
Enthalpies of Adduct Formation between Boron Trifluoride and Selected Organic Bases in Solution: Toward an Accurate Theoretical Entry to Lewis Basicity


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Gas Phase Enthalpies
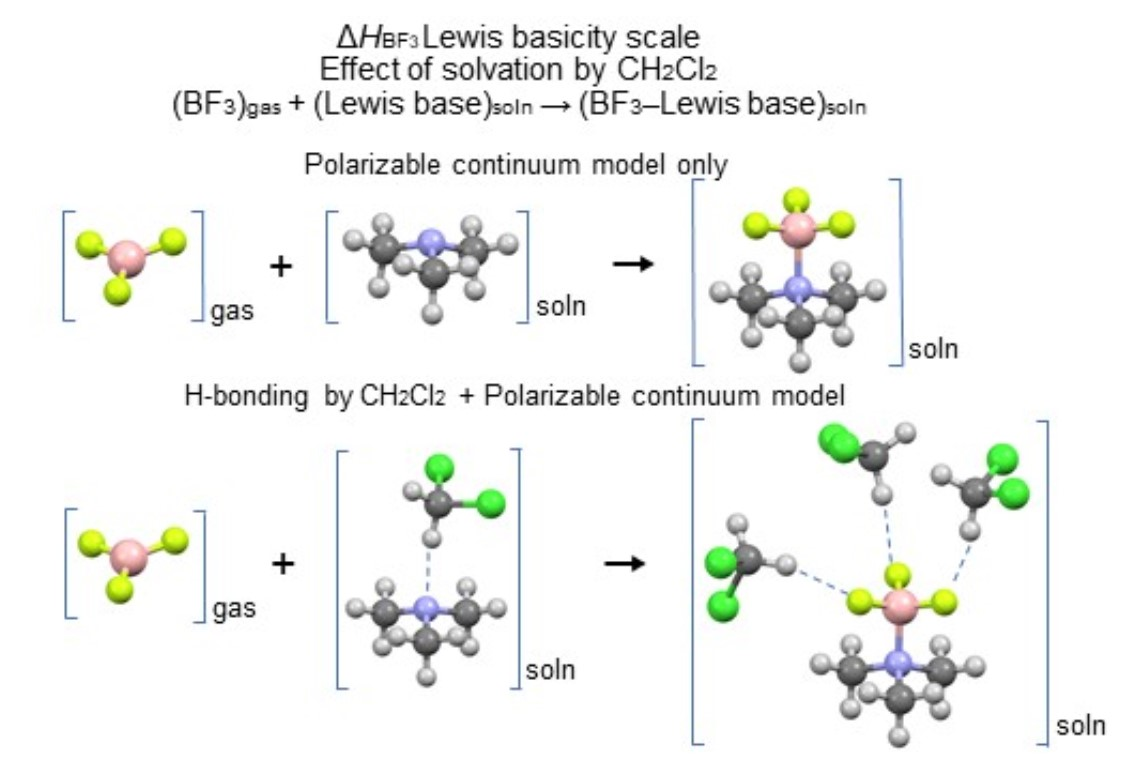
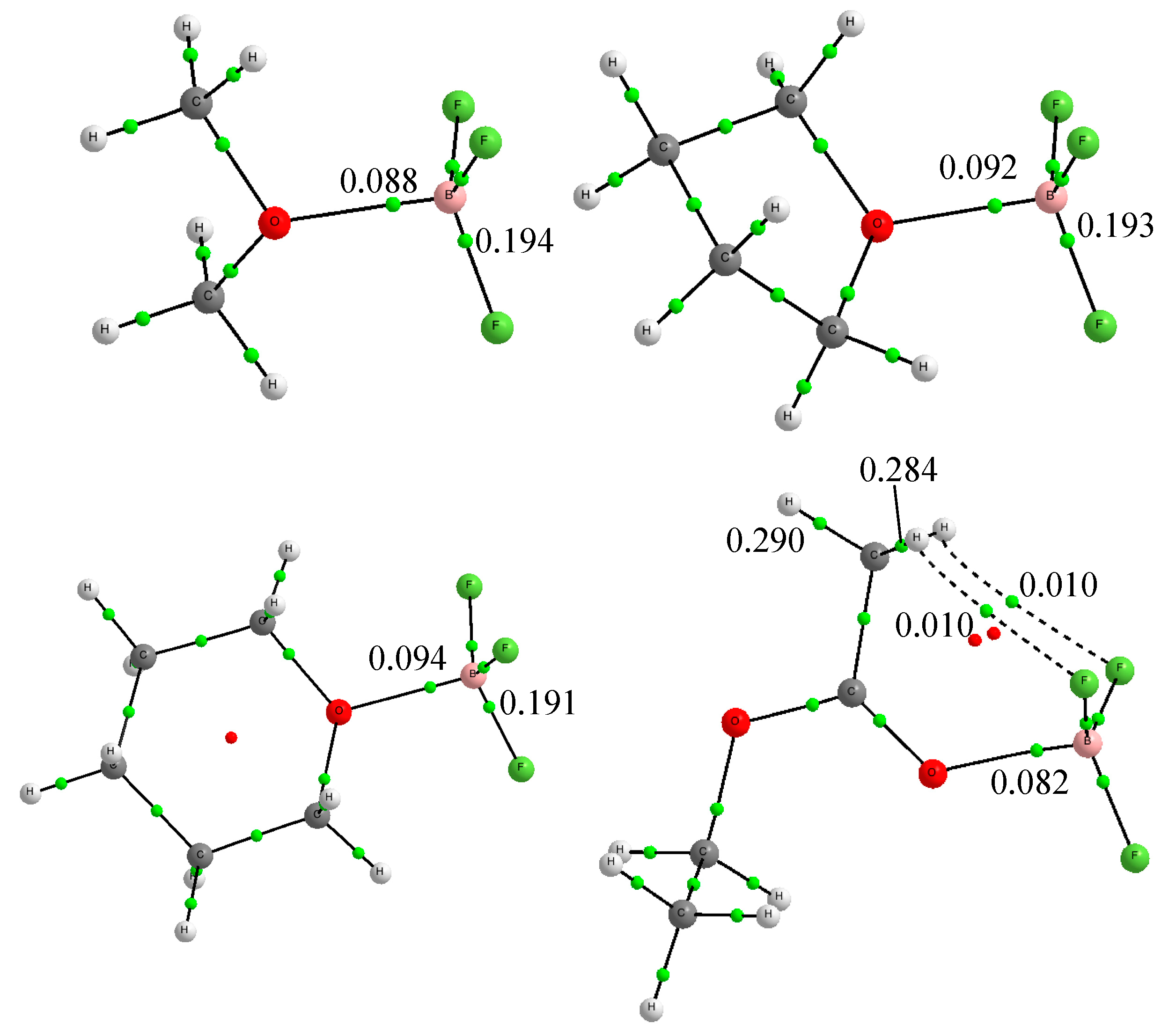
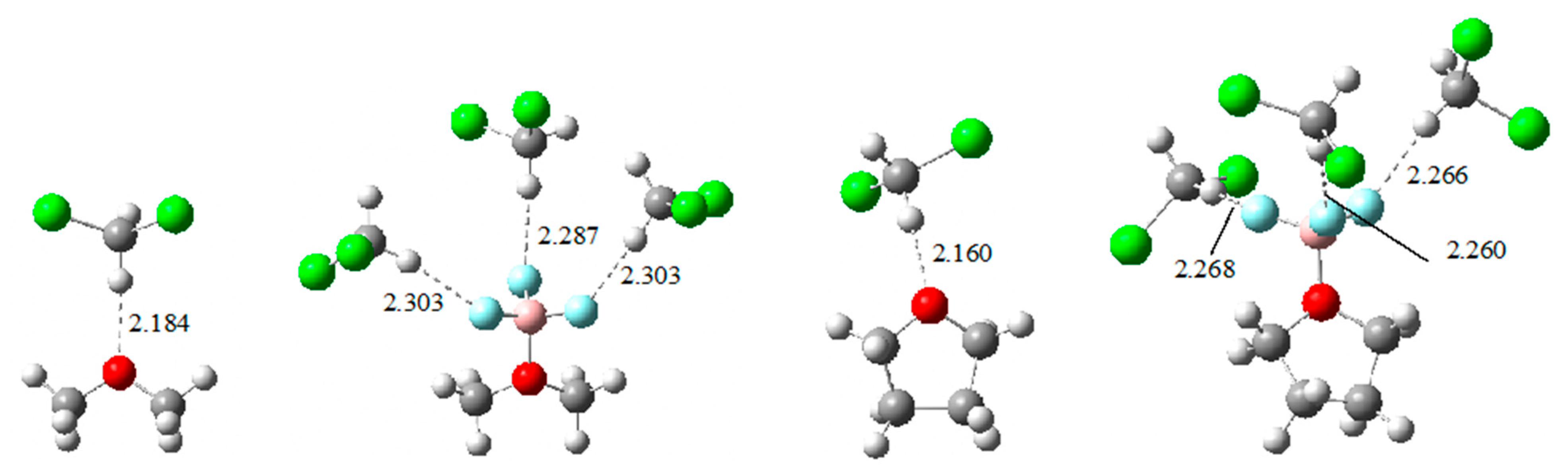
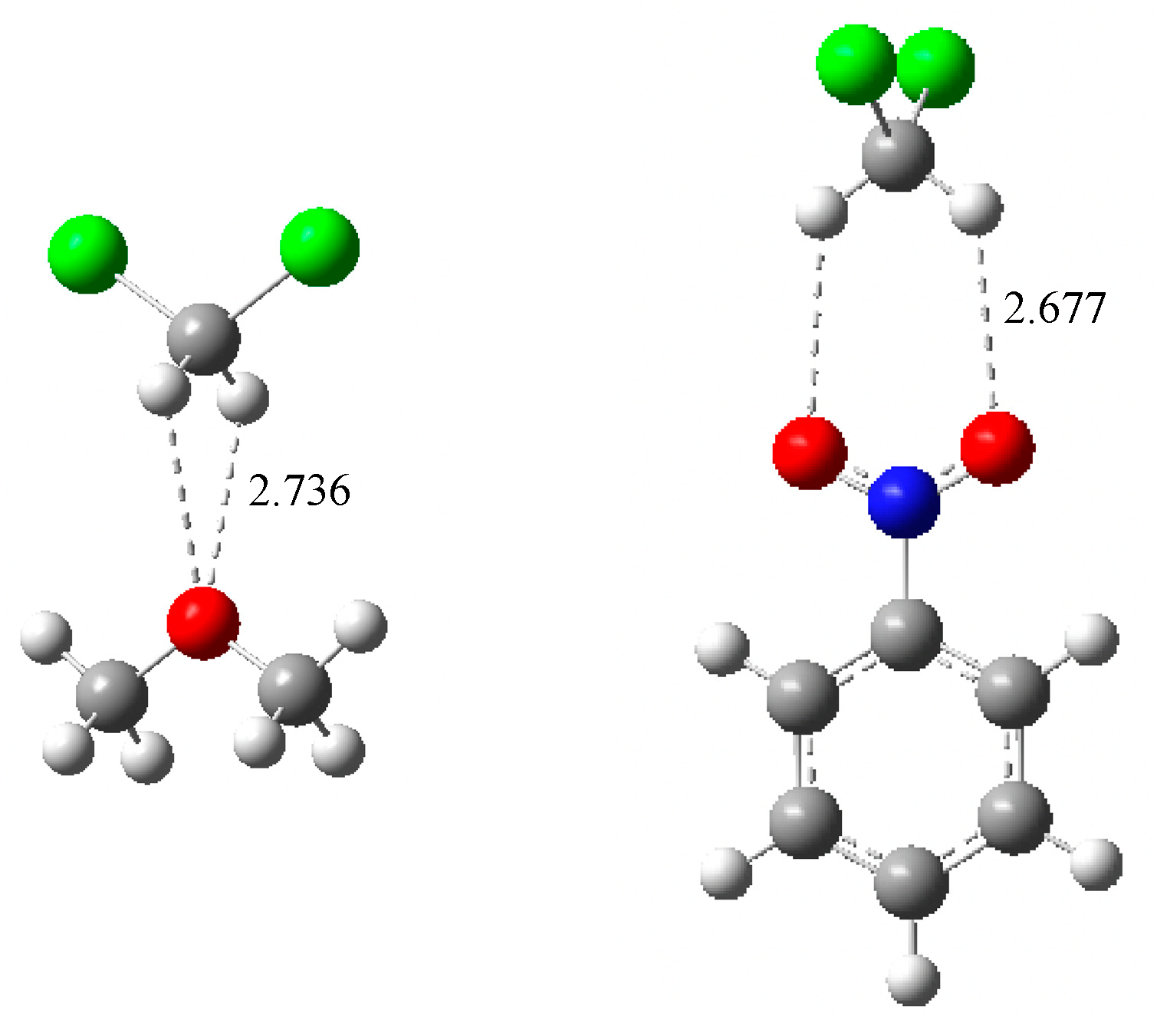
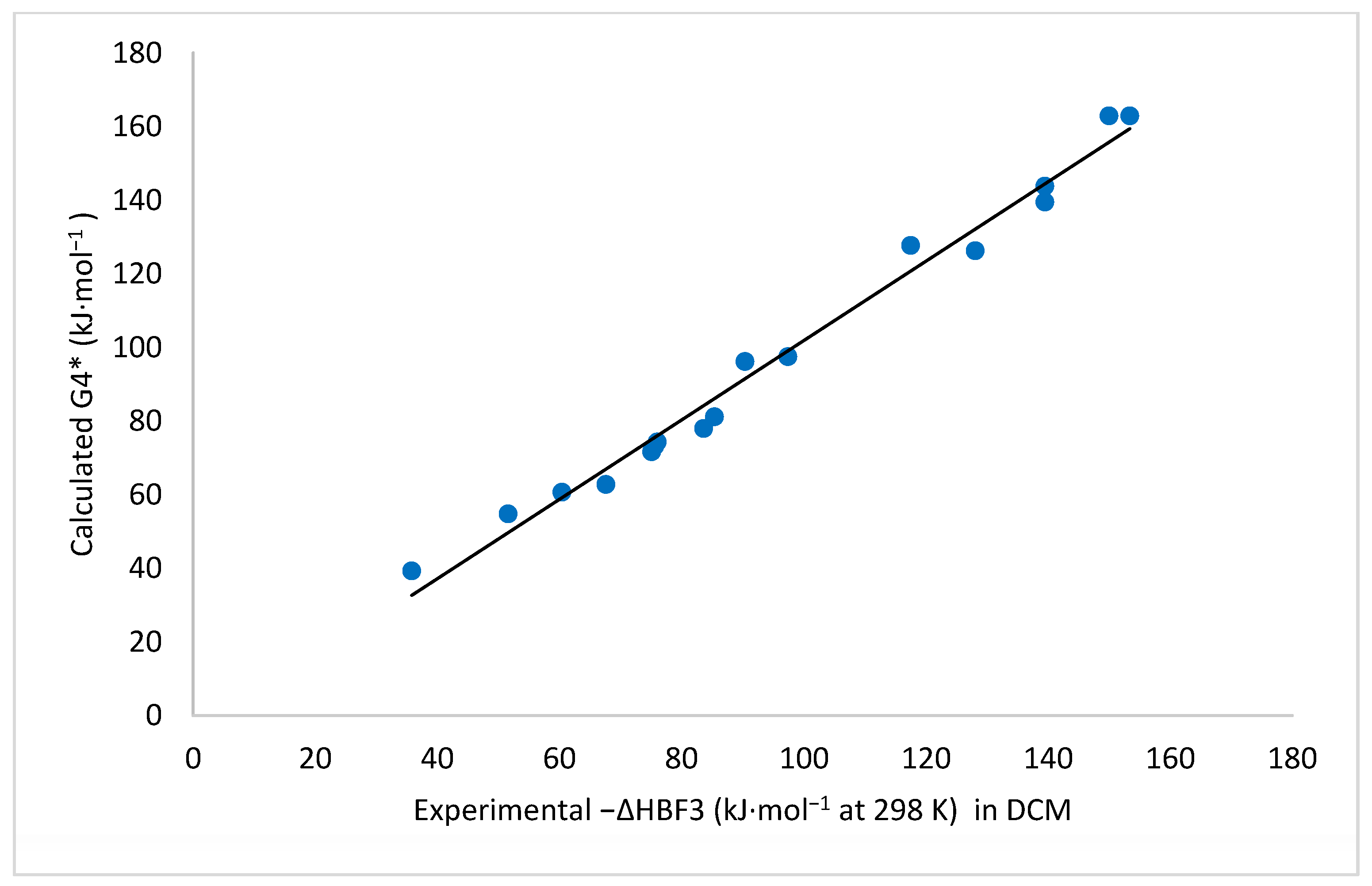
2.2. Enthalpies in Solution
3. Materials and Methods
3.1. Calorimetric Method
3.2. Computational
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gur’yanova, E.N.; Gol’dshtein, I.P.; Romm, I.P. Donor-Acceptor Bond; Wiley: New York, NY, USA, 1975. [Google Scholar]
- Gutmann, V. The Donor-Acceptor Approach to Molecular Interactions; Plenum: New York, NY, USA, 1978. [Google Scholar]
- Jensen, W.B. The Lewis Acid-Base Concepts: An Overview; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Finston, H.L.; Rychtman, A.C. New View of Current Acid–Base Theories; Wiley: New York, NY, USA, 1982. [Google Scholar]
- Laurence, C.; Gal, J.-F. Lewis Basicity and Affinity Scales: Data and Measurement; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Laurence, C.; Graton, J.; Gal, J.-F. An Overview of Lewis Basicity and Affinity Scales. J. Chem. Educ. 2011, 88, 1651–1657. [Google Scholar] [CrossRef]
- Laurence, C.; Graton, J.; Berthelot, M.; Besseau, F.; Le Questel, J.-Y.; Luçon, M.; Ouvrard, C.; Planchat, A.; Renault, E. An Enthalpic Scale of Hydrogen-Bond Basicity. 4. Carbon π Bases, Oxygen Bases, and Miscellaneous Second-Row, Third-Row, and Fourth-Row Bases and a Survey of the 4-Fluorophenol Affinity Scale. J. Org. Chem. 2010, 75, 4105–4123. [Google Scholar] [CrossRef] [PubMed]
- Kone, M.; Illien, B.; Laurence, C.; Graton, J. Can Quantum-Mechanical Calculations Yield Reasonable Estimates of Hydrogen-Bonding Acceptor Strength? The Case of Hydrogen-Bonded Complexes of Methanol. J. Phys. Chem. A 2011, 115, 13975–13985. [Google Scholar] [CrossRef]
