1. Introduction
The kinetic modeling of gases is a relevant issue for the understanding and for the simulation of complex gaseous environments covering Earth and planetary atmospheres [
1,
2], combustion processes, plasma chemistry [
3], and hypersonic aerodynamics [
4,
5], just to mention a few. In many such environments the population of the molecular vibrational states often strongly deviates from the Boltzmann distribution and the detailed description of the gas behavior relies on the knowledge of non-equilibrium energy transfer processes occurring upon collisions. More specifically, the modeling of bulk processes implies the solution of a master equation where the loss/gain of vibrational quanta of energy for molecular species in any initial vibrational quantum state should be included. This means that a very large number of rate coefficients for vibration-to-translation/rotation (V–T/R) and vibration-to-vibration (V–V) energy exchange processes, the most effective for determining the evolution of the vibrational distribution, needs to be known with high accuracy in a large range of temperature.
Unfortunately, experimental measures of such quantities are restricted to a very small number of V–V or V–T/R processes, often involving only low-lying vibrational quantum states. As a consequence, most rates are derived from theoretical calculations, which should be not only reliable but also computationally fast, because of the wealth of processes to be considered. Quasi-classical trajectories (QCT) dynamical treatments are often used, where classical Hamilton equations of motion are propagated in time. In this case, due to the intrinsic quantum nature of the vibrational energy exchange process, a quantum treatment would be highly desirable. However, full quantum mechanical calculations often remain prohibitive for the description of inelastic scattering in four-body systems, because of their computational burden. Mixed quantum-classical (QC) methods, on the other hand, have the advantage of maintaining the computational (and conceptual) simplicity of QCT calculations, while introducing a quantum description for those degrees of freedom which are expected to show a more pronounced quantum behavior in the investigated conditions [
6]. Specifically, for processes involving the transfer of vibrational energy quanta, the diatom vibrations in the quantum-classical method are described by solving the corresponding time-dependent Schrödinger equation, whereas for the remaining degrees of freedom the classical equations of motion are propagated under the influence of an effective potential, obtained as the quantum expectation value [
7,
8,
9]. Such an approach has proved to provide accurate results in a wide temperature range with approximately the same numerical effort of QCT methods.
For this reason, the quantum-classical (QC) approach, sometimes also referred to as semiclassical, has been used over the years for the description of inelastic scattering in a variety of diatom–diatom systems: N
+N
[
10,
11,
12,
13], CO+CO [
14,
15], N
+CO [
11,
16,
17,
18], O
+O
[
19,
20,
21], etc. leading, in some cases, to the creation of large databases for V–V rate coefficients. Databases for V–T/R rate coefficients are instead much more difficult to find, and in fact, the numerical determination of such quantities is often limited to processes involving the loss of one quantum of energy of the first vibrationally excited states. The accurate computation of V–T/R coefficients is computationally more demanding than that of V–V rates: V–T/R rate coefficients are more sensitive to long-range interactions and require larger initial separation distances of the diatoms, up to 70–80 Å, increasing the necessary computation time. Therefore, V–T/R rate coefficients are still often inferred through first-order approaches, like the Schwartz–Slawsky–Herzfeld (SSH) theory [
22], which neglect multiquantum processes and are based on the short-range potential only, leading to approximate values, particularly in the very high or very low-temperature regimes, although the use of scaling procedures can improve their performance. V–T/R rates are believed to play a less important role than V–V ones, particularly when the latter involve nearly resonant energy exchange, but this is usually true in the low-temperature regime only. High-temperature conditions and high vibrational quantum numbers greatly favor V–T/R processes and, as we highlighted in recent investigations on N
-N
[
13] and O
+O
[
21], they can even become the most favorable events, heavily contributing to the overall molecular vibrational distribution. Such temperature regimes are those characterizing, for instance, hypersonic flows around re-entry vehicles. The temperature behind the shock wave is high enough for stimulating inelastic internal energy exchanges (V–T/R, V–V, V–E, and so on) and chemical reactions, and accurate modeling of those physical phenomena is necessary to predict the surface heat flux of the vehicle.
