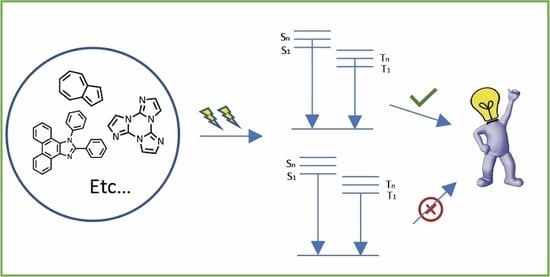
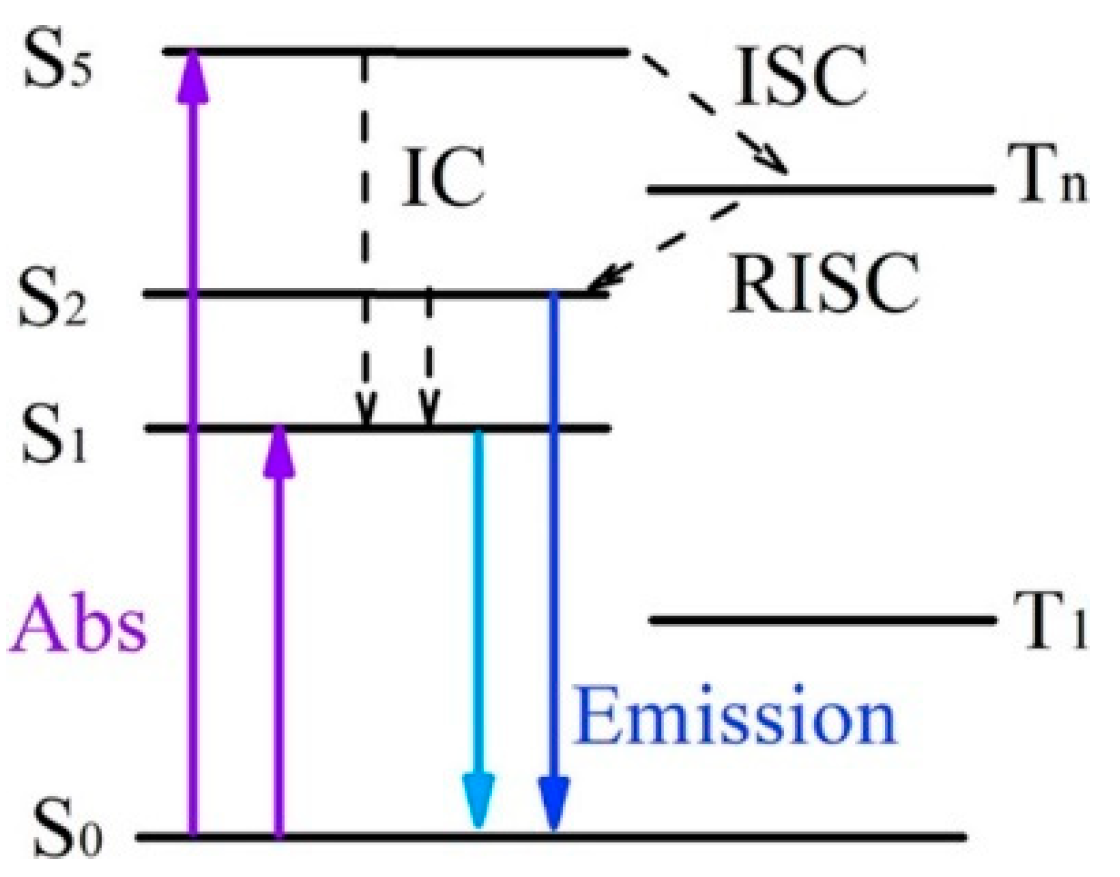
1. Introduction
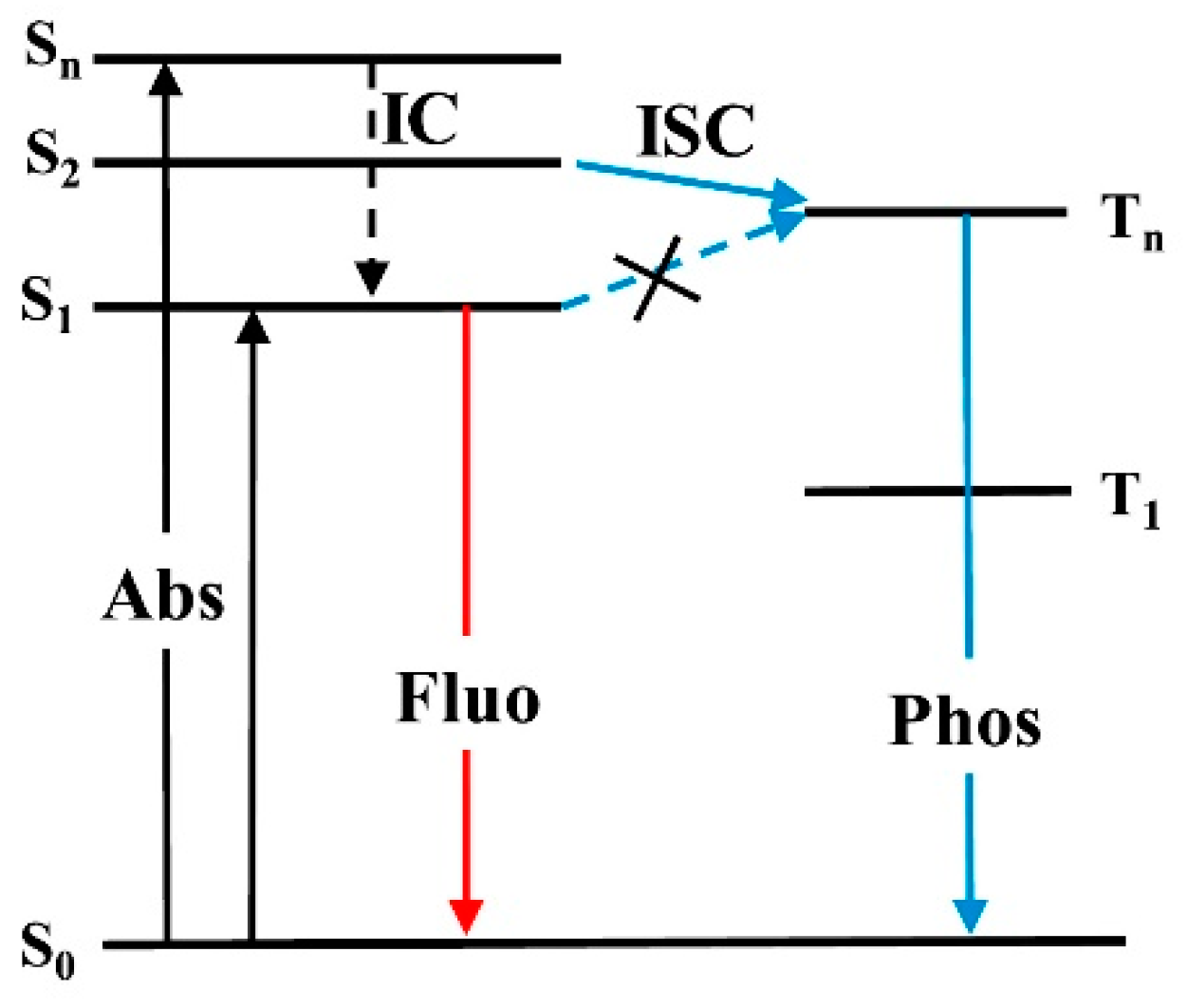
In its original formulation, the Kasha rule states that for complex organic molecules, “regardless of which electronic state of a given multiplicity is excited […], the emitting electronic level of a given multiplicity is the lowest excited level of that multiplicity” [
1]. This rule originates from the widespread feature that the E
1-E
0 energy gap is much larger than the E
n-E
n−1 (
n > 1) ones and the energy gap law. According to this law, the smaller the energy gap between two electronic states is, the more probable is the nonradiative deactivation (by IC and ISC) between them. In the condensed phase, exceptions to Kasha’s rule are possible under various circumstances. In particular, two prototype mechanisms are responsible for fluorescence from S
2: (i) when the energy gap between S
1 and S
2 is large and the oscillator strength (
f) of the S
0-S
2 transition is large, fluorescence from S
2 becomes competitive, a mechanism observed for example for azulene and its derivatives [
2,
3,
4,
5] and thioketones [
6,
7,
8], the difference between the two families being the (π, π*) character of S
1 for the former and (
n, π*) for the latter; (ii) when the S
2-S
1 energy gap and
f of the S
0-S
1 transition are very small but
f of the S
0-S
2 transition is large, fluorescence from S
2 is observed due to thermal population from S
1, a mechanism observed for example for ovalene [
9]. Moreover, fluorescence from higher S
n (
n > 2) can be observed when energy separation between the levels is favorable (necessary condition) and the S
n-S
1 internal conversion is prohibited based on symmetry grounds (sufficient condition) [
10]. Anti-Kasha phosphorescence from high-energy triplet levels is of course less observed. It may be favored by either an easy ISC (by both El-Sayed and energy gap considerations) to T
2 level of proper symmetry, different from the T
1 one so as to give low T
2-T
1 IC efficiency, or T
2 thermal population from T
1.
