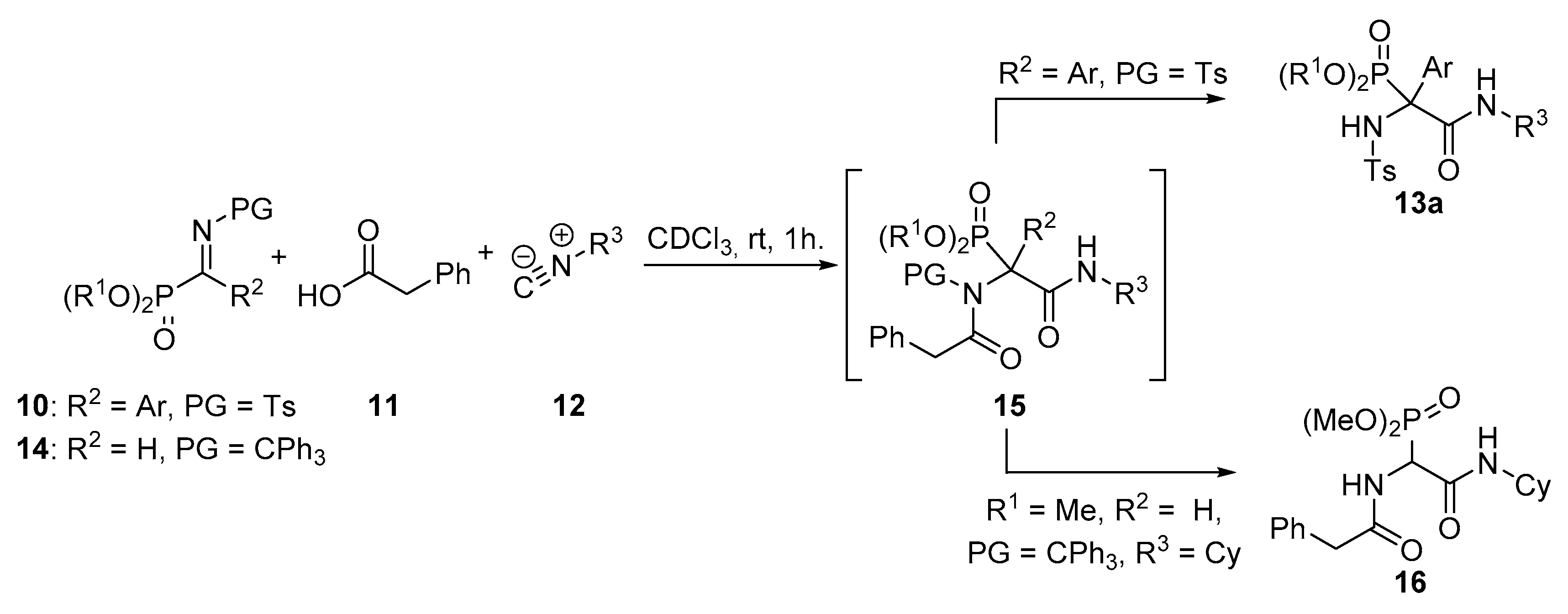
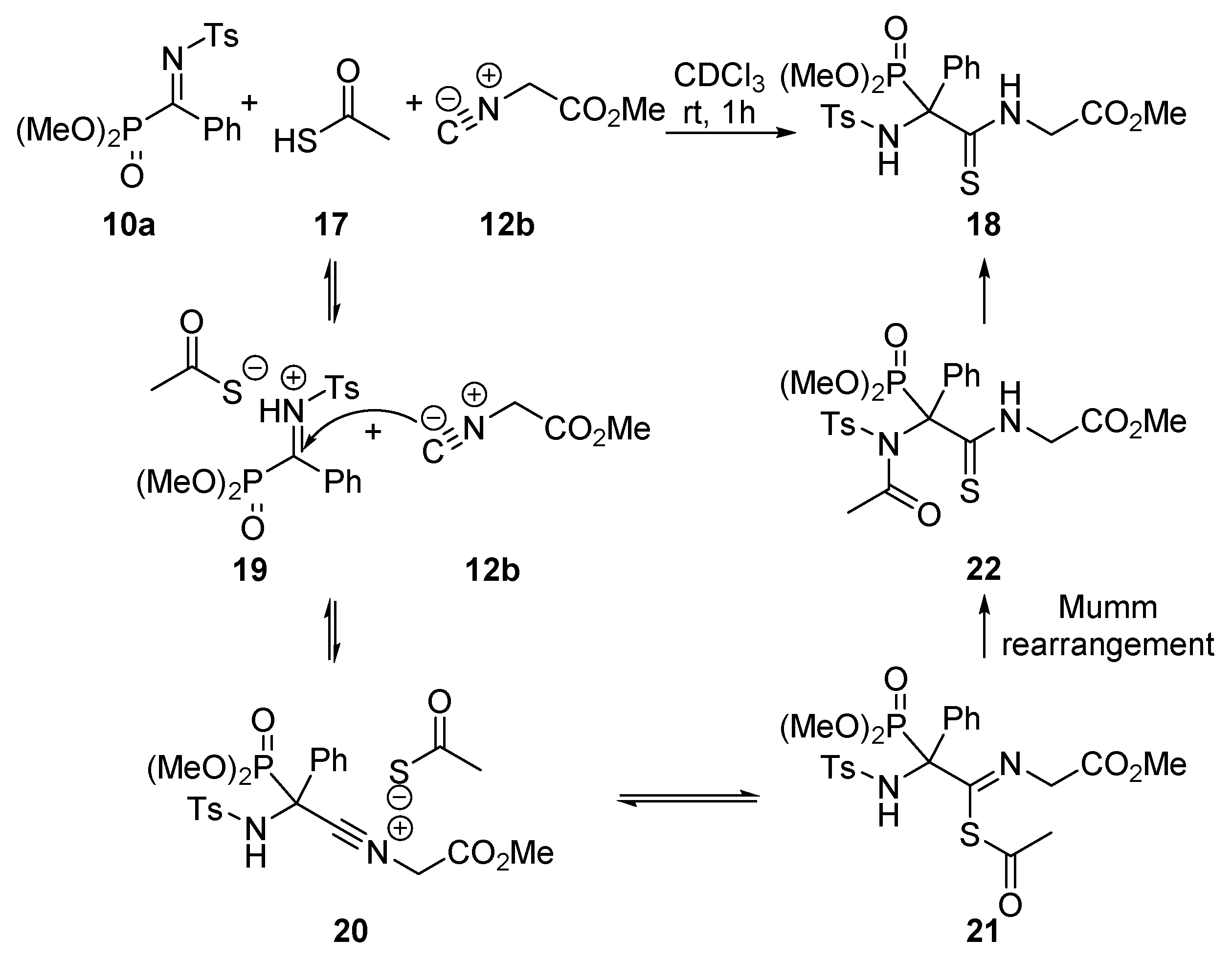
General Procedure for the Ugi Reaction of α-Phosphorated Ketimines 10 and 14
A mixture of α-iminophosphonate 10 or 14 (1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and isocyanide 12 (1.1 mmol) in ichloromethane (3 mL) was stirred at room temperature until disappearance of the starting iminophosphonate 10 as monitored by 31P-NMR. The reaction was concentrated under vacuum and the resulting crude residue was purified by crystallization (Dichomethane/Hexanes 1:3), yielding α-aminophosphonates 13, 15 or 16. In some cases, a purification by column chromatography was necessary as detailed for each compound.