- Laurence, C.; Graton, J.; Berthelot, M.; El Ghomari, M.J. The Diiodine Basicity Scale: Toward a General Halogen-Bond Basicity Scale. Chem. Eur. J. 2011, 17, 10431–10444. [Google Scholar] [CrossRef] [PubMed]
- Maria, P.-C.; Gal, J.-F.; de Franceschi, J.; Fargin, E. Chemometrics of the Solvent Basicity: Multivariate Analysis of the Basicity Scales Relevant to Nonprotogenic Solvents. J. Am. Chem. Soc. 1987, 109, 483–492. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases—The evolution of a chemical concept. Coord. Chem. Rev. 1990, 100, 403–425. [Google Scholar] [CrossRef]
- Drago, R.S. Applications of Electrostatic-Covalent Models in Chemistry; Surfside: Gainesville, FL, USA, 1994. [Google Scholar]
- Gutmann, V.; Mayer, U. Donorstärken in 1,2-Dichloräthan, 3. Mitt. (Donor strengths in 1,2-dichloroethane. Part 3). Monatsh. Chem. 1967, 98, 294–297. [Google Scholar] [CrossRef]
- Gutmann, V. Coordination Chemistry in Non-Aqueous Solutions; Springer: Vienna, Austria, 1968. [Google Scholar]
- Marcus, Y. The Effectivity of Solvents as Electron Pair Donors. J. Solut. Chem. 1984, 13, 599–624. [Google Scholar] [CrossRef]
- Hong, M.; Chen, J.; Chen, E.Y.-X. Polymerization of Polar Monomers Mediated by Main-Group Lewis Acid—Base Pairs. Chem. Rev. 2018, 118, 10551–10616. [Google Scholar] [CrossRef] [PubMed]
- Hamill, J.C., Jr.; Schwartz, J.; Loo, Y.-L. Influence of Solvent Coordination on Hybrid Organic—Inorganic Perovskite Formation. ACS Energy Lett. 2018, 3, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, J.; Huang, Y.; Zhu, D.; Wang, H. Synergetic Effect of Ethyl Methyl Carbonate and Trimethyl Phosphate on BF4− Intercalation into a Graphite Electrode. Langmuir 2019, 35, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Clancy, P. Balancing Multiple Goals and Making