Currently, it is proven that the Kasha rule applies to most of the emissive organic molecules, and despite the increased instrumental sensitivity with respect to that available when Kasha made his observation, it is difficult to recognize exceptions. However, “anti-Kasha” emissions represent interesting cases for both fundamental knowledge and application aspects. In fact, anti-Kasha emitters do not dissipate through IC the excess of electronic energy, which can therefore be exploited in practical applications, such as improved luminescent quantum efficiency, tunable emission colors and lifetimes. The interest in this subject is demonstrated by the increasing number of works appearing in the literature in the last few years. In this regard, in 2012, Itoh reviewed works produced until 2011 on traditional anti-Kasha emitters such as azulene and its derivatives, aromatic acenes, polyenes, thioketones, metalloporphyrins, aromatic carbonyl compounds, quinones and halogenated aromatic compounds [
9]. The reported examples comprise emission from higher singlet and triplet levels.
Anomalous emission includes a large number of photophysical phenomena, among which anti-Kasha behavior represents a very limited subset that manifests either through single or multiple emissions [
11]. The first case may be particularly difficult to discern because low-lying dark excited levels are silent to many experimental investigations. Importantly, when more than one band is displayed by a single molecule due to structural rearrangement and/or chemical transformation, such as “excited state conjugation enhancement” (ESCE), “twisted intramolecular charge transfer” (TICT), “excited state intramolecular proton transfer” (ESIPT) and “optically triggered counterion migration” (OTCM) [
11], dual or multiple emissive behavior is to be used instead of anti-Kasha behavior. Actually, a few examples of ESCE, TICT, ESIPT and OTCM associated with anti-Kasha behavior exist [
12] since the reorganization occurs starting from a higher excited state in the molecule. However, such examples will not be covered in the present review. Even those cases where various emissions are due to the molecule and its aggregates in the same phase (solution, amorphous or crystalline solids) have to be described as Kasha emitters. Of course, trivial mistakes such as the presence of traces of impurities which can be responsible for the appearance of multiple emission bands should be carefully checked before claiming an anti-Kasha behavior [
13,
14].
Here we collect works reporting “rigorous” anti-Kasha emitters that have appeared in the literature since 2011, starting from the seminal analysis from Itoh [
9]. By “rigorous”, we mean systems that radiatively deactivate from S
n or T
n (
n > 1). In fact, in the literature, some works report as anti-Kasha a behavior that instead could be better described as anti-Vavilov [
14,
15]. According to the Vavilov rule, the intensity of the dye emissions is independent of excitation wavelength.
Another important caveat should be presented. Since anti-Kasha behavior is hardly recognizable through an exclusively experimental approach, in particular when states below the emissive S
n or T
n (
n > 1) ones are silent, quantum-chemical computational studies acquire a key role in establishing the presence and the origin of anomalous emission. Owing to the high computational costs associated with accurate
ab initio techniques (e.g., CASPT2 and MRCI), most theoretical investigations aimed at predicting electronic spectra of complex molecules are performed within the TDDFT approach. The performance of this method was always deemed highly reliable for predicting non-charge-transfer excitations, while it is well known that exchange-correlation functionals generally used for ground-state calculations, such as those based on the generalized gradient approximation (GGA) or the hybrid GGA functionals (typically the popular B3LYP one) do not allow for sufficient accuracy to study CT excited states [
16,
17,
18,
19,
20,
21]. Very few exceptions (for example, the PBE0 functional) have been recognized [
22]. Recently, the development of the so-called range-separated hybrid functionals (including CAM-B3LYP, ωB97X and ωB97XD), where the exchange term is split into long- and short-range [
23,
24,
25,
26,
27] and the range-separation parameter can be suitably determined by a self-consistent procedure [
28], has allowed for easy resolution of the CT problem for many systems of practical interest [
29,
30]. Additional functionals have been developed since, such as the hybrid meta-GGA ones (including Mo6-2X) whose performance in electronic spectra calculations has been assessed [
31]. It is therefore to be noted that the cases of anti-Kasha behavior involving CT excited states collected in the present review, especially those where TDDFT calculations have been performed using inadequate functionals, could be confuted by more reliable approaches.
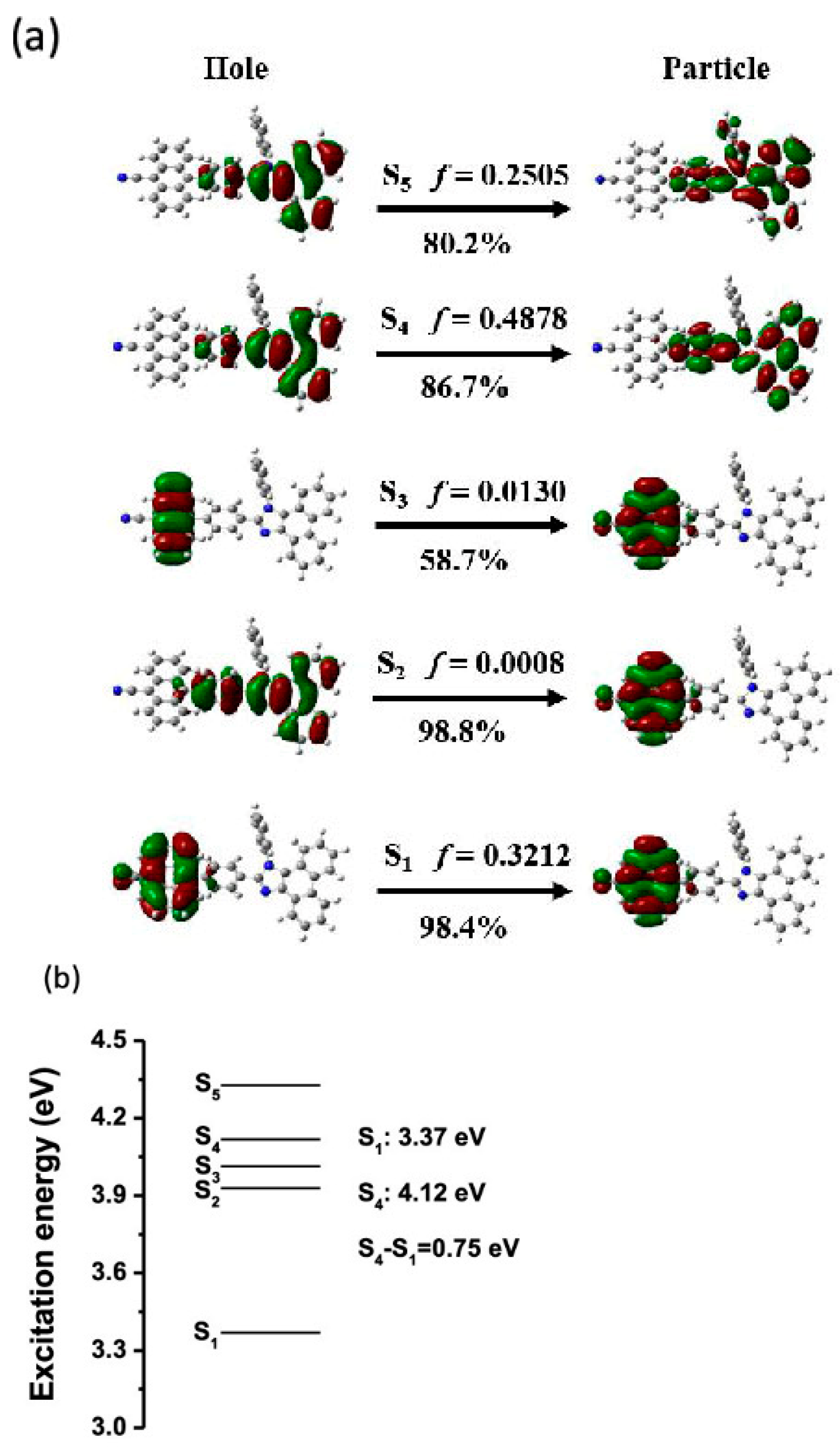
This manuscript is organized by different families of molecules, including azulenes, cyclic triimidazoles, 1,2-diphenylphenanthroimidazole derivatives, cyanines, thiophenes and carbazoles.