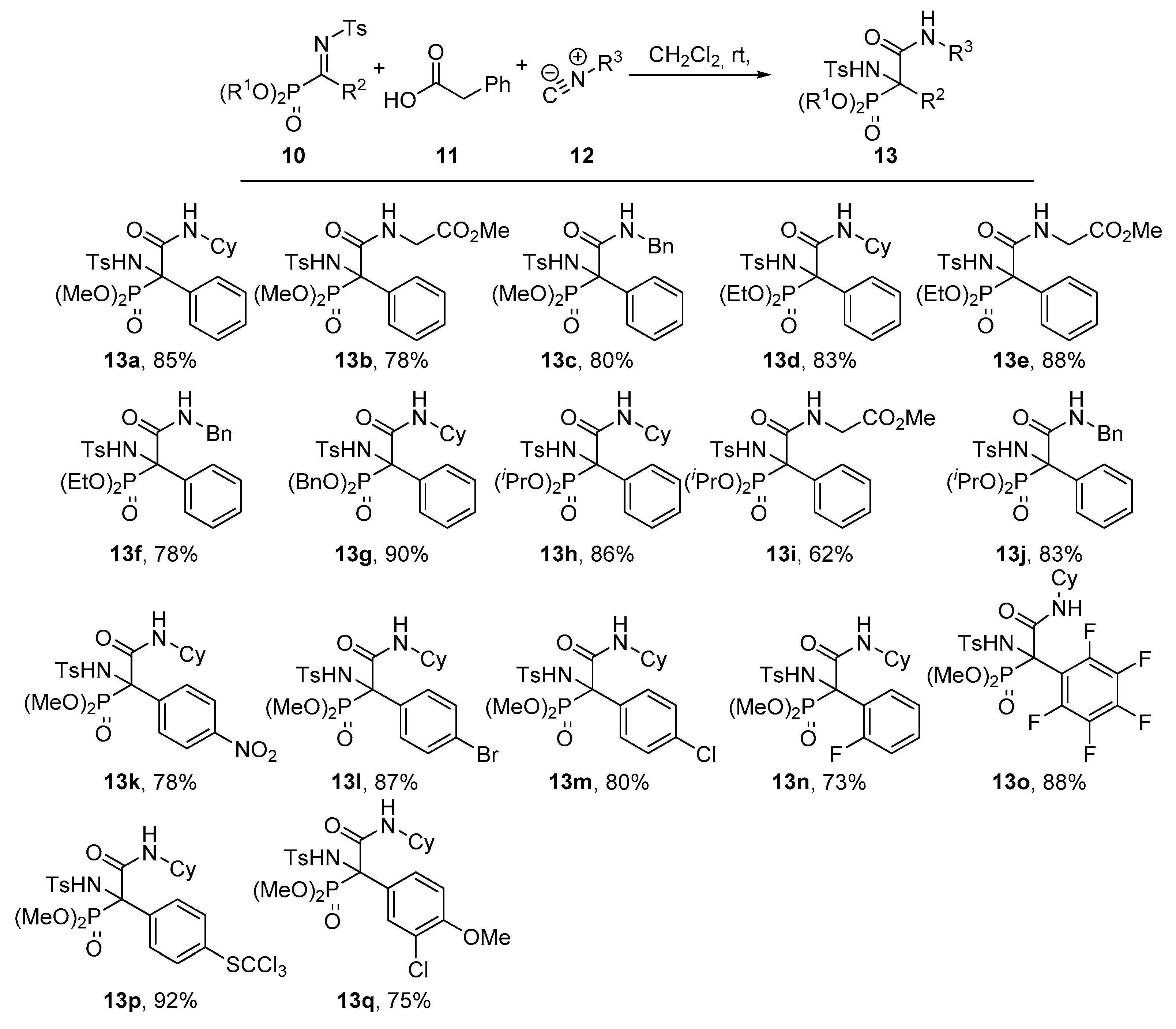
Dimethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13a). The general procedure was followed, using dimethyl (E)-(phenyl(tosylimino)methyl) phosphonate (10a, 367 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1.1 mmol) to afford 420 mg (85%) of 13a after 1 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 206–208 °C. 1H-NMR (400 MHz, CDCl3) δ 7.25–7.15 (m, 3H, 3 × CHAr), 7.16 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.05 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.00 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 6.76 (d, 3JHH = 6.4 Hz, 1H, NHCO), 6.47 (d, 3JPH = 8.2 Hz, 1H, NHTs), 3.99 (d, 3JPH = 10.7 Hz, 3H, OCH3), 3.80 (d, 3JPH = 10.5 Hz, 3H, OCH3), 3.77 (m, 1H, CHCy), 2.33 (s, 3H, CH3Ts), 1.91–1.44 (m, 4H, CH2Cy), 1.39–0.92 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 166.1 (C=O), 142.5 (CquatTs), 139.2 (d, 4JPC = 1.6 Hz, CquatTs), 131.9 (d, 2JPC = 1.8 Hz, Cquat Ph), 130.2 (d, 3JPC = 8.3 Hz, 2 × CHAr Ph), 129.1 (2 × CHAr), 128.7 (CHAr), 127.9 (2 × CHAr), 126.5 (2 × CHAr), 68.5 (d, 1JPC = 157.2 Hz, Cquat-P), 55.8 (d, 2JPC = 8.2 Hz, OCH3), 55.2 (d, 2JPC = 7.5 Hz, OCH3), 49.8 (CHCy), 32.3 (CH2Cy), 32.1 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 20.5 ppm. FTIR (neat) νmax: ν = 3426 (N-H st) 3333 (N-H st) 1678 (C=O st) 1256 (P=O st) 1333 (S=O st sym) 1164 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H32N2O6PS 495.1719, Found 495.1718.
Methyl (2-(dimethoxyphosphoryl)-2-((4-methylphenyl)sulfonamido)-2-phenylacetyl)glycinate (13b). The general procedure was applied starting from dimethyl (E)-(phenyl(tosylimino)methyl)phosphonate (10a, 367 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and methyl 2-isocyanoacetate (12b, 100 μL, 1.1 mmol) to afford 378 mg (78%) of 13b after 1 h as a white solid after a chromatography column (Hexanes/AcOEt 1:1), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 130–132 °C. 1H-NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 2H, 2 × CHAr), 7.23–7.16 (m, 3H, 3 × CHAr), 7.15 (d, 3JHH = 5.5 Hz, 1H, NHCO), 7.11–6.95 (m, 4H, 4 × CHAr), 6.64 (d, 3JPH = 6.8 Hz, 1H, NHTs), 4.00 (d, 3JHH = 5.5 Hz, 2H, CH2), 3.91 (d, 3JPH = 10.8 Hz, 3H, POCH3), 3.78 (d, 3JPH = 10.6 Hz, 3H, POCH3), 3.68 (s, 3H, COCH3), 2.33 (s, 3H, CH3Ts) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 169.2 (C=O), 167.8 (C=O), 142.7 (CquatTs), 139.0 (CquatTs), 131.5 (CquatPh), 130.2 (d, 3JPC = 7.8 Hz, 2 × CHArPh), 129.1 (2 × CHAr), 128.9 (CHAr), 128.0 (2 × CHAr), 126.7 (2 × CHAr), 69.0 (d, 1JPC = 155.3 Hz, Cquat-P), 55.6 (d, 2JPC = 8.0 Hz, POCH3), 55.4 (d, 2JPC = 7.5 Hz, POCH3), 52.5 (COCH3), 42.2 (CH2), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 18.9 ppm. FTIR (neat) νmax: ν = 3437 (N-H st) 3323 (N-H st) 1675 (C=O st) 1266 (P=O st) 1338 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H26N2O8PS 485.1147, Found 485.1149.