The present study aims to provide a large database of V–V and V–T/R rate coefficients for collisions between molecular nitrogen N
and carbon monoxide CO, calculated through the mixed quantum-classical method [
7,
8,
9,
13,
14,
21]. A detailed database for V–V processes, including some multiquantum transitions, calculated through the QC method, is available up to T = 2900 K [
11]. Here we extend the investigation to consider temperatures up to 7000 K and higher vibrational states. To the best of our knowledge, there are no databases for N
+CO V–T/R transitions, even if N
+CO mixtures can be found in many different environments, where high temperatures can be reached and V–T/R processes are essential for their characterization. Indeed, N
and CO are both important components of many atmospheres of our solar system (e.g., Titan, Triton, Pluto, and Mars [
23]) and extrasolar planetary systems. Furthermore, CO quite obviously plays an important role in combustion chemistry and in CO
plasma. Because of the aforementioned difficulty in their determination, presently available repositories for combustion chemistry or astrochemical data contain structural and dynamical values which are often derived from simple extrapolations or oversimplified computations. For this reason we believe that the accurate calculation of V–V and of the unprecedented V–T/R rates for a large variety of initial molecular vibrational states and a wide temperature range, carried out in an internally consistent way on the same potential energy surface, might represent a step forward in the kinetic modeling of a wealth of gaseous environments.
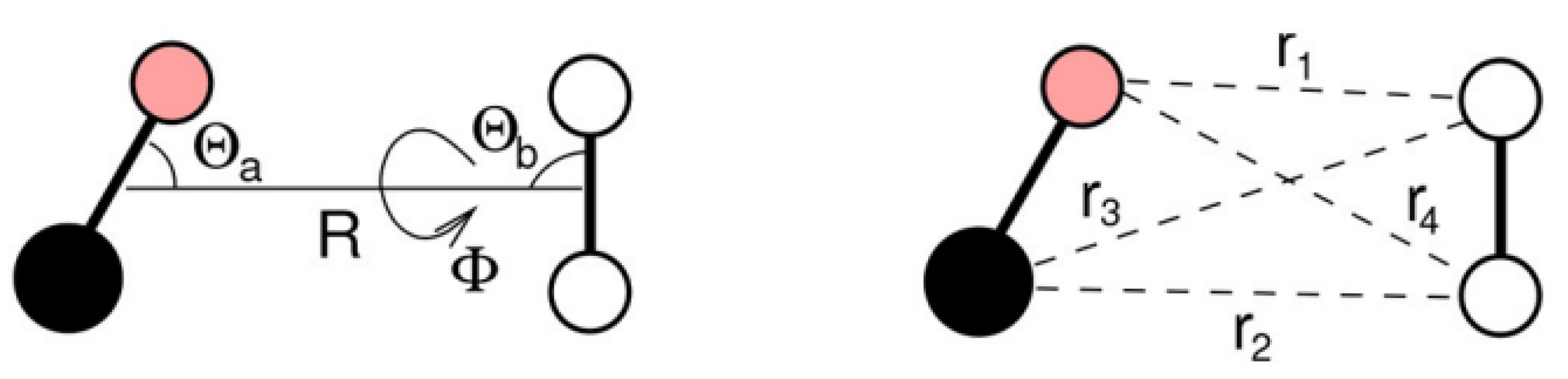
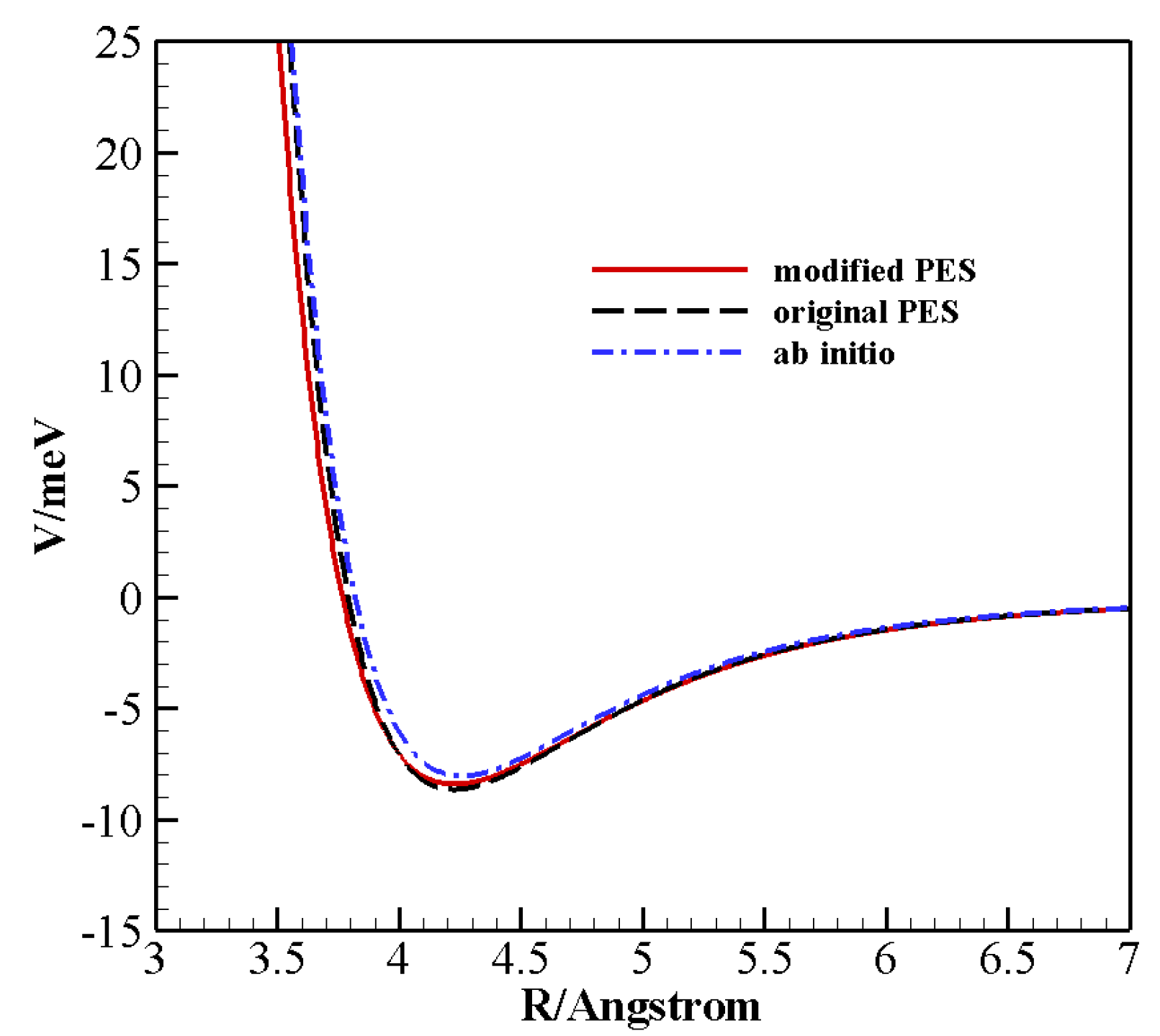
One of the main ingredients to obtain reliable results in the treatment of collisional dynamics is the use of an accurate potential energy surface (PES), capable of describing, in detail, intermolecular energy at long and short range for all possible reciprocal orientations of the diatoms. Reduced dimensionality PESs based on high-level ab initio calculations (at CCSD(T) level coupled to basis sets of quadrupole-
quality) were recently published to describe the N
-CO system [
24,
25,
26]. They have led to a very accurate determination of the roto-vibrational spectrum of the van der Waals complex, but they cannot be used to describe vibrational energy exchange processes with the same confidence, because the intramolecular distance in one or both diatoms is kept frozen. Furthermore, as shown for N
-N
collisions [
27,
28], in general, ab initio based potentials might be inaccurate when very large initial separation distances of the colliding partners are required: the number of ab initio points needed even for an approximate description of all long-range configurations is still prohibitive and interpolation procedures might produce spurious effects [
29]. On the other hand, analytical and/or semiempirical potentials, often simply constructed as a sum of repulsive short-range and attractive long-range components of the interaction potential, have been proposed [
11,
17] and, provided that all interaction regions (long-range, interaction wells, and repulsive walls) are appropriately taken into account, they are found to reproduce V–V and V–T/R rate coefficients quite effectively.
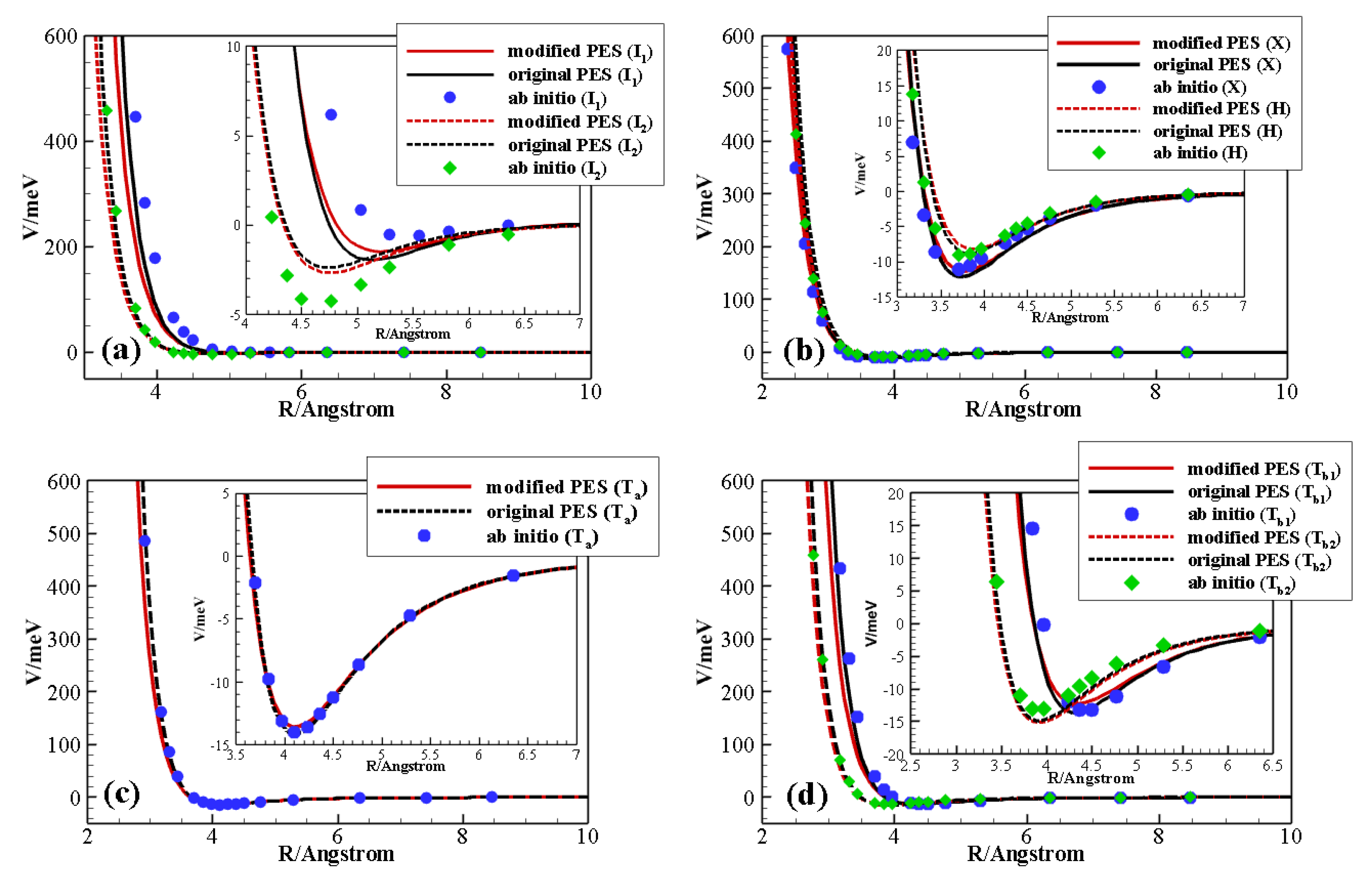
We recently introduced a full dimensional analytical surface [
18] by using a well-established semiempirical approach and tested it to reproduce experimental values, like the second virial coefficients, and against accurate ab initio energies. In the same paper preliminary calculations for selected V–V processes, for which experimental rate coefficients are available, were carried out, which showed an overall good qualitative agreement in a wide temperature range, with some quantitative discrepancies in the low temperature regime.
In order to produce an accurate database, in the present study we start by improving the above PES, according to a procedure we successfully applied in the case of N
-N
[
13], O
-O
[
21], and N
-O [
30] systems. Indeed, one of the main advantages of this analytical formulation, which is far from being a simple fit to ab initio or experimental data, is the possibility of modulating its behavior by modifying the physically meaningful parameters in a limited range. This permits to keep an eye on the physics of the process and on the relevant configurations controlling the dynamics of different phenomena.
The paper is therefore organized as follows. In
Section 2 a short description of the CO+N
PES is given, together with its comparison against experimental and ab initio data and its subsequent improvement against experimental results. A comparison between predictions of QCT and QC methods is also analyzed in this Section.
Section 3 reports a database and a critical discussion of the V–V and V–T/R rate coefficients calculated with the improved PES, aimed to identify the most relevant collisional events taking place at different temperature conditions. Concluding remarks are given in
Section 4.