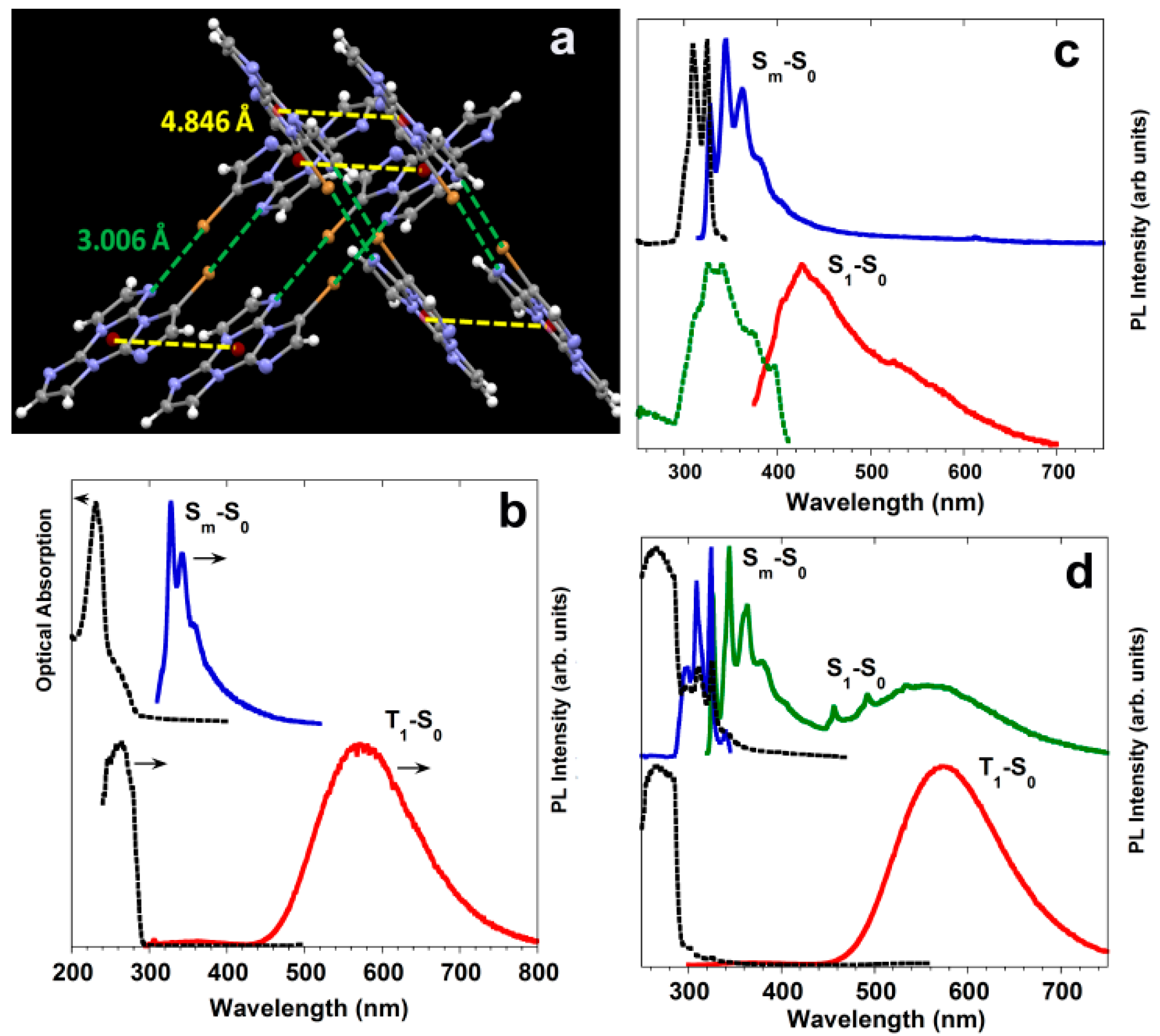
2. Azulenes
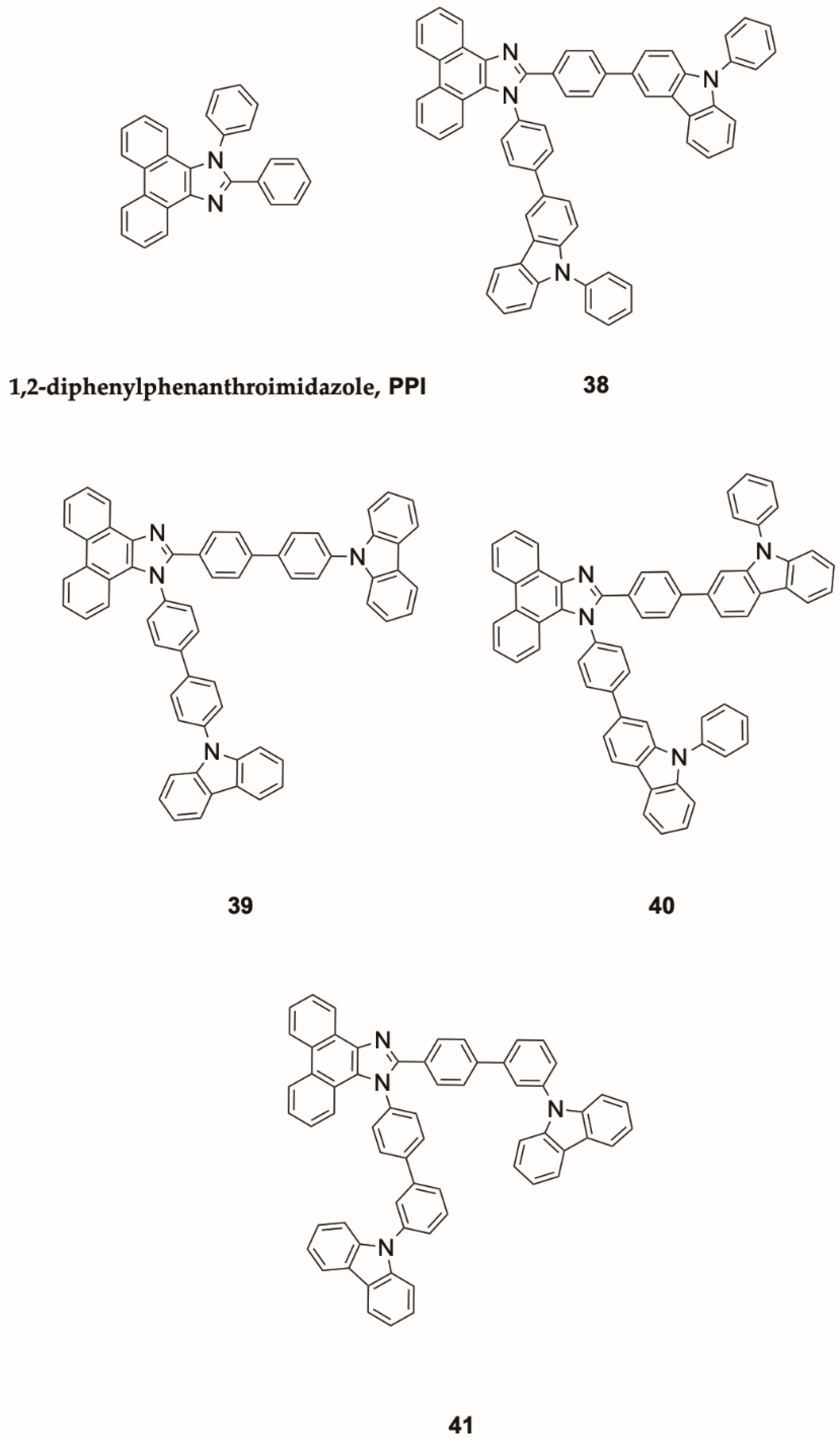
Azulene,
1 (
Scheme 1), can be regarded as the forefather of organic molecules displaying anti-Kasha behavior, namely a fluorescent emission from the S
2 excited state [
9,
32].