Dimethyl (2-(benzylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13c). The general procedure was applied starting from dimethyl (E)-(phenyl(tosylimino)methyl) phosphonate (10a, 367 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and (isocyanomethyl) benzene (12c, 134 μL, 1.1 mmol) to afford 402 mg (80%) of 13c after 1 h as white crystals after a chromatography column (Hexanes/AcOEt 7:3), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 150–152 °C. 1H-NMR (400 MHz, CDCl3) δ 7.32–7.09 (m, 11H, 11 × CHAr), 7.08–6.99 (m, 3H, 3 × CHAr), 6.97 (d, 3JHH = 6.3 Hz, 1H, NHCO), 6.74 (d, 3JPH = 6.6 Hz, 1H, NHTs), 4.51 (dd, 2JHH = 14.9 Hz, 3JHH = 6.2 Hz, 1H, CHACHB), 4.35 (dd, 2JHH = 14.9 Hz, 3JHH = 5.7 Hz, 1H, CHACHB), 3.91 (d, 3JPH = 10.8 Hz, 3H, OCH3), 3.67 (d, 3JPH = 10.6 Hz, 3H, OCH3), 2.34 (s, 3H, CH3Ts) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 167.2 (C=O), 142.7 (CquatTs), 139.2 (CquatTs), 137.2 (CquatPh), 132.0 (CquatPh), 130.1 (d, 3JPC = 8.1 Hz, 2 × CHArPh), 129.1 (2 × CHAr), 128.8 (CHAr), 128.7 (2 × CHAr), 128.1 (2 × CHAr), 127.8 (2 × CHAr), 127.7 (CHAr), 126.6 (2 × CHAr), 68.8 (d, 1JPC = 156.0 Hz, Cquat-P), 55.6 (d, 2JPC = 8.0 Hz, OCH3), 55.1 (d, 2JPC = 7.6 Hz, OCH3), 44.7 (CH2), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 19.3 ppm. FTIR (neat) νmax: ν = 3428 (N-H st) 3341 (N-H st) 1675 (C=O st) 1255 (P=O st) 1332 (S=O st sym) 1160 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H28N2O6PS 503.1406, Found 503.1411.
Diethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13d). The general procedure was applied starting from diethyl (E)-(phenyl(tosylimino)methyl) phosphonate (10b, 395 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1.1 mmol) to afford 434 mg (83%) of 13d after 6 h as white crystals after a chromatography column (Hexanes/AcOEt 8:2), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 122–124 °C. 1H NMR (400 MHz, CDCl3) δ 7.28–7.10 (m, 5H, 5 × CHAr), 7.05–6.95 (m, 4H, 4 × CHAr), 6.73 (d, 3JHH = 6.4 Hz, 1H, NHCO), 6.54 (d, 3JPH = 8.1 Hz, 1H, NHTs), 4.47–4.28 (m, 2H, OCH2), 4.27–4.06 (m, 2H, OCH2), 3.76 (m, 1H, CHCy), 2.33 (s, 3H, CH3Ts), 1.91–1.47 (m, 6H, 3 × CH2Cy), 1.40 (t, 3JHH = 7.0 Hz, 3H, CH3CH2), 1.27 (t, 3JHH = 7.0 Hz, 3H, CH3CH2) 1.30–0.94 (m, 4H, 2 × CH2Cy) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 166.4 (C=O), 142.4 (CquatTs), 139.3 (CquatTs), 132.1 (CquatPh), 130.4 (d, 3JPC = 7.8 Hz, 2 × CHArPh), 129.0 (2 × CHAr), 128.6 (CHAr), 127.8 (2 × CHAr), 126.6 (2 × CHAr), 68.6 (d, 1JPC = 156.2 Hz, Cquat-P), 65.5 (d, 2JPC = 8.3 Hz, OCH2), 65.0 (d, 2JPC = 7.6 Hz, OCH2), 49.7 (CHCy), 32.2 (CH2Cy), 32.2 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.5 (CH3Ts), 16.6 (d, 3JPC = 5.7 Hz, CH3CH2), 16.4 (d, 3JPC = 5.8 Hz, CH3CH2) ppm. 31P-NMR (121 MHz, CDCl3) δ 17.1 ppm. FTIR (neat) νmax: ν = 3432 (N-H st) 3340 (N-H st) 1672 (C=O st) 1253 (P=O st) 1338 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H36N2O6PS 523.2032, Found 523.2034.
Methyl (2-(diethoxyphosphoryl)-2-((4-methylphenyl)sulfonamido)-2-phenylacetyl)glycinate (13e). The general procedure was applied starting from diethyl (E)-(phenyl(tosylimino)methyl) phosphonate (10b, 395 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and methyl 2-isocyanoacetate (12b, 100 μL, 1.1 mmol) to afford 453 mg (88%) of 13e after 6 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 147–149 °C. 1H-NMR (400 MHz, CDCl3) δ 7.36–7.29 (m, 2H, 2 × CHAr), 7.21–7.11 (m, 4H, 3 × CHAr + NHCO), 7.06–6.92 (m, 4H, 4 × CHAr), 6.61 (d, 3JPH = 6.9 Hz, 1H, NHTs), 4.34–4.19 (m, 2H, OCH2), 4.18–4.04 (m, 2H, OCH2), 3.96 (dd, 3JHH = 5.5 Hz, 5JPH = 2.7 Hz, 2H, NCH2), 3.65 (s, 3H, OCH3), 2.29 (s, 3H, CH3Ts), 1.30 (t, 3JHH = 6.9 Hz, 3H, CH3CH2), 1.19 (t, 3JHH = 7.0 Hz, 3H, CH3CH2) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 169.2 (C=O), 167.9 (C=O), 142.6 (CquatTs), 139.09 (d, 4JPC = 1.4 Hz, CquatTs), 131.6 (CquatPh), 130.4 (d, 3JPC = 7.6 Hz, 2 × CHArPh), 129.0 (2 × CHAr), 128.7 (CHAr), 127.8 (2 × CHAr), 126.7 (2 × CHAr), 69.1 (d, 1JPC = 153.9 Hz, Cquat-P), 65.4 (d, 2JPC = 8.0 Hz, OCH2), 65.1 (d, 2JPC = 7.5 Hz, OCH2), 52.4 (OCH3), 42.2 (NCH2), 21.5 (CH3Ts), 16.5 (d, 3JPC = 5.7 Hz, CH3CH2), 16.4 (d, 3JPC = 5.6 Hz, CH3CH2) ppm. 31P-NMR (121 MHz, CDCl3) δ 16.7 ppm. FTIR (neat) νmax: ν = 3425 (N-H st) 3331 (N-H st) 1678 (C=O st) 1256 (P=O st) 1332 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H30N2O8PS 513.1460, Found 513.1462.
Diethyl (2-(benzylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13f). The general procedure was applied starting from diethyl (E)-(phenyl(tosylimino)methyl) phosphonate (10b, 395 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and (isocyanomethyl) benzene (12c, 134 μL, 1.1 mmol) to afford 411 mg (78%) of 13f after 6 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 140–142 °C. 1H-NMR (400 MHz, CDCl3) δ 7.34–7.14 (m, 10H, 10 × CHAr), 7.11–6.97 (m, 5H, 4 × CHAr + NHCO), 6.69 (d, 3JPH = 5.7 Hz, 1H, NHTs), 4.52 (dd, 2JHH = 14.9, 3JHH = 6.2 Hz, 1H, NCHACHB), 4.44–4.22 (m, 3H, NCHACHB + OCH2), 4.17–3.95 (m, 2H, OCH2), 2.35 (s, 3H, CH3Ts), 1.35 (t, 3JHH = 7.0 Hz, 3H, CH3CH2), 1.17 (t, 3JHH = 7.0 Hz, 3H, CH3CH2) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 167.5 (C=O), 142.5 (CquatTs), 139.3 (CquatTs), 137.2 (CquatPh), 132.1 (CquatPh), 130.2 (d, 3JPC = 7.7 Hz, 2 × CHArPh), 129.1 (2 × CHAr), 128.7 (2 × CHAr), 128.6 (CHAr), 128.0 (2 × CHAr), 127.8 (2 × CHAr), 127.7 (CHAr), 126.6 (2 × CHAr), 68.9 (d, 1JPC = 155.0 Hz, Cquat-P), 65.5 (d, 2JPC = 8.2 Hz, OCH2), 65.0 (d, 2JPC = 7.5 Hz, OCH2), 44.7 (NCH2), 21.6 (CH3Ts), 16.5 (d, 3JPC = 5.7 Hz, CH3CH2), 16.3 d, 3JPC = 5.7 Hz, CH3CH2) ppm. 31P-NMR (121 MHz, CDCl3) δ 16.9 ppm. FTIR (neat) νmax: ν = 3433 (N-H st) 3342 (N-H st) 1679 (C=O st) 1257 (P=O st) 1330 (S=O st sym) 1162 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H32N2O6PS 531.1719, Found 531.1728.