3. Results and Discussion
As mentioned in
Section 1, an existing database of V–V rate coefficients, obtained by using a QC method, is available [
11]. Here we extend the calculations to include a larger number of V–V transitions and a wider temperature range (20–7000 K). Furthermore, we calculated V–T/R single and multiquantum energy exchange rate coefficients, whose determination is a computationally demanding task, because they need larger initial diatomic separation distances and thus much longer simulation times. For this reason, to the best of our knowledge, this is the first V–T/R rates database for CO-N
collisions covering a wealth of excited states and temperatures. Because of the high dissociation energies of the two molecules, reactive channels for these collisions are likely to play a role even at the highest temperature investigated here. Dissociation rate coefficients calculated for the similar N
-N
system [
53] show that reactivity starts to be significant for temperatures much above 8000 K.
V–V rate coefficients involving highly excited vibrational states were calculated by coupling 121 initial vibrational states while the other settings are the same of the (0,1) → (1,0) calculations (i.e. by starting from an initial diatoms separation distance equal to 15 Å and 45 different values of total classical energies, comprised between 35 cm and 80,000 cm). For the calculation of V–T/R rate coefficients, 81 vibrational states were coupled for and 121 for , the larger number being needed for higher v because of the close spacing between the levels. In this case, an initial diatoms separation distance equal to 80 Å has been considered, needed for V–T/R processes to avoid artificial contributions from the long-range part of the multipole moments.
The dependence of the present PES on the intramolecular distance has been tested in ref. [
18]: molecular polarizabilities of both molecules, N
quadrupole moment and CO permanent dipole moment depend on the corresponding bond lengths. Its accuracy might, however, slightly decrease for very elongated monomers. Furthermore, Morse potential and Morse wavefunctions also lose accuracy when describing very high vibrational states. Rate coefficients for processes involving vibrational states with
might therefore present an overall uncertainty larger than 20%. However, we believe them to be sufficiently reliable (more than those available by extrapolation or first-order treatments) for such processes and to provide at least the correct qualitative trend in their variation.
A comparison between some exemplary symmetric single quantum and asymmetric multiquantum near-resonant V–V rate coefficients calculated on the present PES and those of Ref. [
11] can be found in
Table 4. The present values are a factor two larger ca., the discrepancy growing larger with temperature, which could be connected to the more repulsive short-range character of the present PES.
In many applications, nitrogen molecules only populate the lower vibrational levels because of a fast energy transfer from N
to CO molecules [
54]. Therefore, the rate coefficients for single quantum V–V processes CO
CO
(
Table 5) and near-resonant asymmetric V–V processes CO
CO
(
Table 6) and CO
CO
(
Table 7) are of particular importance. The rate coefficients for processes CO
CO
are also reported in
Table 5.
Figure 6 shows the QC calculated V–V rate coefficients (symbols) for the exothermic CO
CO
processes and for CO
CO
processes as a function of the vibrational quantum number
v at 100 K, 300 K, 3000 K, and 7000 K. The analytical approximation for the rate coefficients obtained by the modified SSH theory using the present QC results (see
Appendix B) is also reported in the figure (dashed lines).
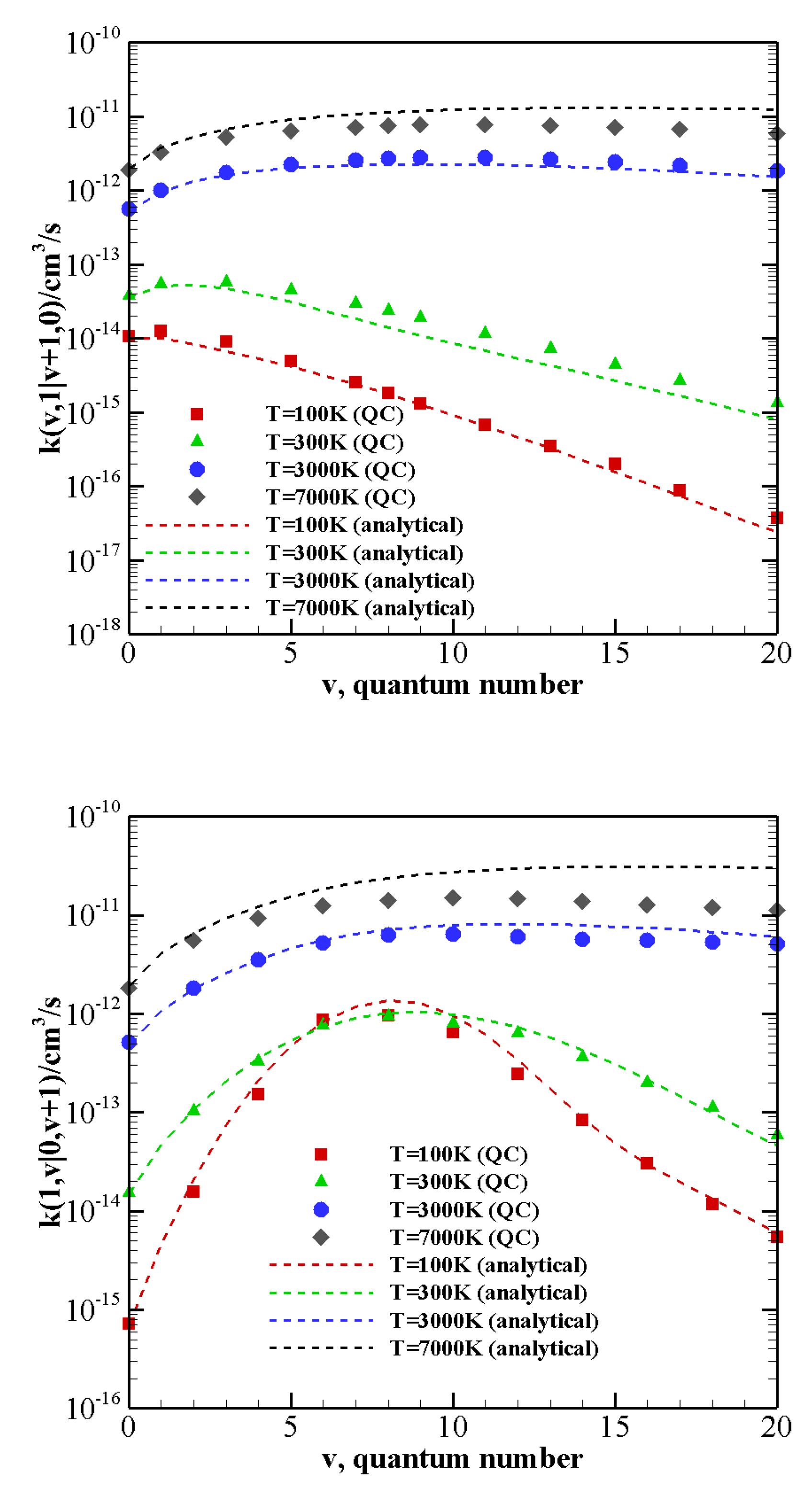
At high temperatures the quenching rate of N (or CO) stimulated by CO (or N) depends little on v. Although the reaction energy increases with the vibrational quantum number, the probability of quenching the first excited state remains not negligible even for the highest vibrationally excited states investigated here. At low temperatures, quasi resonant processes CO CO, with , are the most active to promote quenching and show orders of magnitude differences with the other processes.
The comparison between the QC and analytical rates is quite good at low temperature and for small energy mismatches. At high temperatures, however, the difference grows and the analytical formulation, consistently with the known drawbacks of the SSH theory, fails to correctly reproduce rate coefficients.
The above-described behavior both at high and low temperature is enhanced by the vibrational anharmonicity for the near-resonant asymmetric transitions occurring in N
-CO collisions with vibrationally excited CO, i.e., CO
CO
and CO
CO
. The corresponding rate coefficients are reported in
Figure 7 as a function of the vibrational quantum number
v at 100 K, 300 K, 3000 K, and 7000 K. At high temperature, rate coefficients for such transitions are practically independent on the CO vibrational quantum number and are therefore expected to play an important role in the vibrational kinetics of highly excited CO molecules [
55]. At low temperature rates, coefficients strongly grow as the transition becomes more resonant. For nearly resonant transitions in the low-temperature regime a marked anti-Arrhenius behavior, i.e., rate coefficients getting smaller with temperature, is found, as reported in
Figure S1 in the SI for the CO
CO
transition.