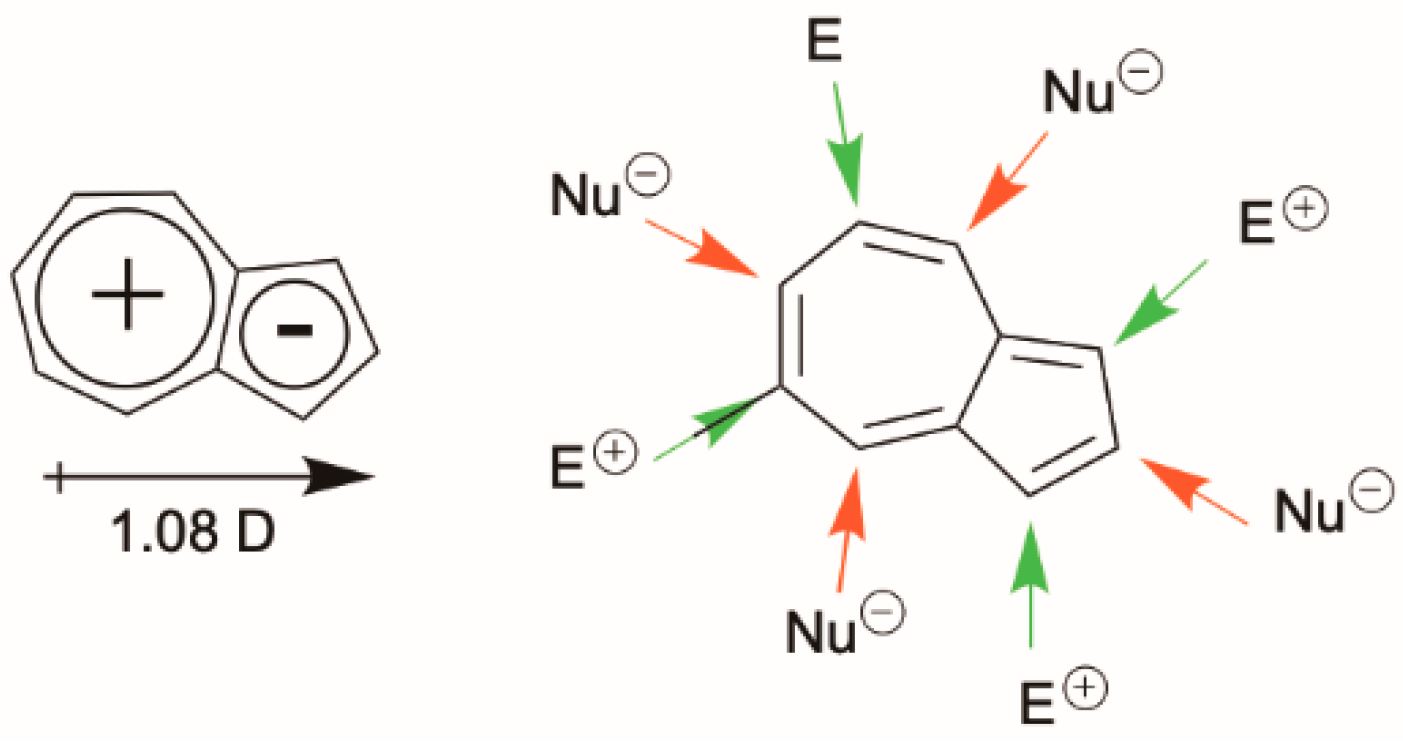
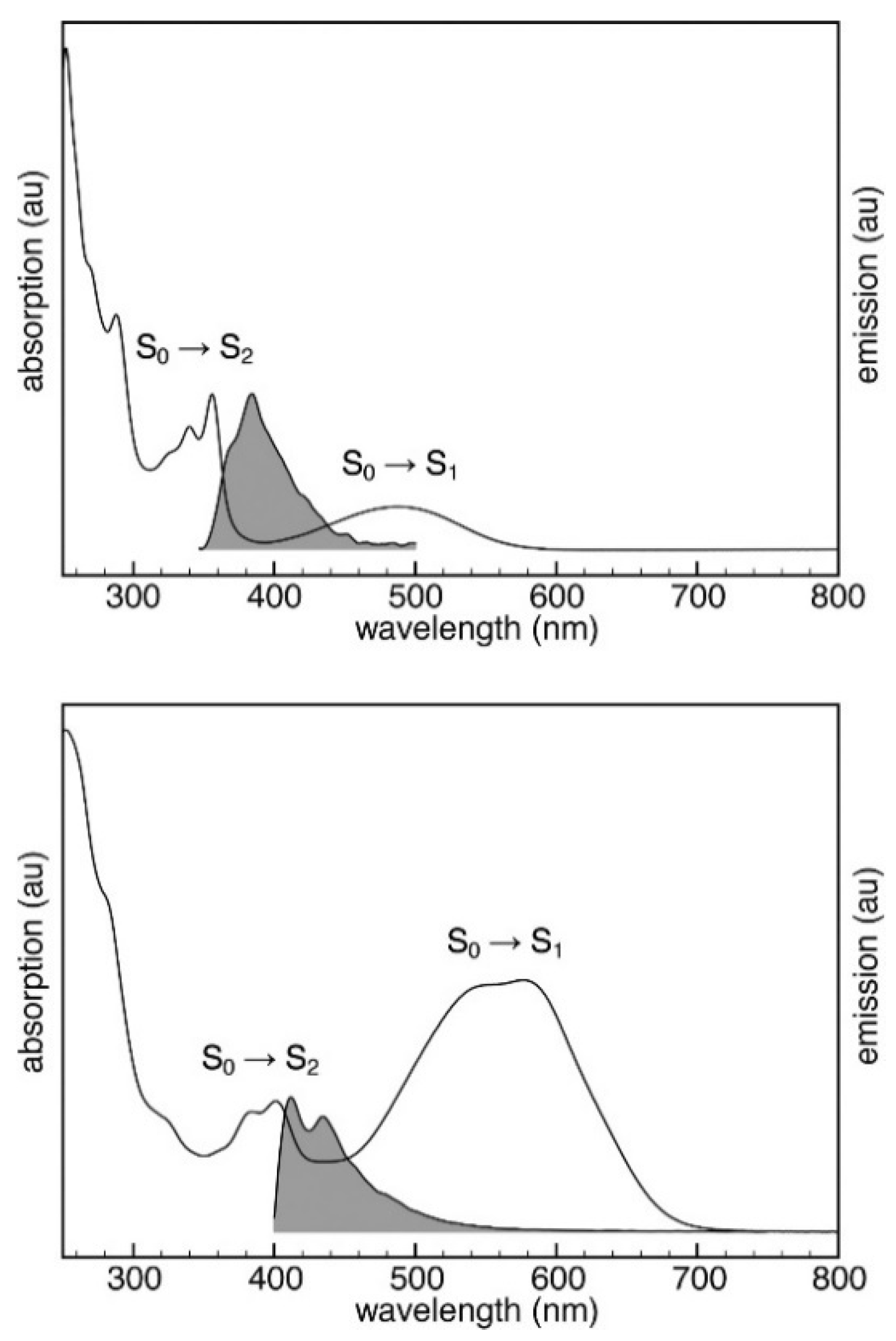
This feature derives from the unique molecular structure of azulene, different from other traditional aromatic compounds and characterized by an electron-rich five-membered ring fused with an electron-poor seven-membered ring with HOMO density localized mainly on the odd positions and LUMO density located on the even positions [
33] (
Figure 1). This leads to an unusually low-lying first electronic excited state (S
1), favoring a large S
2-S
1 energy gap (>1.2 eV) and, according to the energy-gap law, resulting in fluorescence from S
2 rather than IC to S
1. Similar emissive behavior is found for variously substituted azulenes, polyazulenes and pseudoazulenes (having a −CH=CH− group in the heptagonal ring of azulene replaced by a heteroatom). It has been shown that HOMO and LUMO energy levels can be modified by proper choice of electron-donor or electron-withdrawing functional groups. In particular, the insertion of electron-donor substituents at the 1, 3, 5 or 7 ring positions usually increases the S
2-S
1 gap while electron-withdrawing substituents have the opposite effect. A reverse behavior is found for substitutions at the 2, 4, 6 and 8 positions, resulting in an S
2-S
1 energy gap increased by electron-withdrawing groups and lowered by electron-donor substituents. In some cases, for derivatives characterized by a relatively small S
2-S
1 energy gap, IC to S
1 becomes competitive with fluorescence from S
2, and fluorescence from both excited states is observed [
9,
32]. Azulene and a series of its derivatives have been the subject of accurate theoretical benchmark studies to validate state-of-the-art computational protocols predicting anti-Kasha photoluminescence [
34,
35].