It Work for Materials Research. ACS Cent. Sci. 2020, 6, 464–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnida, P.; Libera, M.; Pająk, A.; Schab-Balcerzak, E. Examination of the Effect of Selected Factors on the Photovoltaic Response of Dye-Sensitized Solar Cells. Energy Fuels 2020, 34, 14344–14355. [Google Scholar] [CrossRef]
- Baek, M.; Shin, H.; Char, K.; Choi, J.W. New High Donor Electrolyte for Lithium–Sulfur Batteries. Adv. Mater. 2020, 32, 2005022. [Google Scholar] [CrossRef]
- Romiluyi, O.; Eatmon, Y.; Ni, R.; Rand, B.P.; Clancy, P. The efficacy of Lewis affinity scale metrics to represent solvent interactions with reagent salts in all-inorganic metal halide perovskite solutions. J. Mater. Chem. A 2021, 9, 13087–13099. [Google Scholar] [CrossRef]
- Lee, D.-K.; Lim, K.-S.; Leeb, J.-W.; Park, N.-G. Scalable perovskite coating via anti-solvent-free Lewis acid–base adduct engineering for efficient perovskite solar modules. J. Mater. Chem. A 2021, 9, 3018–3028. [Google Scholar] [CrossRef]
- Gupta, A.; Bhargav, A.; Manthiram, A. Evoking High-Donor-Number-Assisted and Organosulfur-Mediated Conversion in Lithium−Sulfur Batteries. ACS Energy Lett. 2021, 6, 224–231. [Google Scholar] [CrossRef]
- Maria, P.-C.; Gal, J.-F. A Lewis basicity scale for nonprotogenic solvents: Enthalpies of complex formation with boron trifluoride in dichloromethane. J. Phys. Chem. 1985, 89, 1296–1304. [Google Scholar] [CrossRef]
- Maria, P.-C.; Gal, J.-F.; Elégant, L.; Azzaro, M. A microcalorimetric method for the measurement of the enthalpies of solution of gases in liquids. Thermochim. Acta 1987, 115, 67–81. [Google Scholar] [CrossRef]
- Hamill, J.C., Jr.; Romiluyi, O.; Thomas, S.A.; Cetola, J.; Schwartz, J.; Toney, M.F.; Clancy, P.; Loo, Y.-L. Sulfur-Donor Solvents Strongly Coordinate Pb2+ in Hybrid Organic−Inorganic Perovskite Precursor Solutions. J. Phys. Chem. C 2020, 124, 14496–14502. [Google Scholar] [CrossRef]