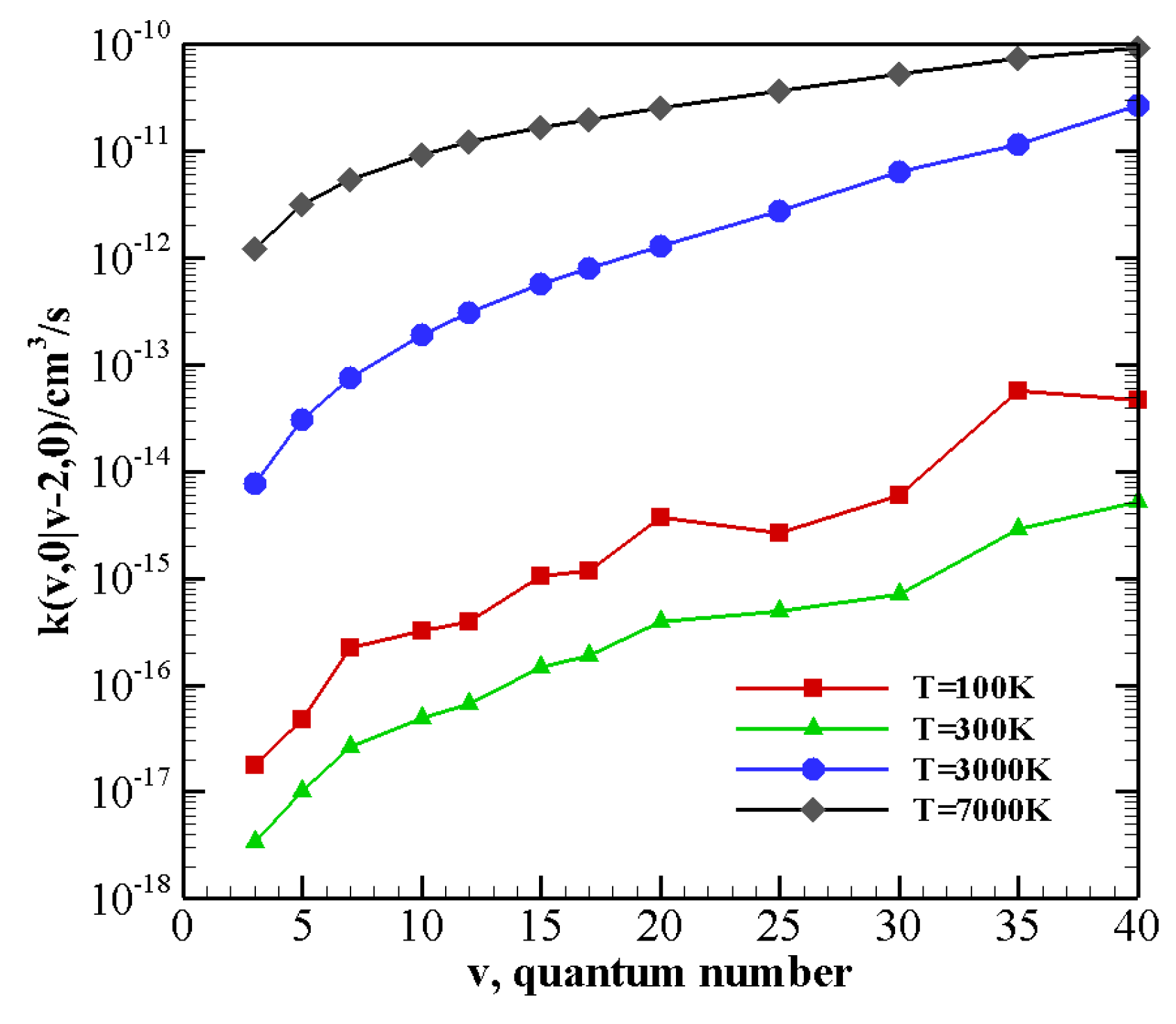
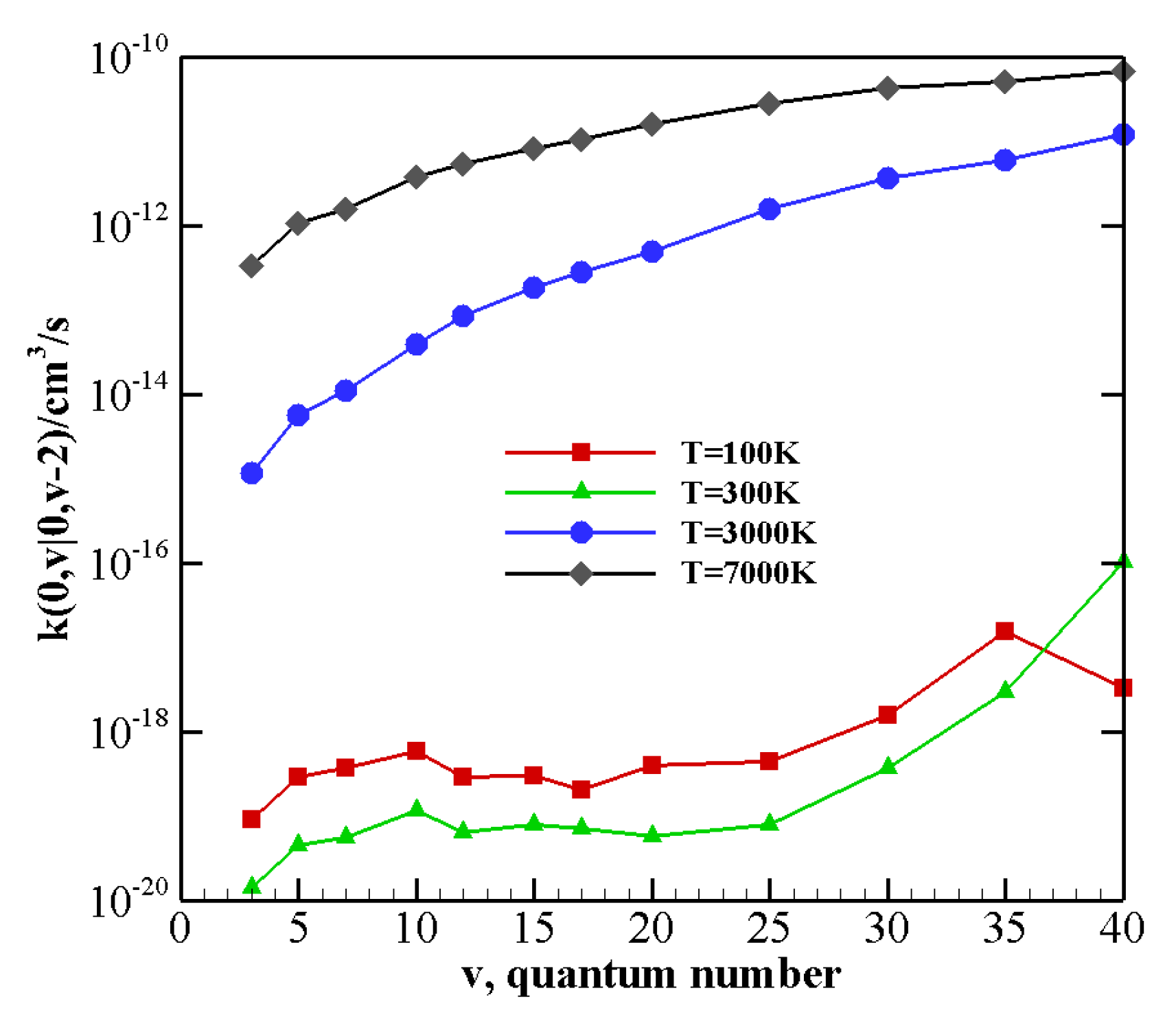
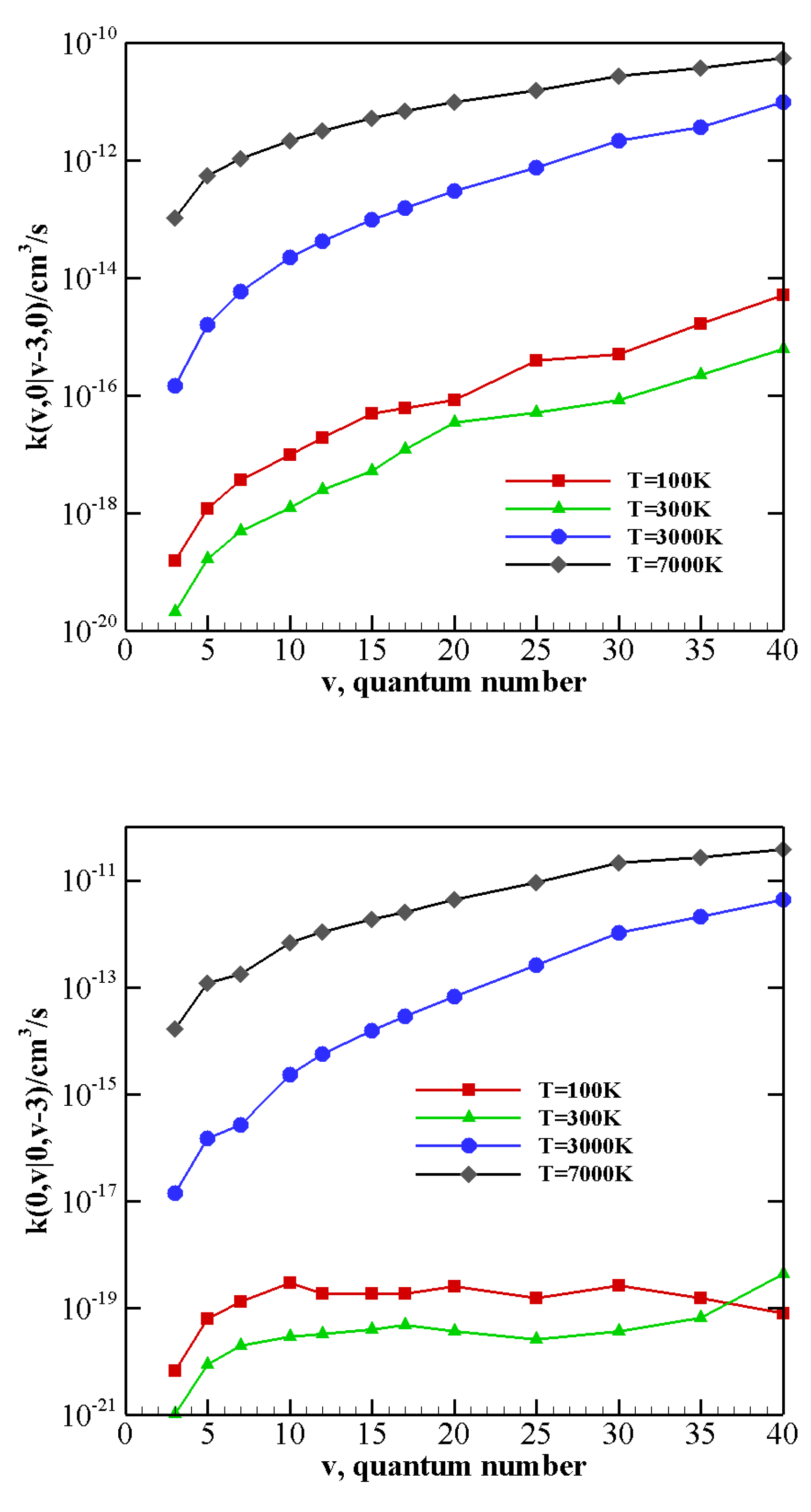
V–T/R rate coefficients for processes where vibrational relaxation occurs due to the collision between N
(CO) in its vibrational ground state and a vibrationally excited CO (N
) molecule, with the loss of a single quantum of vibrational energy are collected in
Table 8 (
Table 9). V–T/R rate coefficients corresponding to the loss of two or three vibrational quanta are reported in
Table 10 and
Table 11. They show that, though the rate of V–T/R processes is very small at low temperature (being generally some orders of magnitude smaller than V–V processes for the same initial vibrational states, see also the following), their efficiency rapidly grows by increasing the temperature. Indeed, V–T/R rates become comparable to V–V ones at high temperatures, and in some cases, they even correspond to the most efficient energy transfer events.
Table 10 and
Table 11 show that even multiquantum V–T/R transitions at high temperature are not negligible, especially for highly excited molecules.