Due to these intriguing photophysical properties, there is a growing interest in the development of azulene-based materials for optoelectronic applications, even though the preparation of new derivatives is hampered by multistep and low-yield synthetic procedures [
32,
33,
36].
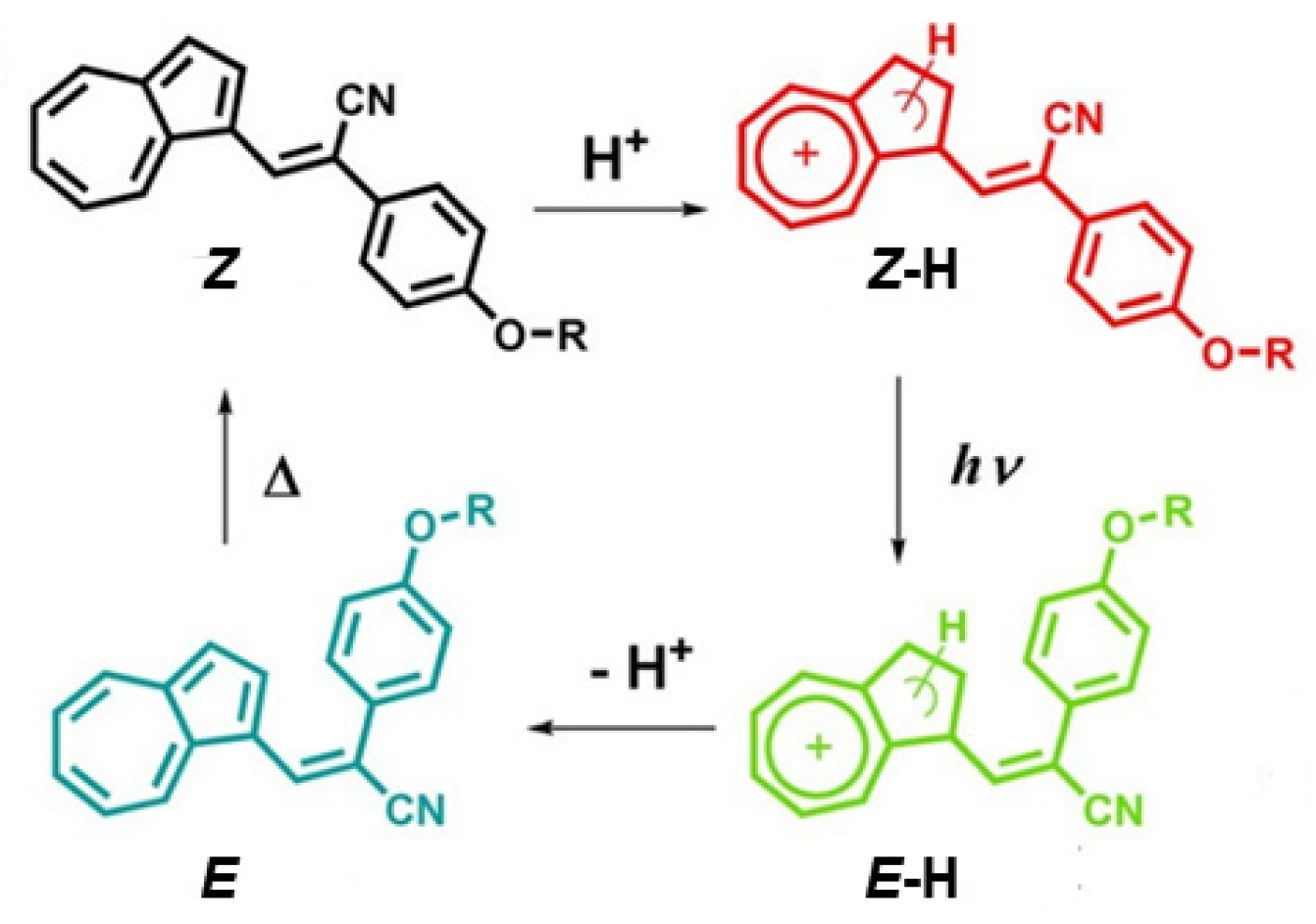
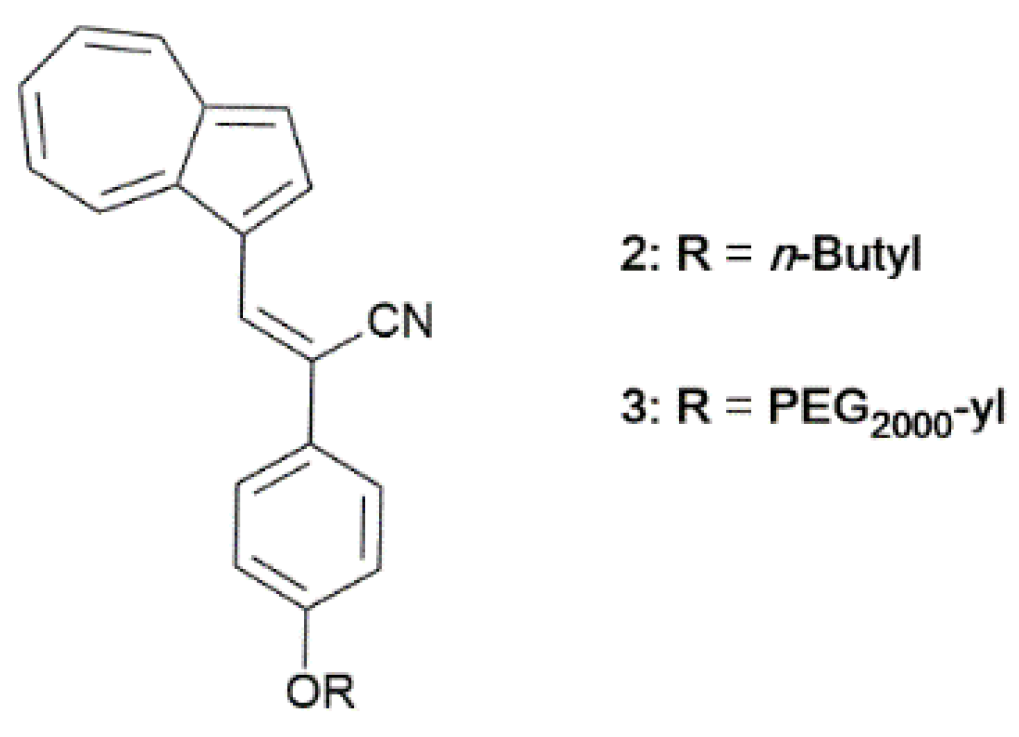
In this regard, Zhou et al. [
37] reported on two azulene derivatives,
2 and
3 (
Scheme 2), substituted at the 3 position with cyanostyryl moieties that, under UV or visible irradiation in acidic conditions, photoisomerize from Z- to E-form (
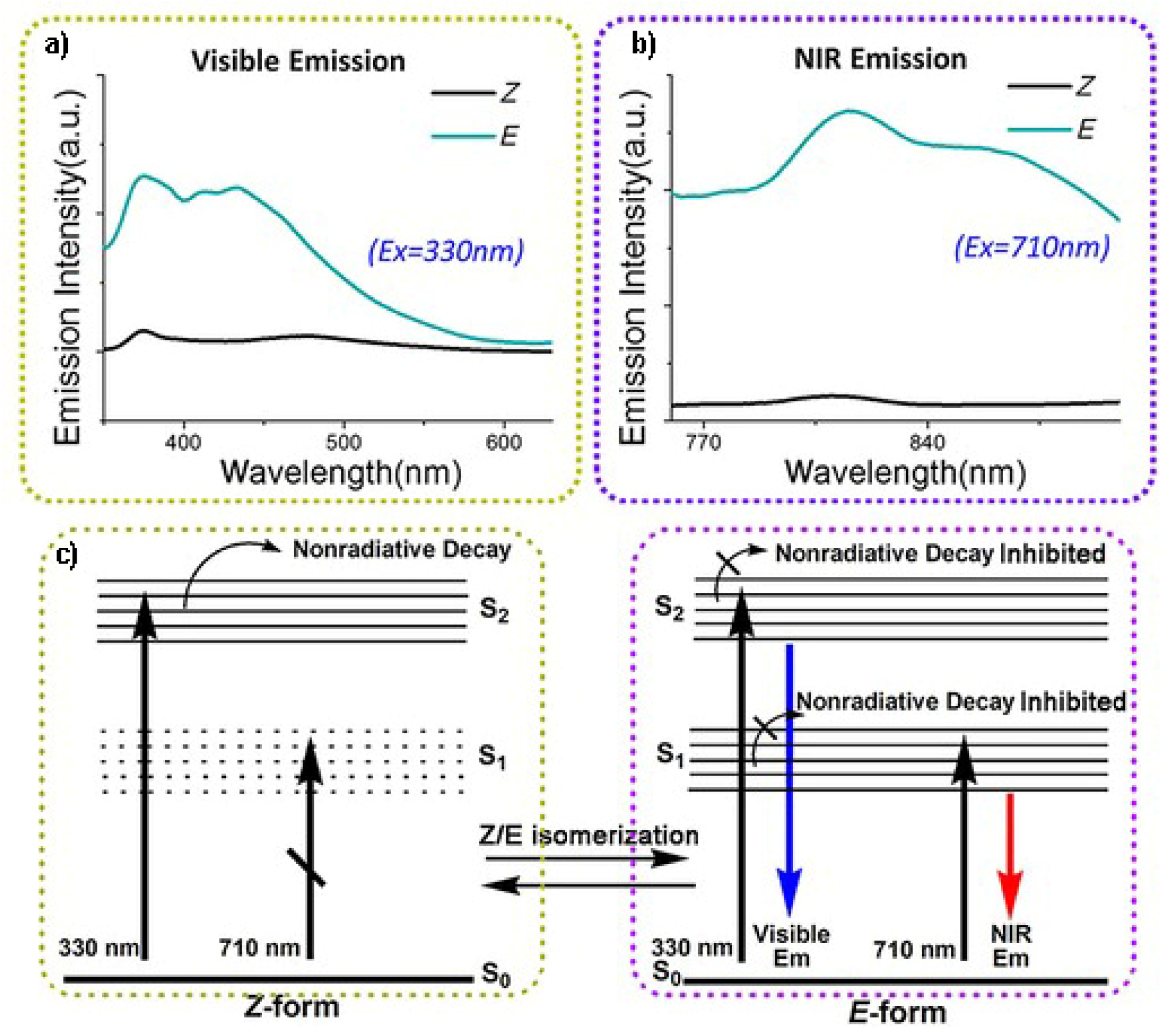
Figure 2). The process is accompanied by a strong modification of the photoluminescent features. In particular, nonradiative