Dibenzyl (2-(benzylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13g). The general procedure was applied starting from dibenzyl (E)-(phenyl(tosylimino)methyl) phosphonate (10c, 520 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1.1 mmol) to afford 583 mg (90%) of 13g after 6 h as white crystals after a chromatography column (Hexanes/AcOEt 8:2), followed by crystallization (Ether/Pentane 1:3). M.p.: 80–82 °C. 1H-NMR (400 MHz, CDCl3) δ 7.44–7.25 (m, 10H, 10 × CHAr), 7.24–7.13 (m, 5H, 5 × CHAr), 7.04 (t, 3JHH = 7.7 Hz, 2H, 2 × CHAr), 6.96 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 6.82 (d, 3JHH = 6.6 Hz, 1H, NHCO), 6.44 (d, 3JPH = 8.0 Hz, 1H, NHTs), 5.31–5.09 (m, 2H, OCH2), 5.06–4.91 (m, 2H, OCH2), 3.77–3.63 (m, 1H, CHCy), 2.33 (s, 3H, CH3Ts), 1.80–0.81 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 166.1 (C=O), 142.5 (CquatTs), 139.2 (CquatTs), 135.9 (d, 3JPC = 5.2 Hz, CquatBn), 135.7 (d, 3JPC = 6.0 Hz, CquatBn), 131.9 (CquatPh), 129.1 (2 × CHAr), 128.8–128.5 (m, 9 × CHAr), 128.3 (2 × CHAr), 128.2 (2 × CHAr), 127.9 (2 × CHAr), 126.6 (2 × CHAr), 70.6 (d, 2JPC = 8.0 Hz, OCH2), 70.1 (d, 2JPC = 7.5 Hz, OCH2), 68.9 (d, 1JPC = 156.4 Hz, Cquat-P), 49.8 (CHCy), 32.1 (CH2Cy), 32.0 (CH2Cy), 25.4 (CH2Cy), 24.5 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ (ppm): 17.7 ppm. FTIR (neat) νmax: ν = 3417 (N-H st) 3324 (N-H st) 1677 (C=O st) 1259 (P=O st) 1336 (S=O st sym) 1162 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C35H40N2O6PS 647.2346, Found 647.2348.
Di-iso-propyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13h). The general procedure was applied starting from di-iso-propyl (E)-(phenyl(tosylimino)methyl) phosphonate (10d, 423 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1.1 mmol) to afford 474 mg (86%) of 13h after 14 h as white crystals after a chromatography column (Hexanes/AcOEt 8:2), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 141–143 °C. 1H-NMR (400 MHz, CDCl3) δ 7.32–7.27 (m, 2H, 2 × CHAr), 7.21–7.09 (m, 3H, 3 × CHAr), 7.05–6.93 (m, 4H, 4 × CHAr), 6.63 (d, 3JHH = 7.3 Hz, 1H, NHCO), 6.58 (d, 3JPH = 8.0 Hz, 1H, NHTs), 4.88 (m, 1H, CH3CH), 4.76 (m, 1H, CH3CH), 3.77 (m, 1H, CHCy), 2.32 (s, 3H, CH3Ts), 1.93–1.44 (m, 6H, 3 × CH2Cy), 1.37 (d, 3JHH = 6.1 Hz, 3H, CH3CH), 1.36 (d, 3JHH = 6.2 Hz, 3H, CH3CH), 1.24 (d, 3JHH = 6.1 Hz, 3H, CH3CH), 1.15 (d, 3JHH = 6.2 Hz, 3H, CH3CH), 1.40–0.97 (m, 4H, 2 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 166.4 (C=O), 142.3 (CquatTs), 139.4 (CquatTs), 132.4 (CquatPh), 130.3 (d, 3JPC = 7.7 Hz, 2 × CHAr), 129.0 (2 × CHAr), 128.4 (CHAr), 127.7 (2 × CHAr), 126.7 (2 × CHAr), 73.9 (d, 2JPC = 7.8 Hz, CH3CH), 73.8 (d, 2JPC = 8.2 Hz, CH3CH), 68.8 (d, 1JPC = 156.0 Hz, Cquat-P), 49.6 (CHCy), 32.2 (2 × CH2Cy), 25.5 (CH2Cy), 24.5 (d, 3JPC = 7.2 Hz, CH3CH), 24.3 (d, 3JPC = 6.5 Hz, CH3CH), 24.2 (2 × CH2Cy), 23.9 (d, 3JPc = 5.5 Hz, CH3CH), 23.5 (d, 2JPc = 5.8 Hz, CH3CH), 21.5 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 15.8 ppm. FTIR (neat) νmax: ν = 3424 (N-H st) 3345 (N-H st) 1680 (C=O st) 1258 (P=O st) 1335 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H40N2O6PS 551.2345, Found 551.2348.
Methyl (2-(di-iso-propoxyphosphoryl)-2-((4-methylphenyl)sulfonamido)-2-phenylacetyl)glycinate (13i). The general procedure was applied starting from diisopropyl (E)-(phenyl(tosylimino)methyl) phosphonate (10d, 423 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and methyl 2-isocyanoacetate (12b, 100 μL, 1.1 mmol) to afford 335 mg (62%) of 13i after 14 h as white crystals after a chromatography column (Hexanes/AcOEt 6:4), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 146–148 °C. 1H-NMR (400 MHz, CDCl3) δ 7.44–7.35 (m, 2H, 2 × CHAr), 7.25–7.14 (m, 4H, 3 × CHAr + NHCO), 7.08–6.98 (m, 4H, 4 × CHAr), 6.43 (d, 3JPH = 7.8 Hz, 1H, NHTs), 4.82 (hept, 3JHH = 6.0, 1H, CHCH3), 4.74 (hept, 3JHH = 6.3 Hz, 1H, CHCH3), 4.03 (d, 3JHH = 5.2 Hz, 2H, CH2), 3.72 (s, 3H, OCH3), 2.33 (s, 3H, CH3Ts), 1.34 (d, 3JHH = 6.0 Hz, 3H, CH3CH), 1.31 (d, 3JHH = 6.3 Hz, 3H, CH3CH), 1.23 (d, 3JHH = 6.3 Hz, 3H, CH3CH), 1.11 (d, 3JHH = 6.0 Hz, 3H, CH3CH) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 169.3 (C=O), 167.9 (d, 2JPC = 1.3 Hz, C=O), 142.6 (CquatTs), 139.0 (d, 4JPC = 1.3 Hz, CquatTs), 131.8 (d, 2JPC = 1.1 Hz, CquatPh), 130.3 (d, 3JPC = 7.5 Hz, 2 × CHAr), 129.1 (2 × CHAr), 128.6 (CHAr), 127.7 (2 × CHAr), 126.8 (2 × CHAr), 74.4 (d, 2JPC = 7.9 Hz, CH), 74.1 (d, 2JPC = 8.0 Hz, CH), 69.5 (d, 1JPC = 152.4 Hz, Cquat-P), 52.5 (OCH3), 42.2 (CH2), 24.3 (d, 3JPC = 5.0 Hz, CH3CH), 24.2 (d, 3JPC = 4.5 Hz, CH3CH), 23.8 (d, 3JPC = 5.7 Hz, CH3CH), 23.4 (d, 3JPC = 6.0 Hz, CH3CH), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 15.1 ppm. FTIR (neat) νmax: ν = 3425 (N-H st) 3334 (N-H st) 1674 (C=O st) 1254 (P=O st) 1330 (S=O st sym) 1163 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H34N2O8PS 541.1773, Found 541.1778.