- Smiatek, J. Enthalpic contributions to solvent–solute and solvent–ion interactions: Electronic perturbation as key to the understanding of molecular attraction. J. Chem. Phys. 2019, 150, 174112. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Hampel, N.; Ofial, A.R. Lewis Acidic Boranes, Lewis Bases, and Equilibrium Constants: A Reliable Scaffold for a Quantitative Lewis Acidity/Basicity Scale. Chem. Eur. J. 2021, 27, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Quintana, R.A.; Smiatek, J. Calculation of donor numbers: Computational estimates for the Lewis basicity of solvents. J. Mol. Liq. 2021, 322, 114506. [Google Scholar] [CrossRef]
- Gal, J.-F.; Maria, P.-C.; Yáñez, M.; Mó, O. On the Lewis basicity of phosphoramides: A critical examination of their Donor Number through the comparison of enthalpies of adduct formation with SbCl5 and BF3. Chem. Phys. Chem. 2019, 20, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.S.; Bankiewicz, B.; Krygowski, T.M.; Palusiak, M.; Stasyuk, O.A.; Szatylowicz, H. Interactions of polar hydrogen bond donor solvents with ions: A theoretical study. Struct. Chem. 2016, 27, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Sorenson, B.A.; Hong, S.S.; Herbol, H.C.; Clancy, P. How well do implicit solvation models represent intermolecular binding energies in organic-inorganic solutions? Comput. Mat. Sci. 2019, 170, 109138. [Google Scholar] [CrossRef]
- Herbol, H.C.; Hu, W.; Frazier, P.; Clancy, P.; Poloczek, M. Efficient search of compositional space for hybrid organic–inorganic perovskites via Bayesian optimization. NPJ Comput. Mat. 2018, 4, 51. [Google Scholar] [CrossRef]
- Gal, J.-F.; Maria, P.-C. Can the Lewis Basicity of an Isolated Solvent Molecule be Used for Characterizing Solvent Effects? Curr. Anal. Chem. 2021, 17, 328–338. [Google Scholar] [CrossRef]