decays are inhibited by sterical restrictions going from the not emissive Z-isomer to the E-isomer. Moreover, the E-form displays dual luminescence with a major emission band at around 480 nm (assigned to an S
2-S
0 transition) and an NIR emission at around 800 nm (attributed to a radiative decay from S
1,
Figure 3). The conversion between the two isomers is reversible, and the E-isomer can be converted into the Z-isomer by simply heating the sample at a temperature above 65 °C.
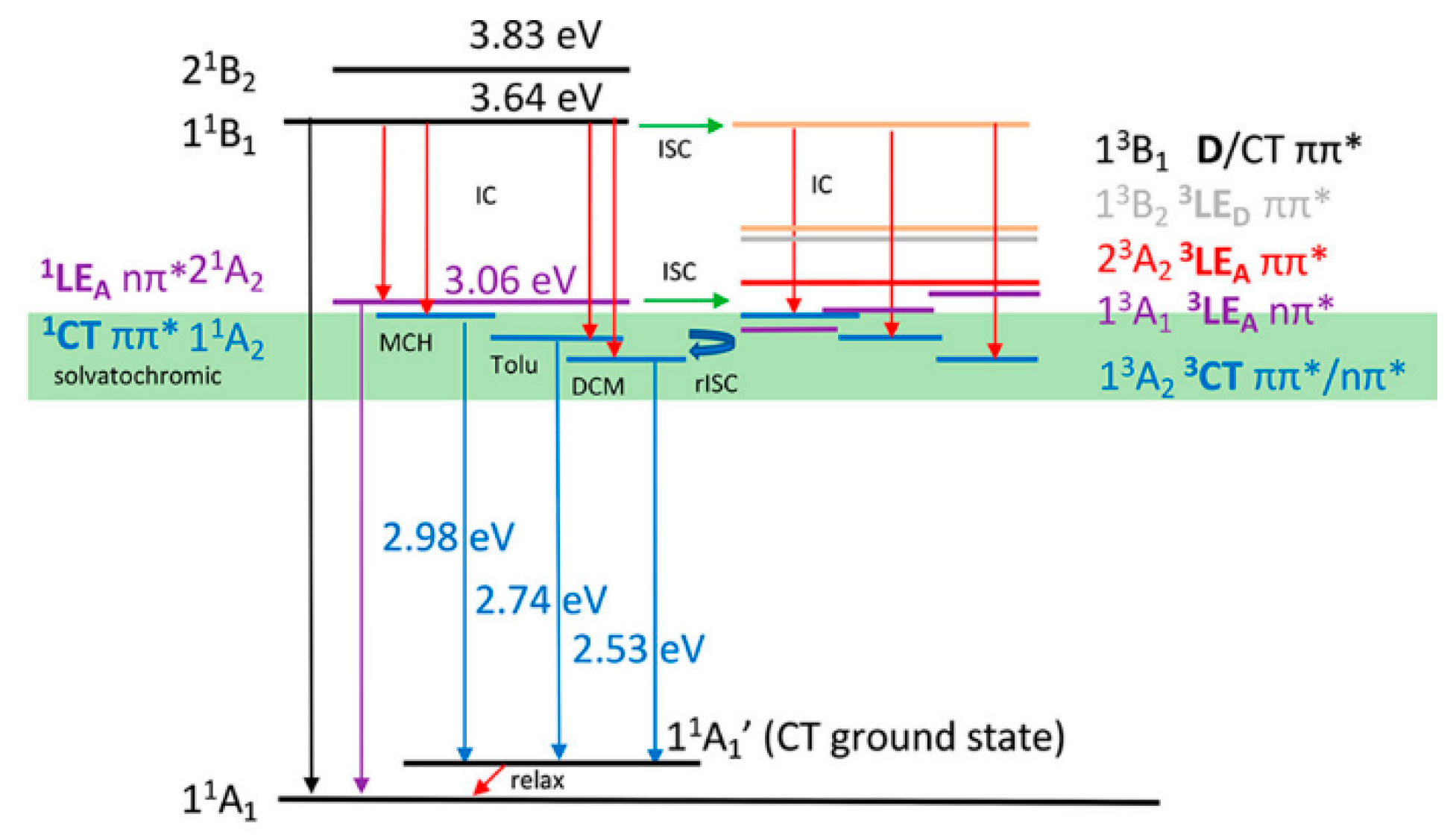
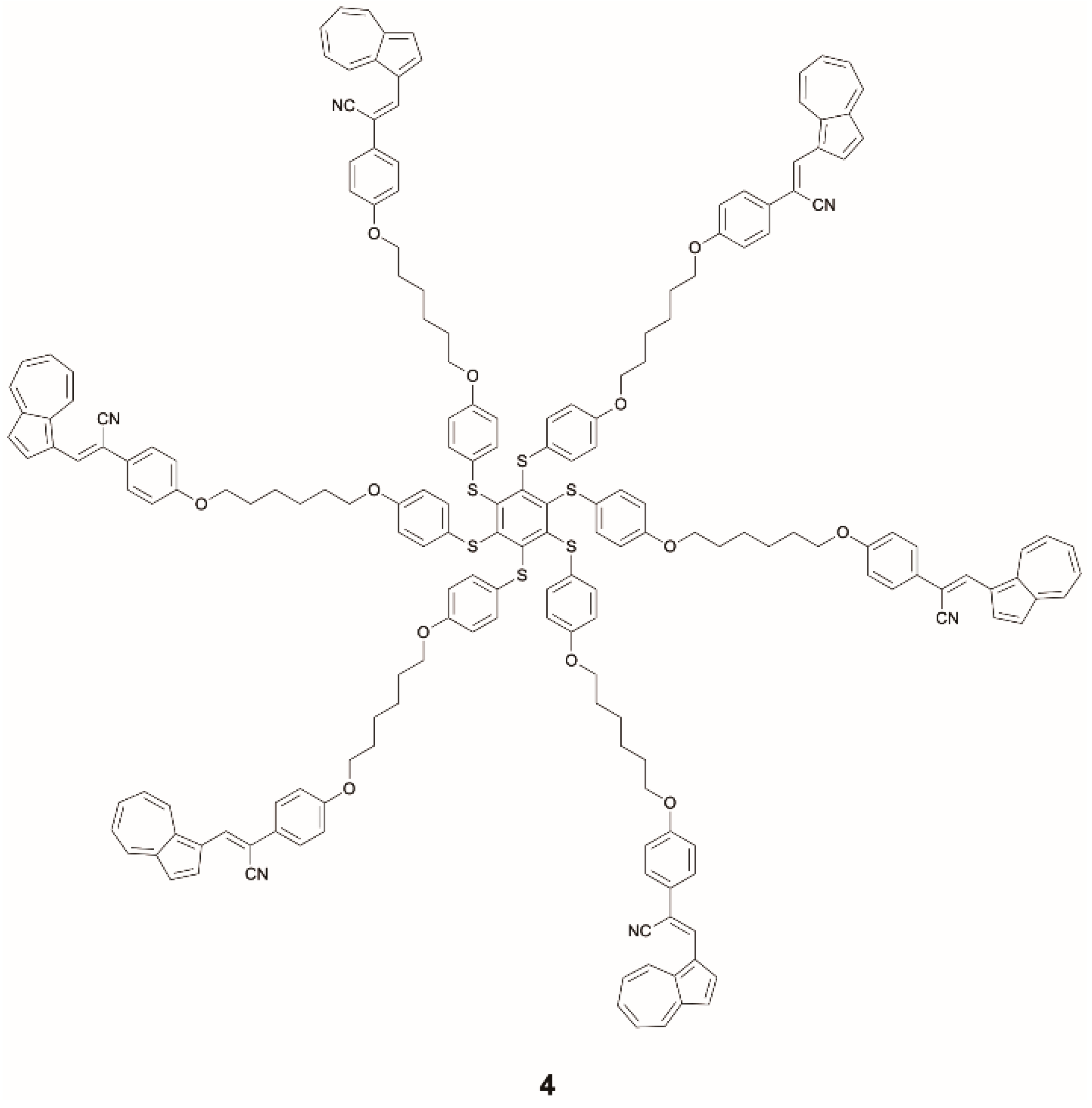
The same research group investigated the photophysics of compound
4 (
Scheme 3) in which dual Vis and NIR emissive cyanostyryl-modified azulene moieties are inserted onto a hexathiobenzene core [
38]. With respect to its cyanostyryl–azulene precursor,
4 displays an increased anti-Kasha emission at 480 nm due to a FRET process from the hexathiobenzene to the azulene. The emission intensity of
4 can be further enhanced in tetrahydrofuran (THF)/H
2O solvent mixtures by an AIE effect, thus leading to a total 15-fold amplification of the QY of this material compared to its precursors. In addition,
4 is a rare example of multiwavelength anti-Kasha emissions since it displays, in addition to the visible S
2-S
0 fluorescence at 480 nm, a UV emission at 360 nm, generated by radiative decay from the higher S
3 excited state. Interestingly, these emissive features are retained in PMMA films, opening the way to possible applications of solid-state anti-Kasha materials.