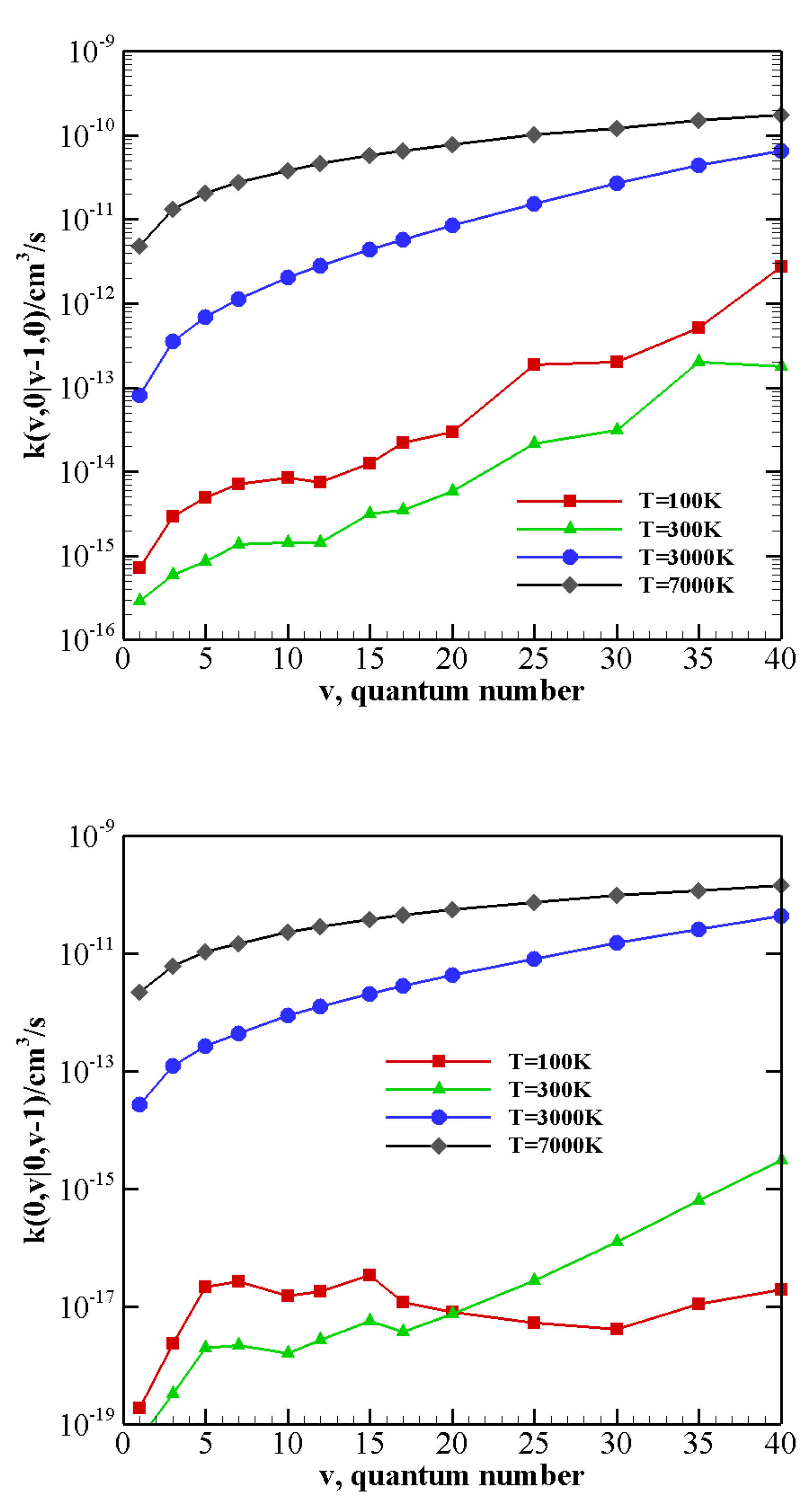
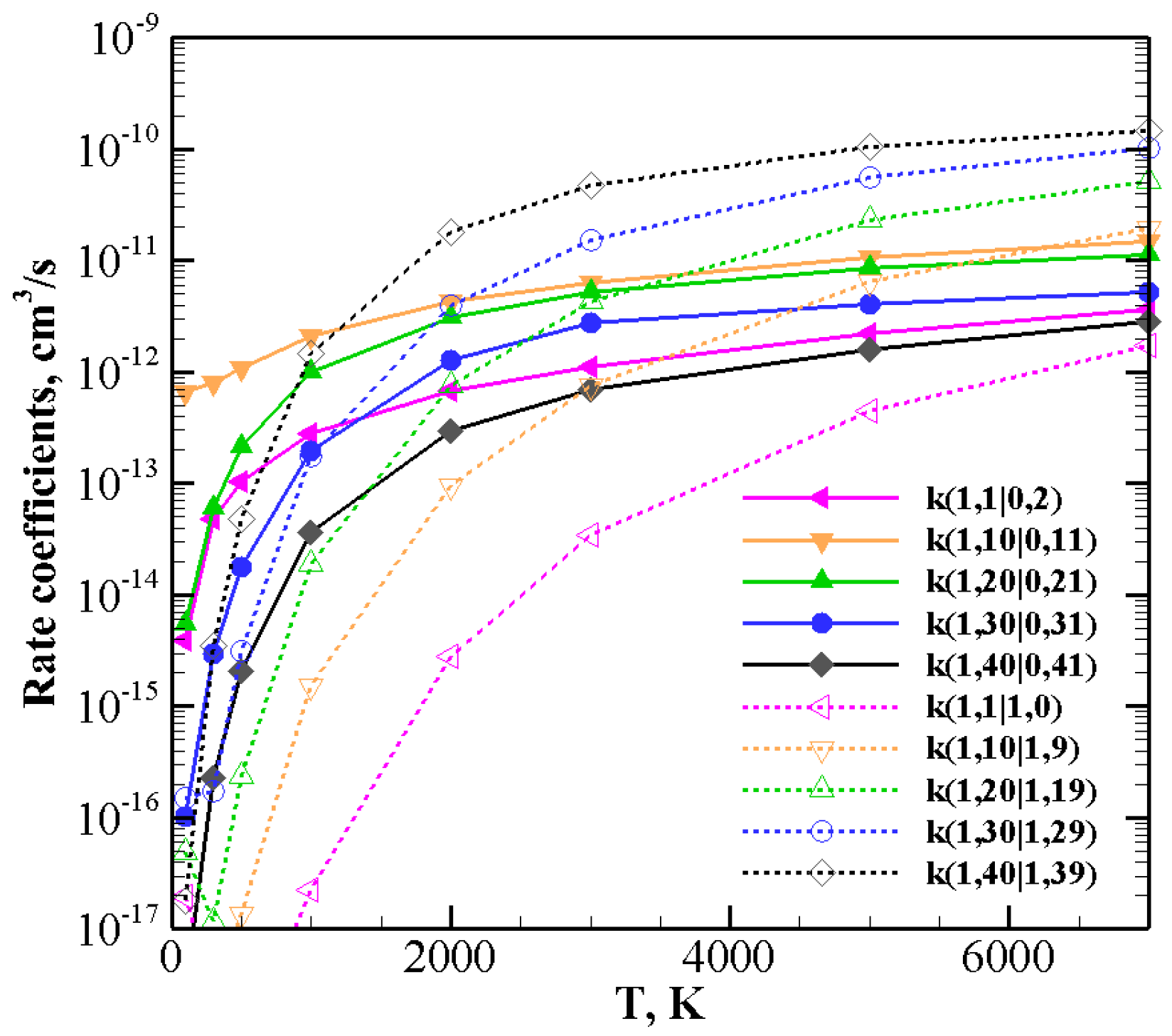
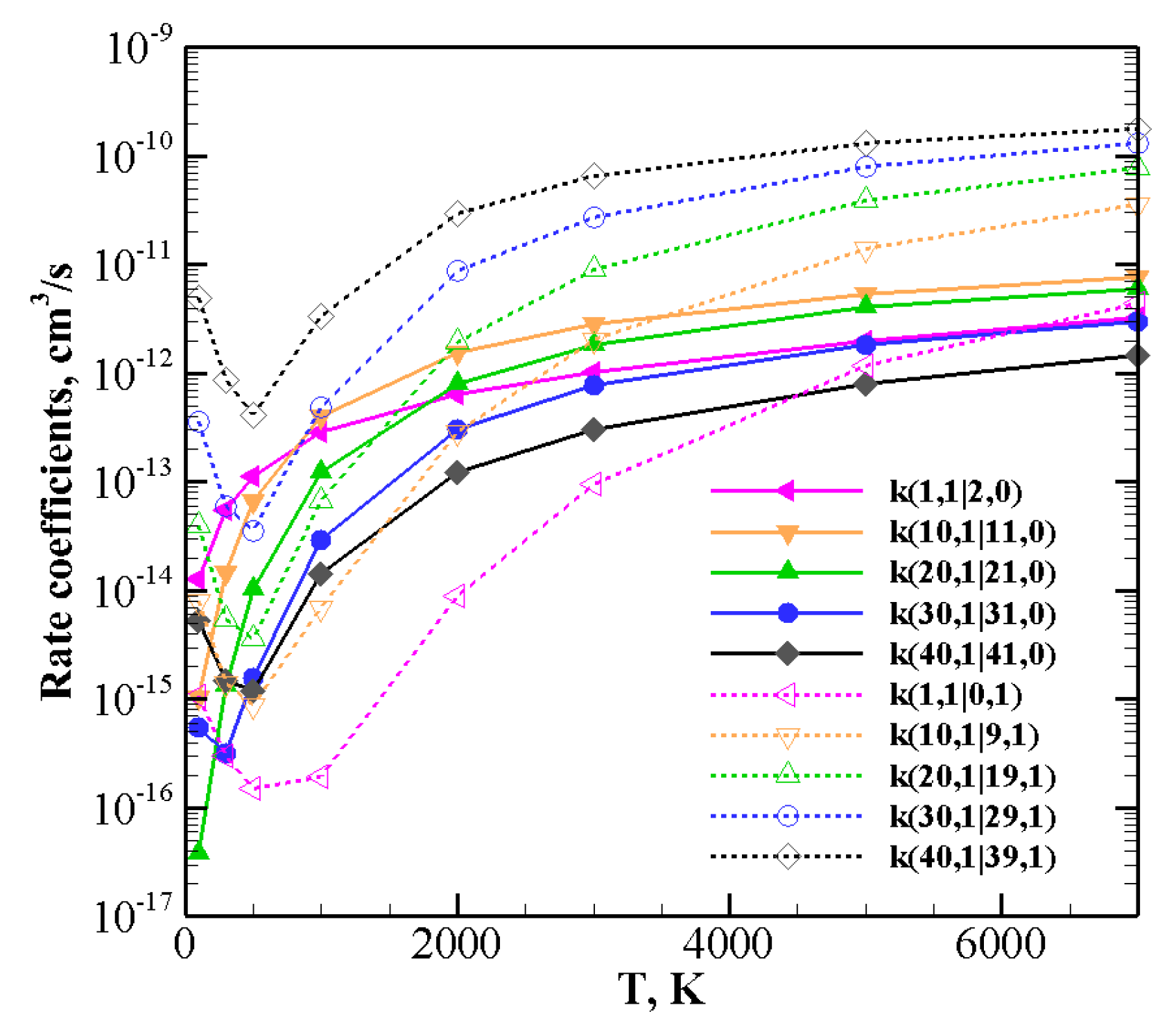
The behavior of rate coefficients for V–T/R processes CO
+ N
(0)→ CO
+ N
(0) and CO
+ N
(
v)→ CO
+N
(
), as a function of the initial quantum number
v at T = 100, 300, 3000, and 7000 K is reported in
Figure 8,
Figure 9 and
Figure 10 for
, respectively. All the figures show a pronounced increase of vibrational relaxation when increasing the initial
v value of either N
or CO
molecule at high temperatures. At low temperatures, V–T/R vibrational relaxation of CO
is more efficient than N
, as a result of the closer vibrational spacing in CO. It is interesting to note that an anti-Arrhenius behavior at low temperatures characterizes the vibrational relaxation of CO(
v) for all
v values investigated here, whereas for the quenching of N
(
v) the standard Arrhenius trend is followed when
v > 20.
The multiquantum V–T/R rate coefficients for CO + N(0)→ CO + N(0) show a very similar qualitative behavior to that of single quantum rates, with correspondingly lower values. For CO + N(v)→ CO + N() processes, at low temperatures, the rate coefficients for the loss of vibrational quanta are practically constant until a sufficiently high value of v is reached. Moreover, with respect to the corresponding single quantum rates, the Arrhenius behavior at low temperatures is restored at higher v values.
It is worth noting that, at low temperatures, many V–T/R rate coefficients are very small and close to the numerical accuracy of the present method, so that such values may not be so accurate as larger ones. However, we believe that they give a correct qualitative indication of the general trend of these processes.
It is important to compare the relative efficiency of V–V and V–T/R processes in different temperature regimes. This is done in
Table 12 where single quantum V–V and V–T/R rate coefficients are reported. V–V processes largely dominate at low temperatures and, though V–T/R rates enhancement with temperature is much more rapid, they remain predominant at the highest temperature investigated here, when V–T/R coefficients are barely the same order of magnitude.