- Shepp, A.; Bauer, S.H. Computation of Entropy Increments in Gaseous Bimolecular Associations. I. Donor-Acceptor Reactions. J. Am. Chem. Soc. 1954, 76, 265–270. [Google Scholar] [CrossRef]
- Bauer, S.H.; McCoy, R.E. Energetics of the Boranes. I. The Heats of Reaction of Diborane with the Methylamines, and of Tetramethyldiborane with Trimethylamine; the Dissociation Energy of Diborane. J. Am. Chem. Soc. 1956, 76, 2061–2065. [Google Scholar]
- Bauer, S.H.; McCoy, R.E. Energetics of the Boranes. III. The Enthalpy and Heat Capacity of Trimethylamine-Trifluoroborane as Determined by the Drop Method. J. Phys. Chem. 1956, 60, 1529–1532. [Google Scholar] [CrossRef]
- McLaughin, D.E.; Tamres, M. The boron trifluoride addition compounds of dimethyl ether and diethyl ether. J. Am. Chem. Soc. 1960, 82, 5618–5621. [Google Scholar] [CrossRef]
- McLaughin, D.E.; Tamres, M.; Searles, S., Jr. The addition compounds of cyclic ethers with boron trifluoride. J. Am. Chem. Soc. 1960, 82, 5621–5625. [Google Scholar] [CrossRef]
- Sacks, L.J.; Drago, S.R.; Eyman, D.P. Gas-phase enthalpies of adduct formation: Dimethylamine–chloroform and ethyl acetate–boron trifluoride. Inorg. Chem. 1968, 7, 1484–1488. [Google Scholar] [CrossRef]
- Brown, H.C. Chemical Effects of Steric Strains. J. Chem. Soc. 1956, 1248–1268. [Google Scholar] [CrossRef]
- Morris, H.L.; Tamres, M.; Searles, S. The addition compounds of some phosphines with boron trifluoride, borane, and trimethylboron. Inorg. Chem. 1966, 5, 2156–2160. [Google Scholar] [CrossRef]
- Morris, H.L.; Kulevsky, I.N.; Tamres, M.; Searles, S., Jr. The addition compounds of some sulfides with boron trifluoride and with boron trichloride. Inorg. Chem. 1966, 5, 124–130. [Google Scholar] [CrossRef]
- Davydova, E.I.; Sevastianova, T.N.; Suvorov, A.V.; Timoshkin, A.Y. Molecular complexes formed by halides of group 4,5,13–15 elements and the thermodynamic characteristics of their vaporization and dissociation found by the static tensimetric method. Coord. Chem. Rev. 2010, 254, 2031–2077. [Google Scholar] [CrossRef]