Di-iso-propyl (2-(benzylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-phenylethyl)phosphonate (13j). The general procedure was applied starting from di-iso-propyl (E)-(phenyl(tosylimino)methyl) phosphonate (10d, 423 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and (isocyanomethyl) benzene (12c, 134 μL, 1.1 mmol) to afford 464 mg (83%) of 13j after 14 h as white crystals after a chromatography column (Hexanes/AcOEt 8:2), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 100–102 °C. 1H-NMR (400 MHz, CDCl3) δ 7.39–7.31 (m, 2H, 2 × CHAr), 7.30–7.13 (m, 8H, 7 × CHAr + NHCO), 7.06–6.98 (m, 5H, 5 × CHAr), 6.59 (d, 3JPH = 7.4 Hz, 1H, NHTs), 4.85 (hept, 3JHH = 6.2 Hz, 1H, CH), 4.64 (hept, 3JHH = 6.1 Hz, 1H, CH), 4.55 (dd, 2JHH = 14.9, 3JHH = 6.3 Hz, 1H, CHACHB), 4.33 (dd, 2JHH = 14.9, 3JHH = 5.3 Hz, 1H, CHACHB), 2.33 (s, 3H, CH3Ts), 1.33 (d, 3JHH = 6.1 Hz, 3H, CH3CH), 1.30 (d, 3JHH = 6.2 Hz, 3H, CH3CH), 1.14 (d, 3JHH = 6.1 Hz, 3H, CH3CH), 1.06 (d, 3JHH = 6.2 Hz, 3H, CH3CH) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 167.5 (C=O), 142.5 (CquatTs), 139.3 (d, 4JPC = 1.3 Hz, CquatTs), 137.2 (CquatPh), 132.4 (CquatPh), 130.2 (d, 3JPC = 7.6 Hz, 2 × CHAr), 129.0 (2 × CHAr), 128.7 (2 × CHAr), 128.5 (CHAr), 127.8 (2 × CHAr), 127.8 (2 × CHAr), 127.7 (CHAr), 126.8 (2 × CHAr), 74.1 (d, 2JPC = 8.0 Hz, 2 × CH), 69.1 (d, 1JPC = 154.6 Hz, Cquat-P), 44.6 (CH2), 24.3 (d, 2JPC = 3.6 Hz, CH3CH), 24.1 (d, 2JPC = 3.0 Hz, CH3CH), 23.8 (d, 2JPc = 5.8 Hz, CH3CH), 23.5 (d, 2JPC = 5.7 Hz, CH3CH), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 15.4 ppm. FTIR (neat) νmax: ν = 3423 (N-H st) 3336 (N-H st) 1678 (C=O st) 1256 (P=O st) 1331 (S=O st sym) 1162 (S=O st as) cm−1.HRMS (ESI-TOF) m/z: [M + H]+ calcd for C28H36N2O6PS 559.2032, Found 559.2038.
Dimethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-1-(4-nitrophenyl)-2-oxoethyl)phosphonate (13k). The general procedure was applied starting from dimethyl (E)-((4-nitrophenyl)(tosylimino)methyl)phosphonate (10e, 412 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1mmol) to afford 418 mg (78%) of 13k after 1 h as white crystals after a chromatography column (Hexanes/AcOEt 7:3), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 100–102 °C. 1H-NMR (400 MHz, CDCl3) δ 7.88–7.84 (m, 2H, 2 × CHAr), 7.52 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.22 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.08–7.01 (m, 2H, 2 × CHAr), 6.79 (d, 3JHH = 8.2 Hz, 1H, NHCO), 6.77 (d, 3JPH = 8.1 Hz, 1H, NHTos), 3.96 (d, 3JPH = 10.9 Hz, 3H, OCH3), 3.76 (d, 3JPH = 10.7 Hz, 3H, OCH3), 3.76 (m, 1H, CHCy), 2.37 (s, 3H, CH3Ts), 1.93–1.51 (m, 4H, 2 × CH2Cy), 1.41–0.97 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.0 (C=O), 147.5 (Cquat-NO2), 143.6 (Cquat Ts), 139.7 (Cquat Ts), 138.8 (CquatAr), 130.9 (d, 3JPC = 7.6 Hz, 2 × CHAr), 129.3 (2 × CHAr), 126.5 (2 × CHAr), 122.8 (2 × CHAr), 67.8 (d, 1JPC = 153.9 Hz, Cquat-P), 56.0 (d, 2JPC = 8.0 Hz, OCH3), 55.6 (d, 2JPC = 7.6 Hz, OCH3), 50.0 (CHCy), 32.3 (CH2Cy), 32.2 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 18.8 ppm. FTIR (neat) νmax: ν = 3422 (N-H st) 3346 (N-H st) 1677 (C=O st) 1263 (P=O st) 1348 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H31N3O8PS 540.1569, Found 540.1571.
Dimethyl (1-(4-bromophenyl)-2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxoethyl)phosphonate (13l). The general procedure was applied starting from dimethyl (E)-((4-bromophenyl)(tosylimino)methyl)phosphonate (10f, 446 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 495 mg (87%) of 13l after 1 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 150–152 °C. 1H-NMR (400 MHz, CDCl3) δ 7.16 (d, 3JHH = 8.3 Hz, 2 × CHAr), 7.09 (m, seen as s, 4H, 4 × CHAr), 7.04 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 6.77 (d, 3JHH = 7.4 Hz, 1H, NHCO), 6.60 (d, 3JPH = 8.2 Hz, 1H, NHTs), 3.96 (d, 3JPH = 10.8 Hz, 3H, OCH3), 3.77 (d, 3JPH = 10.6 Hz, 3H, OCH3), 3.74 (m, 1H, CHCy), 2.36 (s, 3H, CH3Ts), 1.91–1.48 (m, 4H, 2 × CH2Cy), 1.41–0.95 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.7 (C=O), 143.0 (CquatTs), 138.9 (CquatTs), 131.8 (d, 3JPC = 8.1 Hz, 2 × CHAr), 131.1 (CquatAr), 130.9 (2 × CHAr), 129.2 (2 × CHAr), 126.5 (2 × CHAr), 123.4 (Cquat-Br), 67.9 (d, 1JPC = 155.9 Hz, Cquat-P), 55.9 (d, 2JPC = 8.2 Hz, OCH3), 55.4 (d, 2JPC = 7.5 Hz, OCH3), 49.8 (CHCy), 32.3 (CH2Cy), 32.2 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 19.2 ppm. FTIR (neat) νmax: ν = 3421 (N-H st) 3320 (N-H st) 1670 (C=O st) 1250 (P=O st) 1343 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H31BrN2O6PS 573.0824, Found 573.0830.