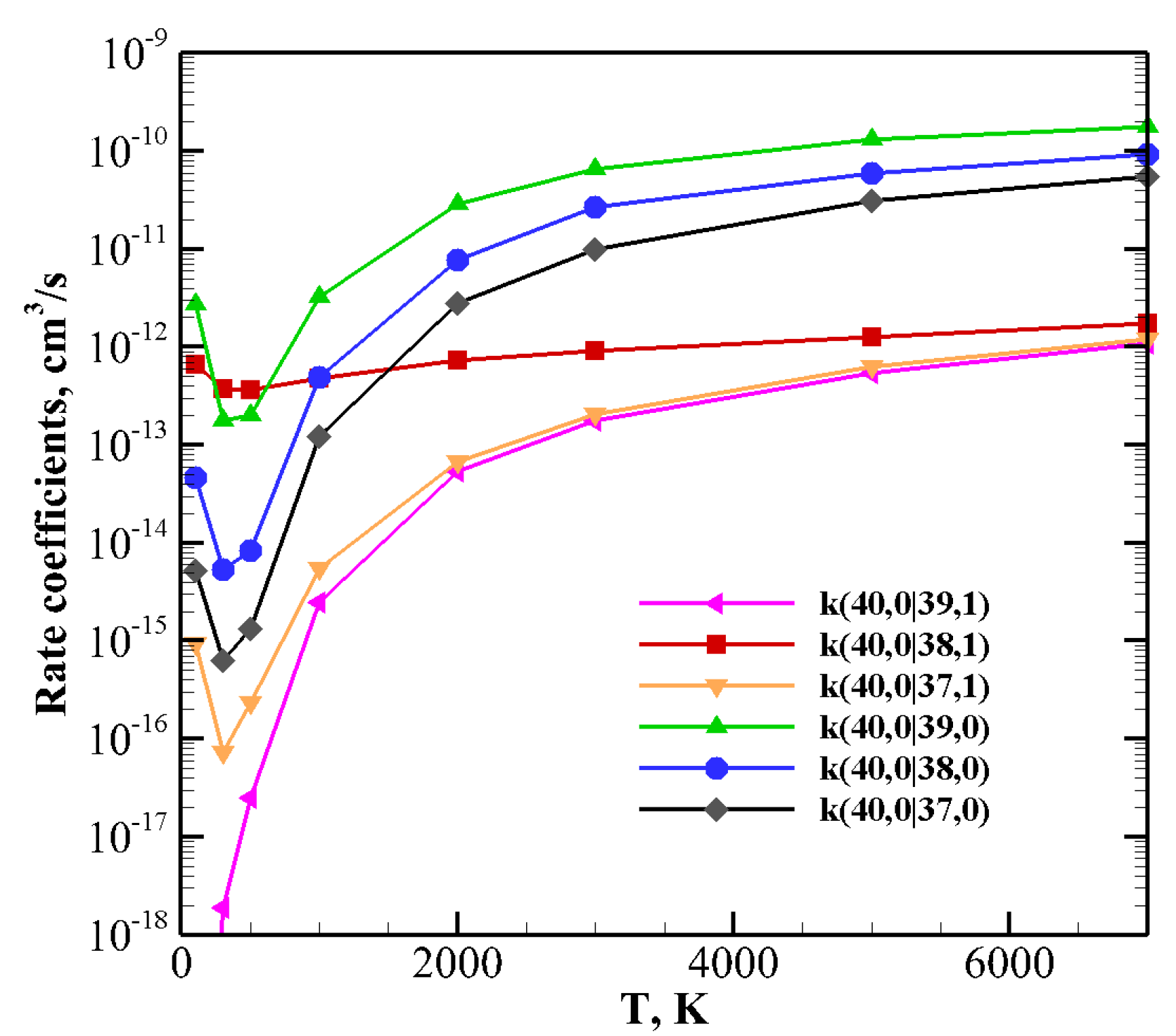
Things change when one of the molecules is highly vibrationally excited and the other one is in the ground (
Figure 11) or in the lowest vibrational excited state
(
Figure 12 and
Figure 13).
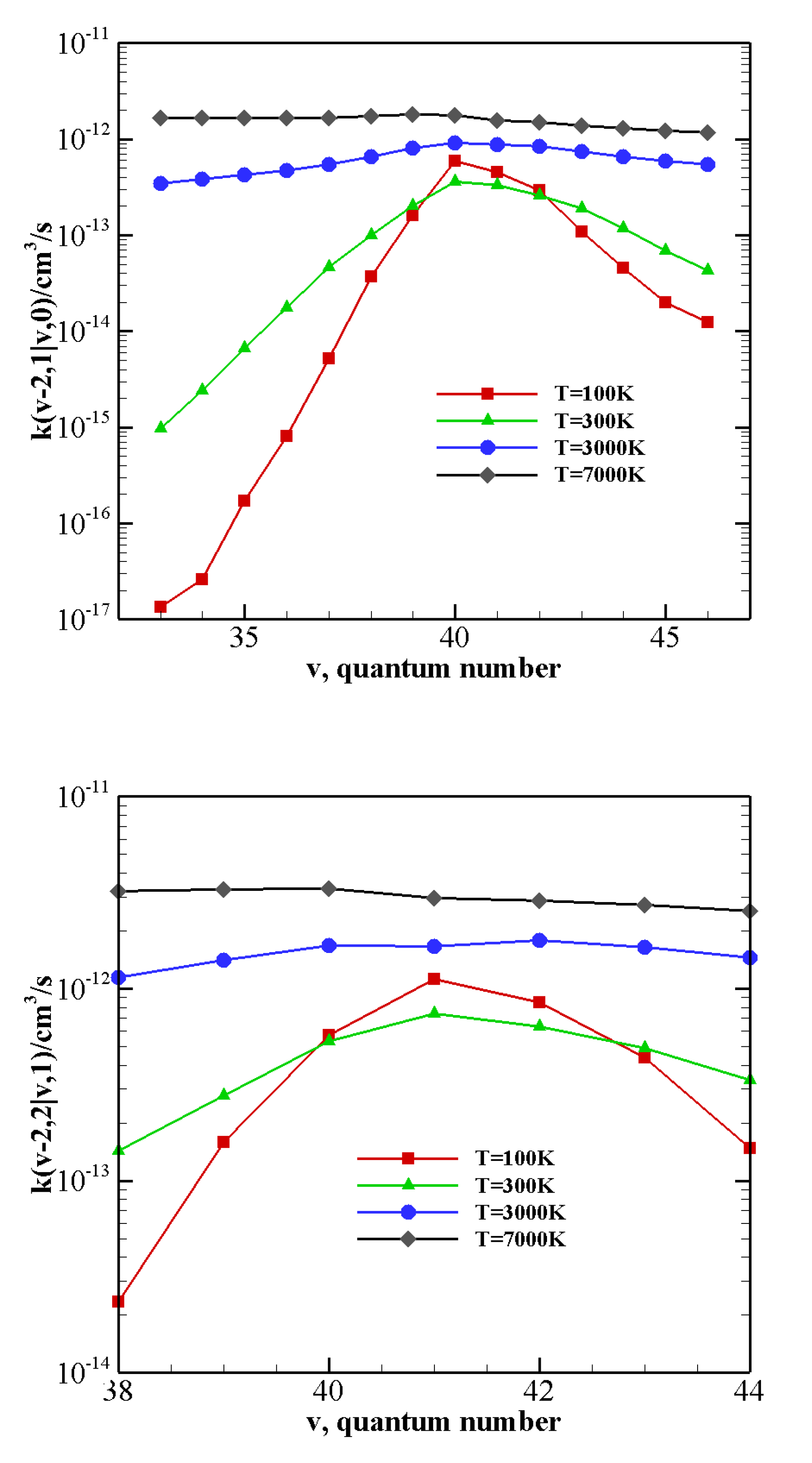
For collision between CO(1) and highly excited N
(
v), rate coefficients for V–V (
Table 5) and V–T/R (
Table 13) exothermic processes are reported in
Figure 12: for the lowest
v value (
) V–V processes always predominate. However, as
v grows, V–T/R processes are faster at high temperatures and they become the most probable events for the highest value investigated here,
, in the whole temperature range. When collisions between N
(1) and highly excited CO(
v) (
Table 5 and
Table 14), corresponding to exothermic processes, are considered (
Figure 13), the above behavior is enhanced. V–T/R always predominate at the highest temperature, and they often do in the low-temperature regime as well. In fact, the anti-Arrhenius behavior occurring at the lowest temperature tends to favor V–T/R processes over V–V ones.
The same indication is found in
Figure 11, which reports the rate coefficients of the collision between a highly vibrationally excited CO
molecule and N
at its ground state. Here both endothermic and exothermic processes are included: V–T/R relaxation tends to be predominant in the whole temperature range, with the exception of the quasi-resonant CO(40) + N
(0) → CO(38) + N
(1) process, which, at low temperatures, is most efficient.
These results suggest that, in the high-temperature range and/or when highly excited states are considered, V–T/R relaxation processes are competitive or more effective than V–V ones. These conditions are those commonly found in plasmas, hypersonic flows, and other situations of interest in aerospace science, therefore V–T/R databases are useful not only for numerical simulation of the above scenarios but also a valuable source for comparison and interpretation of laboratory-based experiments.
4. Conclusions
A recent analytical PES for non-reactive collisions between CO and N was critically improved to be used for the construction of a large reliable database of V–V and V–T/R rate coefficients. This is possible thanks to the ILJ formulation of the potential, which gives the opportunity of modulating physically meaningful parameters by looking at the stereodynamical behavior of the system and by checking and analyzing the performance of the PES against high-level ab initio and experimental data.