- Chickos, J.S.; Acree, W.E. Enthalpies of sublimation of organic and organometallic compounds. 1910–2001. J. Phys. Chem. Ref. Data 2002, 31, 537–698. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook. NIST Standard Reference Database No. 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2014. Available online: http://webbook.nist.gov/chemistry (accessed on 14 September 2021).
- Fletcher, E.A. A Study of the Steric Consequences of Planar Boron—The Base Strengths of the Methylphosphines. Ph.D. Thesis, Purdue University, West Lafayette, IN, USA, 1952. [Google Scholar]
- Rauk, A.; Hunt, I.R.; Keay, B.A. Lewis acidity and basicity: An ab initio study of proton and BF3 affinities of oxygen-containing organic compound. J. Org. Chem. 1994, 59, 6808–6916. [Google Scholar] [CrossRef]
- Allerhand, A.; von Schleyer, P.R. A Survey of C-H Groups as Proton Donors in Hydrogen Bonding. J. Am. Chem. Soc. 1963, 85, 1715–1723. [Google Scholar] [CrossRef]
- Drago, R.S.; Nusz, J.A.; Courtright, R.C. Solvation Contributions to Enthalpies Measured in Methylene Chloride. J. Am. Chem. Soc. 1974, 96, 2082–2086. [Google Scholar] [CrossRef]
- Drago, R.S.; Dadmun, A.P.; Vogel, G.C. Addition of New Donors to the E and C Model. Inorg. Chem. 1993, 32, 2473–2479. [Google Scholar] [CrossRef]
- Cerón-Carrasco, J.P.; Jacquemin, D.; Laurence, C.; Planchat, A.; Reichardt, C.; Sraïdi, K. Determination of a solvent hydrogen-bond acidity scale by means of the solvatochromism of pyridinium-N-phenolate betaine dye 30 and PCM-TD-DFT calculations. J. Phys. Chem. B 2014, 118, 4605–4614. [Google Scholar] [CrossRef]
- Rakipov, I.T.; Petrov, A.A.; Akhmadiyarov, A.A.; Khachatrian, A.A.; Mukhametzyanov, T.A.; Solomonov, B.N. Thermochemistry of Solution, Solvation, and Hydrogen Bonding of Cyclic Amides in Proton Acceptor and Donor Solvents. Amide Cycle Size Effect. Molecules 2021, 26, 1411. [Google Scholar] [CrossRef] [PubMed]