Dimethyl (1-(4-chlorophenyl)-2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxoethyl)phosphonate (13m). The general procedure was applied starting from dimethyl (E)-((4-chlorophenyl)(tosylimino)methyl) phosphonate (10g, 402 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 422 mg (80%) of 13m after 1 h as white crystals after a chromatography column (Hexanes/AcOEt 7:3), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 165–167 °C. 1H-NMR (400 MHz, CDCl3) δ 7.19–7.12 (m, 4H, 4 × CHAr), 7.04 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.95 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.76 (d, 3JHH = 7.2 Hz, 1H, NHCO), 6.58 (d, 3JPH = 8.1 Hz, 1H, NHTs), 3.97 (d, 3JPH = 10.8 Hz, 3H, OCH3), 3.78 (d, 3JPH = 10.6 Hz, 3H, OCH3), 3.75 (m, 1H, CHCy), 2.36 (s, 3H, CH3Ts), 1.91–0.95 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.7 (C=O), 142.9 (CquatTs), 139.0 (CquatTs), 135.0 (CquatAr), 131.5 (d, 3JPC = 7.7 Hz, 2 × CHAr), 130,6 (Cquat-Cl), 129.1 (2 × CHAr), 128.0 (2 × CHAr), 126.5 (2 × CHAr), 67.8 (d, 1JPC = 155.9 Hz, Cquat-P), 55.9 (d, 2JPC = 8.2 Hz, OCH3), 55.4 (d, 2JPC = 7.5 Hz, OCH3), 49.8 (CHCy), 32.3 (CH2Cy), 32.2 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ (ppm): 19.2 ppm. FTIR (neat) νmax: ν = 3417 (N-H st) 3319 (N-H st) 1670 (C=O st) 1258 (P=O st) 1353 (S=O st sym) 1160 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H31ClN2O6PS 529.1331, Found 529.1335.
Dimethyl (2-(cyclohexylamino)-1-(2-fluorophenyl)-1-((4-methylphenyl)sulfonamido)-2-oxoethyl)phosphonate (13n). The general procedure was applied starting from dimethyl (E)-((2-fluorophenyl)(tosylimino)methyl) phosphonate (10h, 385 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 370 mg (73%) of 13n after 1 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 203–205 °C. 1H-NMR (400 MHz, CDCl3) δ 7.88 (t, 3JFH = 7.3 Hz, 1H, CHAr), 7.22 (m, 1H, CHAr), 7.18–7.09 (m, 3H, 3 × CHAr), 7.03–6.93 (m, 3H, 3 × CHAr), 6.41 (m, 1H, NHCO), 5.98 (d, 3JPH = 8.2 Hz, 1H, NHTs), 4.03 (d, 3JPH = 10.7 Hz, 3H, OCH3), 3.87 (d, 3JPH = 10.5 Hz, 3H, OCH3), 3.75 (m, 1H, CHCy), 2.32 (s, 3H, CH3Ts), 1.88–1.44 (m, 4H, 2 × CH2Cy), 1.38–0.78 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.3 (C=O), 160.6 (dd, 1JFC = 252.7, 3JPC = 13.1 Hz, CF). 142.7 (CquatTs), 138.4 (d, CquatTs), 134.2 (d, 3JFC = 5.2 Hz, CHAr), 131.5 (d, 3JFC = 9.1 Hz, CHAr), 129.0 (2 × CHAr), 126.6 (2 × CHAr), 123.9 (d, 4JFC = 3.4 Hz, CHAr), 119.9 (dd, 2JFC = 10.8, 2JPC = 4.6 Hz, CquatAr), 115.7 (d, 2JFC = 22.5 Hz, CHAr), 64.6 (d, 1JPC = 158.9 Hz, Cquat-P), 56.2 (d, 2JPC = 8.5 Hz, OCH3), 55.6 (d, 2JPC = 7.7 Hz, OCH3), 50.0 (CHCy), 32.2 (2 × CH2Cy), 25.4 (CH2Cy), 24.7 (CH2Cy), 24.6 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 17.8 ppm. 19F-NMR (282 MHz, CDCl3) δ −105.5 ppm. FTIR (neat) νmax: ν = 3417 (N-H st) 3319 (N-H st) 1672 (C=O st) 1268 (P=O st) 1352 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H31FN2O6PS 513.1624, Found 513.1630.
Dimethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-(perfluorophenyl)ethyl)phosphonate (13o). The general procedure was applied starting from dimethyl (E)-((perfluorophenyl)(tosylimino)methyl) phosphonate (10i, 457 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 514 mg (88%) of 13o after 1 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 189–191 °C. 1H-NMR (400 MHz, CDCl3) δ 7.31 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 6.92 (d, 3JHH = 7.3 Hz, 1H, NHCO), 6.40 (br s, 1H, NHTs), 4.01 (d, 3JPH = 10.9 Hz, 3H, OCH3), 3.82 (d, 3JPH = 10.7 Hz, 3H, OCH3), 3.79–3.69 (m, 1H, CHCy), 2.36 (s, 3H, CH3Ts), 1.96–0.94 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 164.0 (C=O), 146.4 (m, 2 × CF), 144.0 (CquatTs), 142.1 (m, 2 × CF), 137.8 (CquatTs), 137.5 (m, CF), 129.2 (2 × CHAr), 126.3 (2 × CHAr), 108.9 (m, CquatAr), 61.2 (d, 1JPC = 159.7 Hz, Cquat-P), 56.4 (d, 2JPC = 8.6 Hz, OCH3), 55.9 (d, 2JPC = 8.1 Hz, OCH3), 50.3 (CHCy), 32.3 (CH2Cy), 31.9 (CH2Cy), 25.4 (CH2Cy), 24.7 (CH2Cy), 24.6 (CH2Cy), 21.5 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 15.9 ppm. 19F-NMR (282 MHz, CDCl3) δ −129.4, −151.7, −162.3 ppm. FTIR (neat) νmax: ν = 3417 (N-H st) 3316 (N-H st) 1677 (C=O st) 1268 (P=O st) 1330 (S=O st sym) 1165 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H27F5N2O6PS 585.1249, Found 584.1166.
Dimethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-(4-((trichloromethyl)thio)phenyl)ethyl)phosphonate (13p). The general procedure was applied starting from dimethyl (E)-((tosylimino)(4-((trichloromethyl)thio)phenyl)methyl) phosphonate (10j), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 580 mg (92%) of 13p after 14 h as white crystals after a chromatography column (Hexanes/AcOEt 7:3), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 148–150 °C. 1H-NMR (400 MHz, CDCl3) δ 7.49 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.42 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.24 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 7.06 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 6.75 (d, 3JHH = 6.8 Hz, 1H, NHCO), 6.58 (d, 3JPH = 8.2 Hz, 1H, NHTs), 3.96 (d, 3JPH = 10.8 Hz, 3H, OCH3), 3.77 (d, 3JPH = 10.6 Hz, 3H, OCH3), 3.71 (m, 1H, CHCy), 2.33 (s, 3H, CH3Ts), 1.91–1.45 (m, 4H, 2 × CH2Cy), 1.39–0.94 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.4 (C=O), 143.1 (CquatTs), 139.1 (CquatTs), 136.4 (2 × CHAr), 136.2 (CquatAr), 131.2 (CquatAr) 130.8 (d, 3JPC = 7.9 Hz, 2 × CHAr), 129.3 (2 × CHAr), 126.5 (2 × CHAr), 98.2 (CCl3), 68.0 (d, 1JPC = 155.9 Hz, Cquat-P), 55.9 (d, 2JPC = 8.1 Hz, OCH3), 55.4 (d, 2JPC = 7.5 Hz, OCH3), 49.8 (CHCy), 32.2 (CH2Cy), 32.0 (CH2Cy), 25.4 (CH2Cy), 24.5 (CH2Cy), 24.4 (CH2Cy), 21.6 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ (ppm) 19.0 ppm. FTIR (neat) νmax: ν = 3412 (N-H st) 3337 (N-H st) 1677 (C=O st) 1265 (P=O st) 1353 (S=O st sym) 1160 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H31Cl3N2O6PS2 643.0427, Found 643.0430.