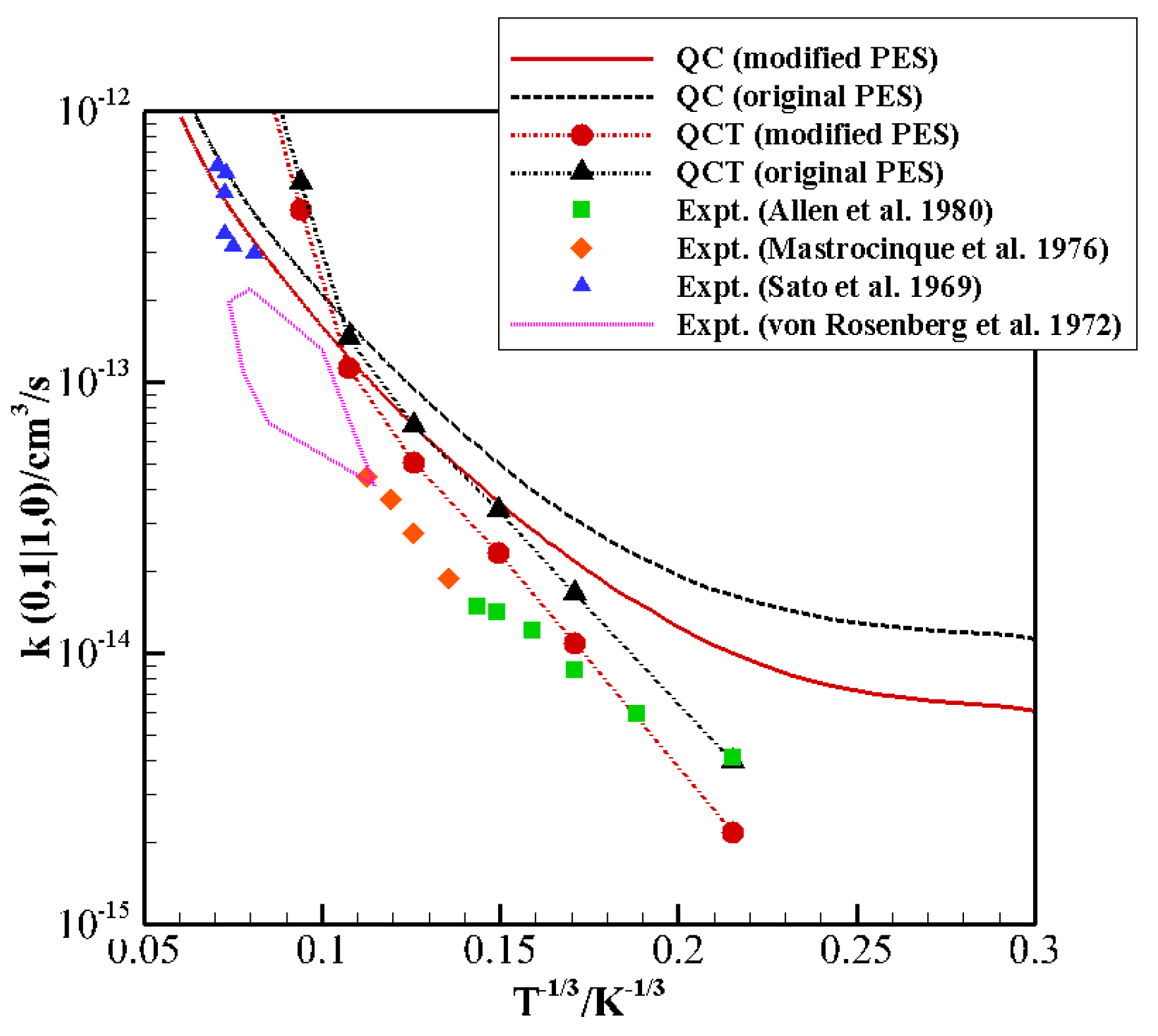
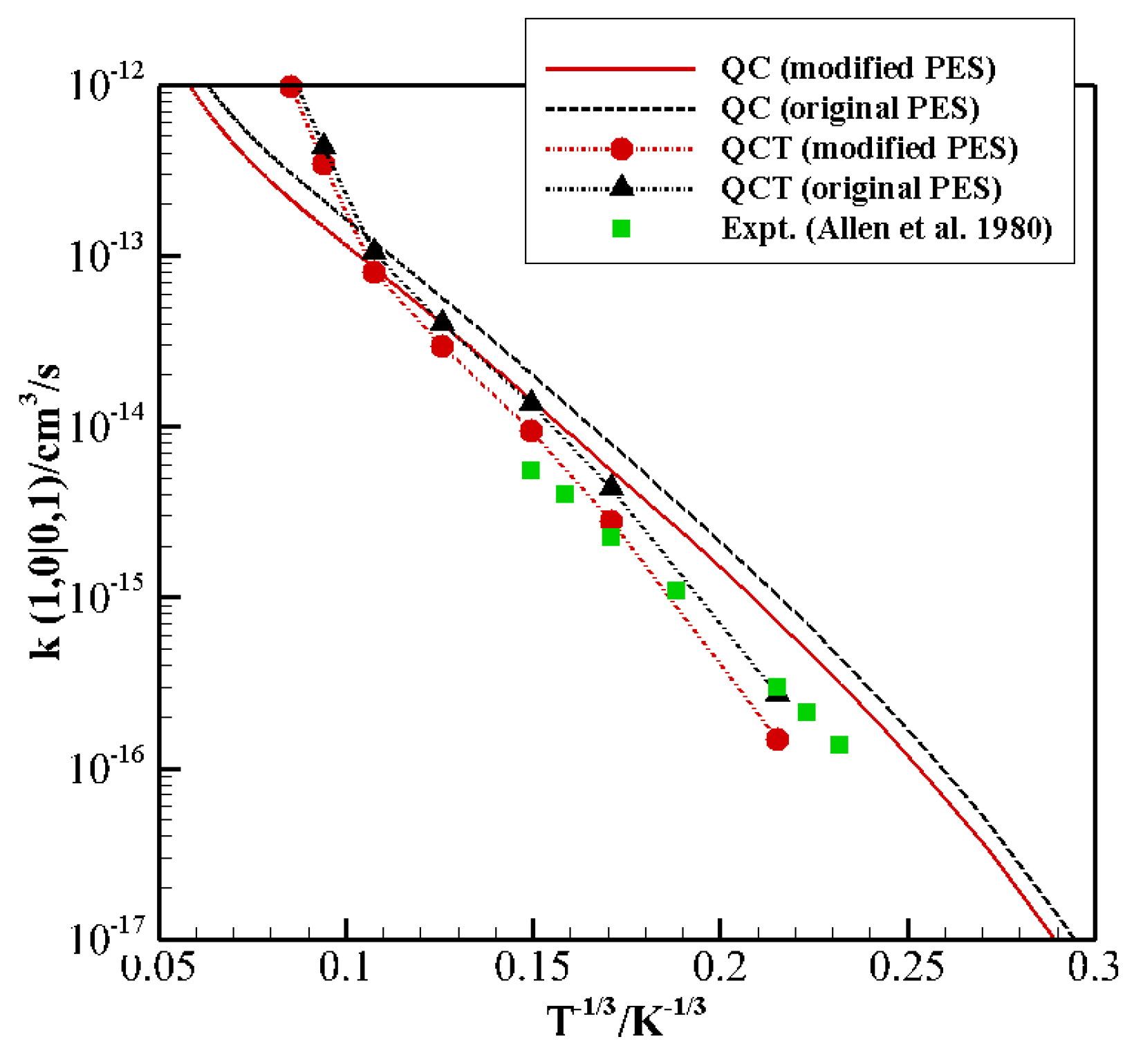
QCT and QC rate coefficients for (1,0) → (0,1) and (0,1) → (1,0) transitions were determined, for which experimental data are available, showing a better agreement than the original PES, particularly at low temperature. QCT calculations show an excellent agreement at low temperature, but fail to reproduce the correct trend in the whole temperature range, whereas QC ones provide the correct slope of the Landau–Teller plots and a more accurate overall behavior. In fact, the new potential improves the description of the inelastic scattering dynamics also in the high temperature regime, most important for the modeling of hypersonic flows and aerospace applications.
We find that, in the above-mentioned high-temperature conditions, V–T/R coefficients, whose calculation represents an additional challenge, due to the required computational time, are generally comparable or even larger to V–V ones. This makes the determination of these quantities an important step towards the accurate modeling of combustion processes, satellite or spacecraft re-entry conditions, etc. The present database is at the date the first one containing a large number of V–T/R rate coefficients.
We would like to conclude by pointing out that the present investigation is one of a series where a systematic approach is used to build PESs in an internally coherent fashion, i.e., by using a physically meaningful analytical formulation of the potential performing well in wide temperature ranges, tested against ab initio calculations and available experimental properties of different kinds. Such potentials are then used for the determination of large bodies of inelastic scattering cross-sections, involving vibrational energy transfer. Work is still in progress to extend such methodology to other diatom–diatom and diatom–atom systems.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}