- Mooibroek, T.J.; Gamez, P. Halogen bonding versus hydrogen bonding: What does the Cambridge Database reveal? Cryst. Eng. Comm. 2013, 15, 4565–4570. [Google Scholar] [CrossRef]
- Mills, J.E.; Maryanoff, C.A.; Cosgrove, R.M.; Scott, L.; McComsey, D.F. The reactions of amines with methylene chloride. A brief review. Org. Prep. Proc. Int. 1984, 16, 97–114. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 84108. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Herbert, J.M. Dielectric continuum methods for quantum chemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1519. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Pomogaeva, A.V.; Timoshkin, A.Y. Influence of the solvent on the Lewis acidity of antimony pentahalides and group Lewis acids toward acetonitrile and pyridine. J. Comput. Chem. 2021, 42, 1792–1802. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lewis Base | G4*-Calculated a | Experimental | Δ(Calc. − Exp.) |
|---|---|---|---|
| Trimethylamine | 126.4 | 111.3 ± 8.4 b [36,37,38] | 15.1 |
| Dimethyl ether | 62.4 | 57.1 ± 0.8 [39] | 5.3 |
| Tetrahydrofuran | 72.5 | 70.3 ± 0.8 [40] | 2.2 |
| Tetrahydropyran | 69.0 | 64.5 ± 0.8 [40] | 4.5 |
| Ethyl acetate | 55.9 | 53.6 ± 2.9 [41] | 2.3 |
| Trimethylphosphine | 66.1 | 79.1 [42,43] | −13.0 |
| Tetrahydrothiophene | 37.9 | 21.8 ± 1.7 [44] | 16.1 |
| Solvent CH2Cl2 (DCM) | Solvent PhNO2 (NB) | Gas Phase | |||||
|---|---|---|---|---|---|---|---|
| Lewis Base | Experimental in DCM a | G4* + Discrete Solvation Model b | Δ = Calc − Exp | Experimental in NB a | G4* + Continuous Solvation Model c | Δ = Calc − Exp | G4* |
| Trimethylamine | 139.5 ± 1.8 | 145.7 | 6.2 | 126.4e | |||
| N-Methylpyrrolidine | 139.5 ± 0.8 | 143.8 | 4.3 | 125.2 | |||