Dimethyl (1-(3-chloro-4-methoxyphenyl)-2-(cyclohexylamino)-1-((4-methylphenyl)-sulfonamido)-2-oxoethyl)-phosphonate (13q). The general procedure was followed, using dimethyl (E)-((3-chloro-4-methoxyphenyl)(tosylimino)-methyl) phosphonate (10k, 431 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 418 mg (75%) of 13q after 14 h as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 115–117 °C. 1H-NMR (400 MHz, CDCl3) δ 7.45 (m, 1H, CHAr), 7.34 (d, 3JHH = 4.4 Hz, 1H, NHCO), 7.15 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.02 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 6.78–6.29 (m, 2H, 2 × CHAr), 6.53 (d, 3JPH = 7.5 Hz, 1H, NHTs), 4.04 (d, 3JPH = 10.7 Hz, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.83 (d, 3JPH = 10.4 Hz, 3H, OCH3), 3.79 (m, 1H, CHCy), 2.34 (s, 3H, CH3Ts), 1.87–1.02 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.9 (C=O), 155.2 (Cquat), 143.2 (Cquat Ts), 138.7 (Cquat Ts), 132.0 (d, 3JPC = 10.9 Hz, CHAr), 131.0 (d, 3JPC = 6.7 Hz, CHAr), 129.2 (2 × CHAr Ts), 126.4 (2 × CHAr Ts), 124.3 (Cquat), 121.9 (Cquat), 110.2 (CHAr), 67.5 (d, 1JPC = 157.8 Hz, Cquat-P), 56.3 (OCH3), 56.2 (d, 2JPC = 7.4 Hz, OCH3), 55.4 (d, 2JPC = 7.5 Hz, OCH3), 49.8 (CHCy), 32.3 (CH2Cy), 32.2 (CH2Cy), 25.4 (CH2Cy), 24.6 (CH2Cy), 24.5 (CH2Cy), 21.6 (CH3 Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 19.1 ppm. FTIR (neat) νmax: ν = 3434 (N-H st) 3323 (N-H st) 1677 (C=O st) 1258 (P=O st) 1334 (S=O st sym) 1160 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H33ClN2O7PS 559.1436, Found 559.1437.
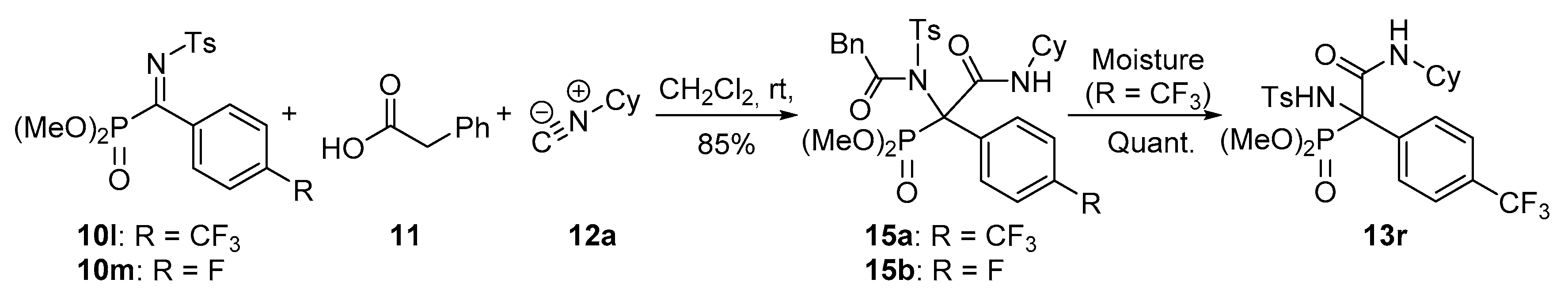
Dimethyl (2-(cyclohexylamino)-2-oxo-1-(2-phenyl-N-tosylacetamido)-1-(4-(trifluoromethyl)phenyl)ethyl)phosphonate (15a). The general procedure was applied starting from dimethyl (E)-((tosylimino)(4-(trifluoromethyl)phenyl)methyl) phosphonate (10l, 435 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 579 mg (85%) of 15a after 1 h as white crystals after a chromatography column (Hexanes/AcOEt 7:3), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 84–86 °C. 1H-NMR (400 MHz, CDCl3) δ 8.20 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.78 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.57 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.36 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.28–7.17 (m, 5H, 5 × CHAr), 5.95 (br s, 1H, NHCO), 4.20 (d, 2JHH = 17.1 Hz, 1H, CHACHB), 3.98 (d, 2JHH = 17.1 Hz, 1H, CHACHB), 3.84 (d, 3JPH = 11.2 Hz, 3H, OCH3), 3.80–3.68 (m, 1H, CHCy), 3.50 (d, 3JPH = 11.5 Hz, 3H, OCH3), 2.44 (s, 3H, CH3Ts), 1.98–0.90 (m, 10H, 6 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 176.6 (C=O), 164.7 (d, 2JPC = 5.2 Hz, C=O), 144.8 (CquatTs), 138.5 Cquat), 138.4 (CquatTs), 133.9 (CquatCF3), 133.6 (CquatAr), 130.2 (2 × CHAr), 129.9 (2 × CHAr), 129.2 (d, 3JPC = 5.3 Hz, 2 × CHAr), 128.4 (2 × CHAr), 128.0 (2 × CHAr), 127.3 (d, 4JFC = 3.8 Hz, 2 × CHAr), 125.1 (CHAr), 123.9 (q, 1JFC = 272.3 Hz, CF3), 79.4 (d, 1JPC = 147.5 Hz, Cquat-P), 55.4 (d, 2JPC = 7.3 Hz, OCH3), 55.1 (d, 2JPC = 7.7 Hz, OCH3), 49.2 (CHCy), 45.7 (CH2Bn), 32.4 (CH2Cy), 31.9 (CH2Cy), 25.5 (CH2Cy), 24.6 (CH2Cy), 24.4 (CH2Cy), 21.8 (CH3Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ (ppm): 22.1 ppm. 19F-NMR (282 MHz, CDCl3) δ −63.1.ppm. FTIR (neat) νmax: ν = 3430 (N-H st) 1676 (C=O st) 1248 (P=O st) 1330 (S=O st sym) 1160 (S=O st as) cm−1.HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H36F3N2O7PS 680.1933, Found 680.1934.
Dimethyl (2-(cyclohexylamino)-1-((4-methylphenyl)sulfonamido)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)phosphonate (13r). Exposure of 15a under air moisture for 48 h yields 13r in quantitative yield as white crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p.: 88–90 °C. 1H-NMR (400 MHz, CDCl3) δ 7.36 (d, 3JHH = 7.8 Hz, 2H, 2 × CHAr), 7.22 (d, 3JHH = 7.8 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 6.98 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 6.84 (d, 3JHH = 7.8 Hz, 1H, NHCO), 6.70 (d, 3JPH = 8.1 Hz, 1H, NHTs), 3.99 (d, 3JPH = 10.8 Hz, 3H, OCH3), 3.80 (m, 1H, CHCy), 3.78 (d, 3JPH = 10.6 Hz, 3H, OCH3), 2.32 (s, 3H, CH3Tos), 1.91–0.94 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 165.4 (C=O), 143.0 (CquatTs), 138.8 (CquatTs), 135.8 (Cquat), 130.6 (q seen as d, 3JFC = 7.9 Hz, 2 × CHAr), 130.7 (d, 2JPC = 32.8 Hz, CquatCF3), 129.2 (2 × CHAr), 126.3 (2 × CHAr), 124.6 (d, 4JFC = 3.7 Hz, 2 × CHAr), 123.7 (q, 1JFC = 272.5 Hz, CF3), 68.0 (d, 1JPC = 155.2 Hz, Cquat-P), 55.8 (d, 2JPC = 8.1 Hz, OCH3), 55.5 (d, 2JPC = 7.6 Hz, OCH3), 49.9 (CHCy), 32.2 (CH2Cy), 32.1 (CH2Cy), 25.3 (CH2Cy), 24.6 (CH2Cy), 24.6 (CH2Cy), 21.4 (CH3Tos) ppm. 31P-NMR (121 MHz, CDCl3) δ (ppm): 20.0 ppm. 19F-NMR (282 MHz, CDCl3) δ −63.5.ppm. FTIR (neat) νmax: ν = 3430 (N-H st) 3332 (N-H st) 1677 (C=O st) 1259 (P=O st) 1326 (S=O st sym) 1165 (S=O st as) cm−1.HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H30F3N2O6PS 562.1514, Found 562.1519.
Dimethyl (2-(cyclohexylamino)-1-(4-fluorophenyl)-2-oxo-1-(2-phenyl-N-tosylacetamido)ethyl)phosphonate (15b). The general procedure was followed, using dimethyl (E)-(4-fluorophenyl phenyl(tosylimino)methyl) phosphonate (10m, 385 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 536 mg (85%) of 15b after 14 h as white crystals after column chromatography (Hexanes/AcOEt 8:2), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 90–92 °C. 1H-NMR (400 MHz, CDCl3) δ 1H-NMR (400 MHz, CDCl3) δ 8.26 (d, 3JHH = 7.9 Hz, 2H, 2 × CHAr), 7.05 (m, 2H, 2 × CHAr), 7.37 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 7.28–7.18 (m, 5H, 5 × CHAr), 7.03 (dd, seen as t, 3JHH = 8.5 Hz, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 5.93 (broad s, 1H, NH), 4.17 (d, 2JHH = 17.1 Hz, 1H, CH2), 3.92 (d, 2JHH = 17.1 Hz, 1H, CH2), 3.82 (d, 3JPH = 11.2 Hz, 3H, OCH3), 3.73 (m, 1H, CHCy), 3.50 (d, 3JPH = 11.5 Hz, 3H, OCH3), 2.44 (s, 3H, CH3Ts), 1.85–0.94 (m, 10H, 5 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CDCl3) δ 176.4 (C=O), 165.3 (d, 2JPC = 4.8 Hz, C=O), 162.4 (d, 1JFC = 248.9 Hz, CAr-F), 144.6 (Cquat), 138.6 (Cquat), 133.8 (Cquat), 131.9 (m, Cquat + 2 × CHAr), 130.2 (2 × CHAr), 129.8 (2 × CHAr), 128.4 (2 × CHAr), 128.0 (2 × CHAr), 127.2 (CHAr), 115.2 (d, 2JFC = 21.6 Hz, 2 × CHAr), 78.9 (d, 1JPC = 148.5 Hz, Cquat-P), 55.2 (d, 3JPc = 7.2 Hz, CH3O), 54.9 (d, 3JPC = 7.7 Hz, CH3O), 48.9 (CH Cy), 45.7 (CH2 Bn), 32.4 (CH2 Cy), 31.9 (CH2 Cy), 25.6 (CH2 Cy), 24.5 (CH2 Cy), 24.4 (CH2 Cy), 21.8 (CH3 Ts) ppm. 31P-NMR (121 MHz, CDCl3) δ 21.8 ppm. 19F NMR (282 MHz, CDCl3): δ −113.9 ppm. FTIR (neat) νmax: ν = 3426 (N-H st) 1677 (C=O st) 1263 (P=O st) 1348 (S=O st sym) 1156 (S=O st as) cm−1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C31H36FN2O7PS 631.2043, Found 631.2040.
Dimethyl (2-(cyclohexylamino)-2-oxo-1-(2-phenylacetamido)ethyl)phosphonate (16). The general procedure was followed, using dimethyl (E)-dimethyl (E)-(phenyl(tritylimino)methyl)phosphonate (14, 455 mg, 1 mmol), phenylacetic acid (11, 136 mg, 1 mmol) and cyclohexyl isocyanide (12a, 136 μL, 1 mmol) to afford 536 mg (65%) of 16 after 14 h as white crystals after column chromatography (Hexanes/AcOEt 8:2), followed by crystallization (Dichloromethane/Hexanes 1:3). M.p.: 121–122 °C. 1H-NMR (300 MHz, CDCl3) δ 1H-NMR (400 MHz, CDCl3) δ 7.43–7.20 (m, 5H, 5 × CHAr), 5.25 (d, 2JPH = 20.6 Hz, 1H, CH-P), 4.91 (br s, 2H, 2 × NH), 3.73 (br s, 3H, OCH3), 3.70 (br s, 3H, OCH3), 3.63 (m, 1H, CHCy), 3.58 (s, 2H, CH2), 1.84–1.65 (m, 4H, 2 × CH2Cy), 1.36–1.13 (m, 6H, 3 × CH2Cy) ppm. 13C-NMR {1H} (101 MHz, CD3OD) δ 174.3 (C=O), 172.0 (d, 2JPC = 5.3 Hz, C=O), 135.3 (Cquat), 129.0 (2 × CHAr), 128.3 (2 × CHAr), 126.7 (CHAr), 53.5 (d, 3JPc = 6.8 Hz, CH3O), 53.3 (d, 3JPc = 6.4 Hz, CH3O), 50.5 (d, 1JPC = 148.0 Hz, Cquat-P), 49.0 (CH Cy), 42.0 (CH2 Bn), 32.2 (CH2 Cy), 32.1 (CH2 Cy), 25.3 (CH2 Cy), 24.7 (CH2 Cy), 24.6 (CH2 Cy) ppm. 31P-NMR (121 MHz, CDCl3) δ 21.0 ppm. FTIR (neat) νmax: ν = 3436 (N-H st) 3334 (N-H st) 1674 (C=O st) 1265 (P=O st) 1342 (S=O st sym) 1160 (S=O st as) cm−1.HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H27N2O5P, 383.1736, Found 383.1741.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
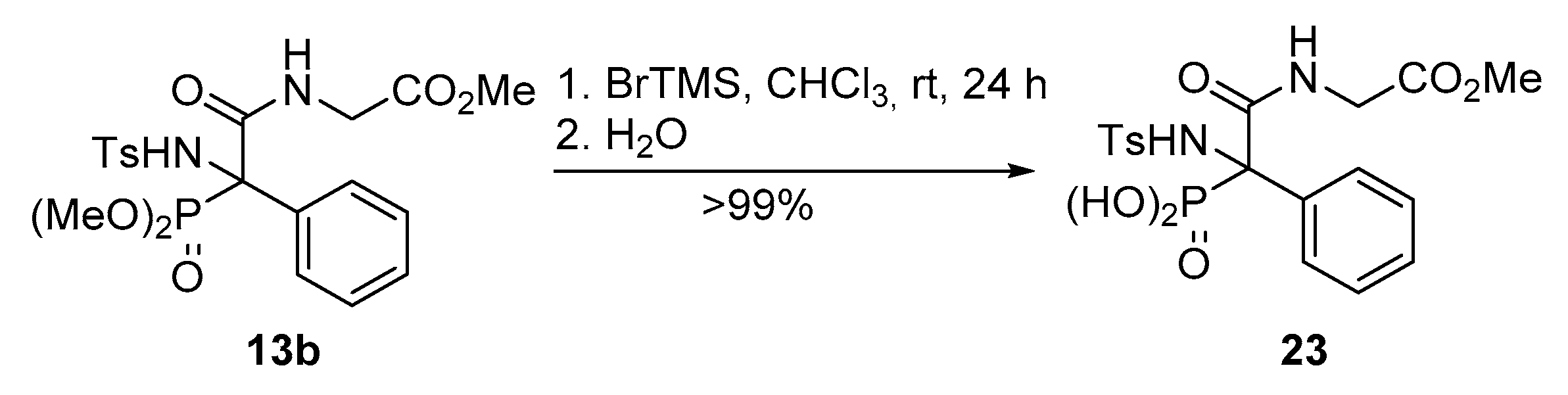{kind=link}