| Quinuclidine | 150.01 ± 3.48 [153.4 ± 0.9] d | 162.9 | 12.9 [9.5] | 160.5 ± 0.9 | 171.2 | 10.7 | 139.1 |
| Pyridine | 128.1 ± 0.5 | 126.3 | −1.8 | 137.9 ± 0.7 | 137.4 | −0.5 | 100.4e |
| Acetonitrile | 60.4 ±0.5 | 60.7 | −0.3 | 32.3 | |||
| Dimethyl ether | 83.6 ± 0.2 | 78.0 | −5.6 | 62.4 | |||
| Tetrahydrofuran | 90.4 ± 0.3 | 96.2 | 5.8 | 93.0 ± 0.3 | 103.9 | 10.9 | 74.8 |
| Tetrahydropyran | 85.4 ± 0.5 | 81.2 | −4.2 | 69.0e | |||
| Acetone | 76.0 ± 0.2 | 74.3 | −1.7 | 78.1 ± 0.3 | 82.7 | 4.6 | 54.2 |
| Ethyl acetate | 75.6 ± 0.3 | 73.2 | −2.4 | 55.9 | |||
| γ-Butyrolactone | 75.1 ± 1.2 | 71.7 | −3.4 | 53.1 | |||
| Dimethyl carbonate | 67.6 ± 0.4 | 62.8 | −4.8 | 30.8 | |||
| Nitrobenzene | [35.8 ± 1.4] d,f | 39.3 | [3.5] | 37.7 ± 1.4 g | 45.3 | 7.6 | 21.0e |
| Hexamethyl-phosphoramide (HMPA) | 117.5 ± 0.5 | 127.7 | 10.2 | 123.1 ± 0.5 | 135.2 (121.9) i | 12.1 (−1.2) | 101.3 h |
| Trimethylphosphine | 97.4 ± 0.3 | 97.5 | 0.1 | 66.1 | |||
| Tetrahydrothiophene | 51.6 ± 0.2 | 54.8 | 3.2 | 37.9 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gal, J.-F.; Maria, P.-C.; Yáñez, M.; Mó, O. Enthalpies of Adduct Formation between Boron Trifluoride and Selected Organic Bases in Solution: Toward an Accurate Theoretical Entry to Lewis Basicity. Molecules 2021, 26, 6659. https://doi.org/10.3390/molecules26216659
Gal J-F, Maria P-C, Yáñez M, Mó O. Enthalpies of Adduct Formation between Boron Trifluoride and Selected Organic Bases in Solution: Toward an Accurate Theoretical Entry to Lewis Basicity. Molecules. 2021; 26(21):6659. https://doi.org/10.3390/molecules26216659
Chicago/Turabian StyleGal, Jean-François, Pierre-Charles Maria, Manuel Yáñez, and Otilia Mó. 2021. "Enthalpies of Adduct Formation between Boron Trifluoride and Selected Organic Bases in Solution: Toward an Accurate Theoretical Entry to Lewis Basicity" Molecules 26, no. 21: 6659. https://doi.org/10.3390/molecules26216659