In an extension of their research, Zhou et al. [
39] reported on a series of mono-, di- and triformyl-substituted azulenes, among which the triformyl derivative
5 (
Scheme 4) shows a remarkable dual fluorescence (photoluminescence QY, Φ, up to 10%) in DMF solution. On the basis of experimental and theoretical investigations, the latter based on TDDFT calculations at the B3LYP/6-31G(d) level of theory, the photoluminescent behavior of
5 was attributed to concomitant radiative deactivations from the upper S
3 and S
2 energy levels with (π, π*) and (
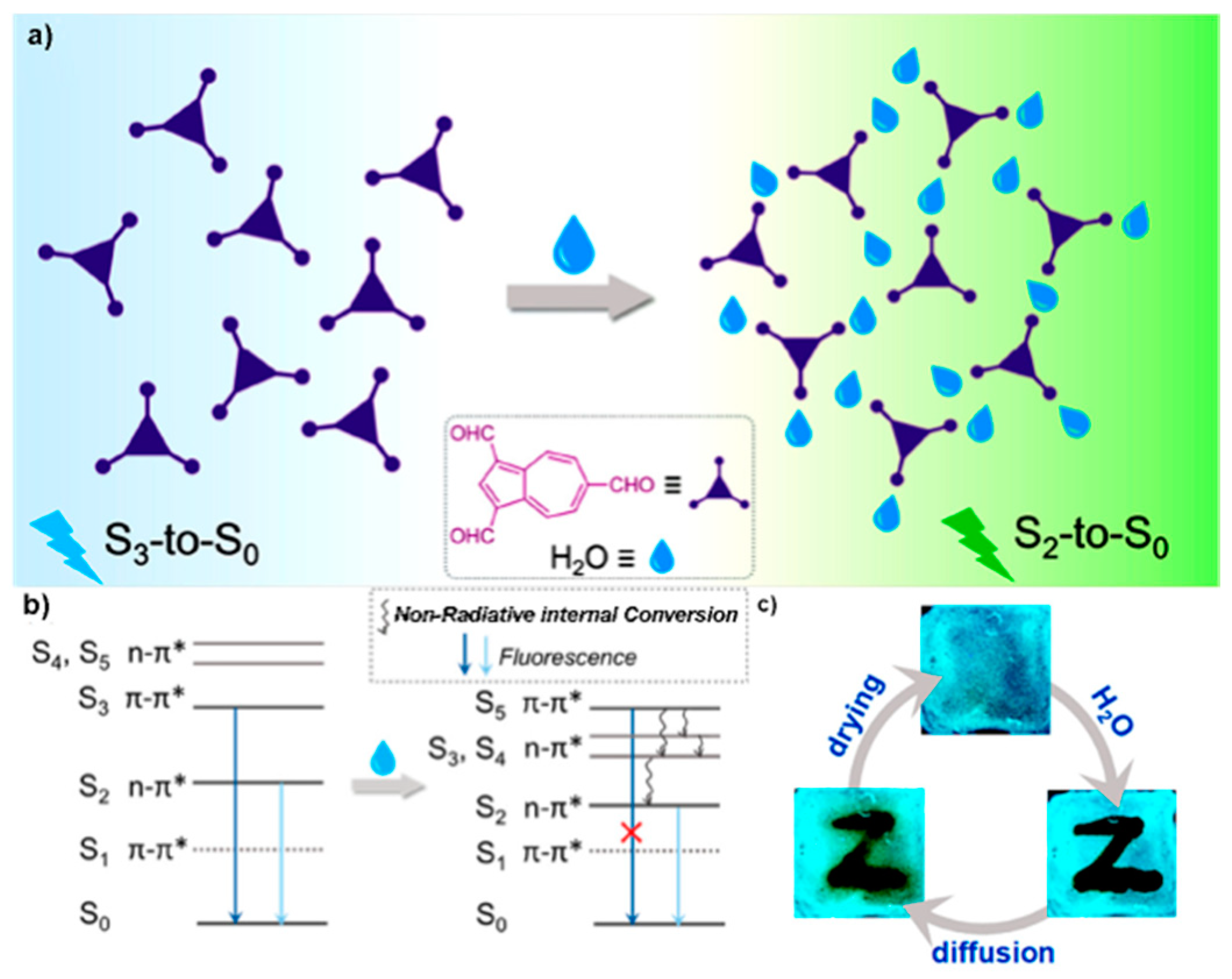
n, π*) character, respectively, and having an energy gap of 0.35 eV which guarantees a slow IC. In the presence of water, intermolecular H-bonds between the aldehydic groups and water result in the red shift of the emission and in the quenching of the S
3 deactivation path due to the stabilization of S
4 and S
5 (
n, π*) levels (see
Figure 4b). Tunable emission color, from blue to cyan and green, by H-bonding control was demonstrated both in solution and in the solid state by using hydrophilic polymers such as polyvinyl alcohol (PVA) and polyacrylic acid sodium salt (PAAS) as matrices to ensure a sufficient moisture sensitivity and the realization of a visual sensor for moisture (
Figure 4c).
Zhang et al. [
40] reported on the synthesis of dicyclohepta[
ij-
kl,
uvwx]rubicene,
6 (
Scheme 5), a polycyclic aromatic hydrocarbon containing two coplanar pentagons and heptagons constituting two formal azulene moieties. The presence of these conjugated rings ensures thermal and air stability together with planarity, as confirmed by crystal structure analysis. A combined photophysical and theoretical investigation revealed the presence in THF solution of two emission bands at 670 and 400 nm which were assigned, according to CASPT2/6-31G(d)/(12,12) calculations based on CASSCF orbitals, to S
1-S
0 and S
2-S
0 radiative decays, respectively. The anti-Kasha emission was established to originate mainly from a contribution of the two formal azulene units present in the molecular structure.
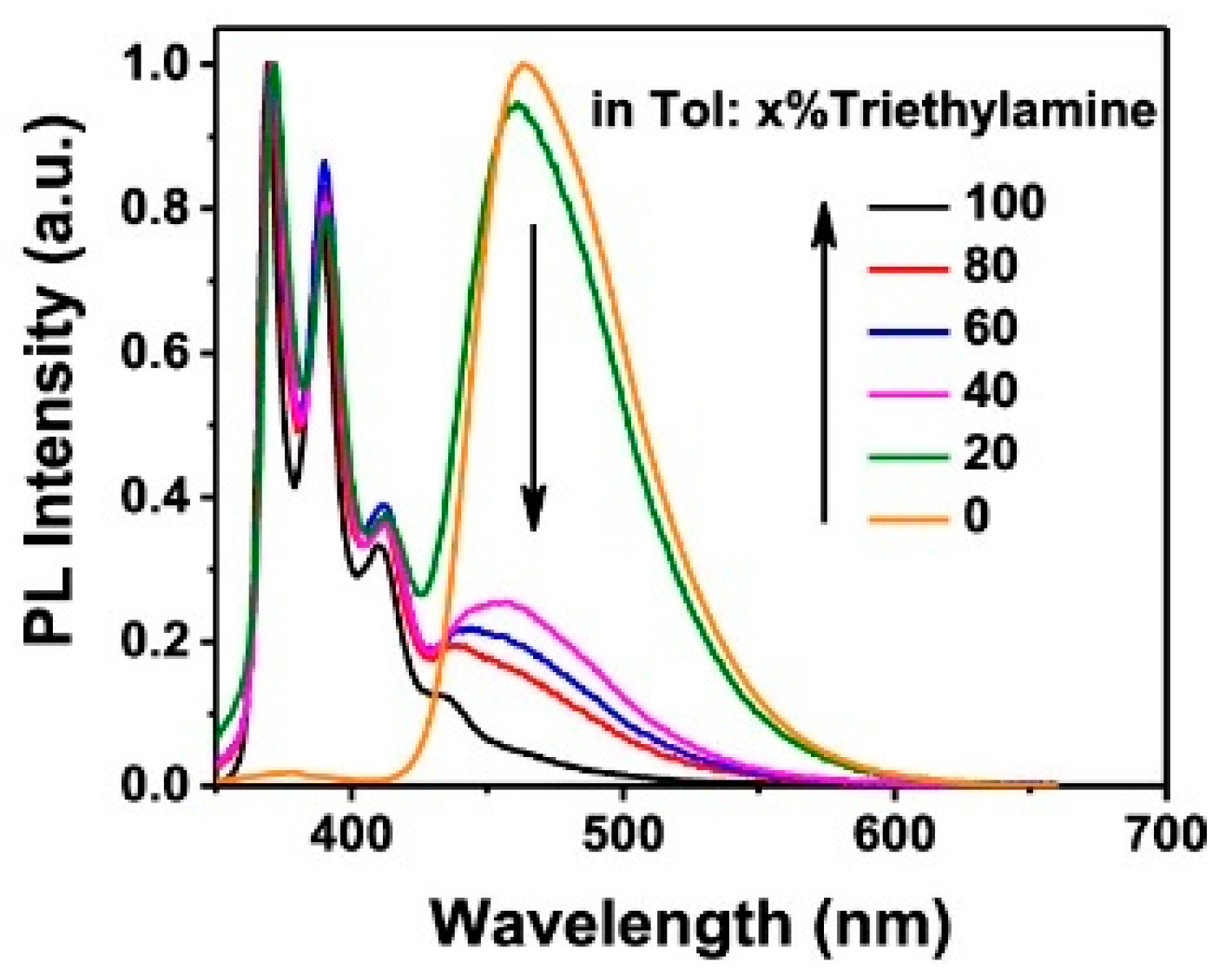
Based on the observation that protonation of azulene can result in a tropylium form with intensified fluorescence due to the suppression of anti-Kasha emission in favor of the S
1 one, Amir et al. [
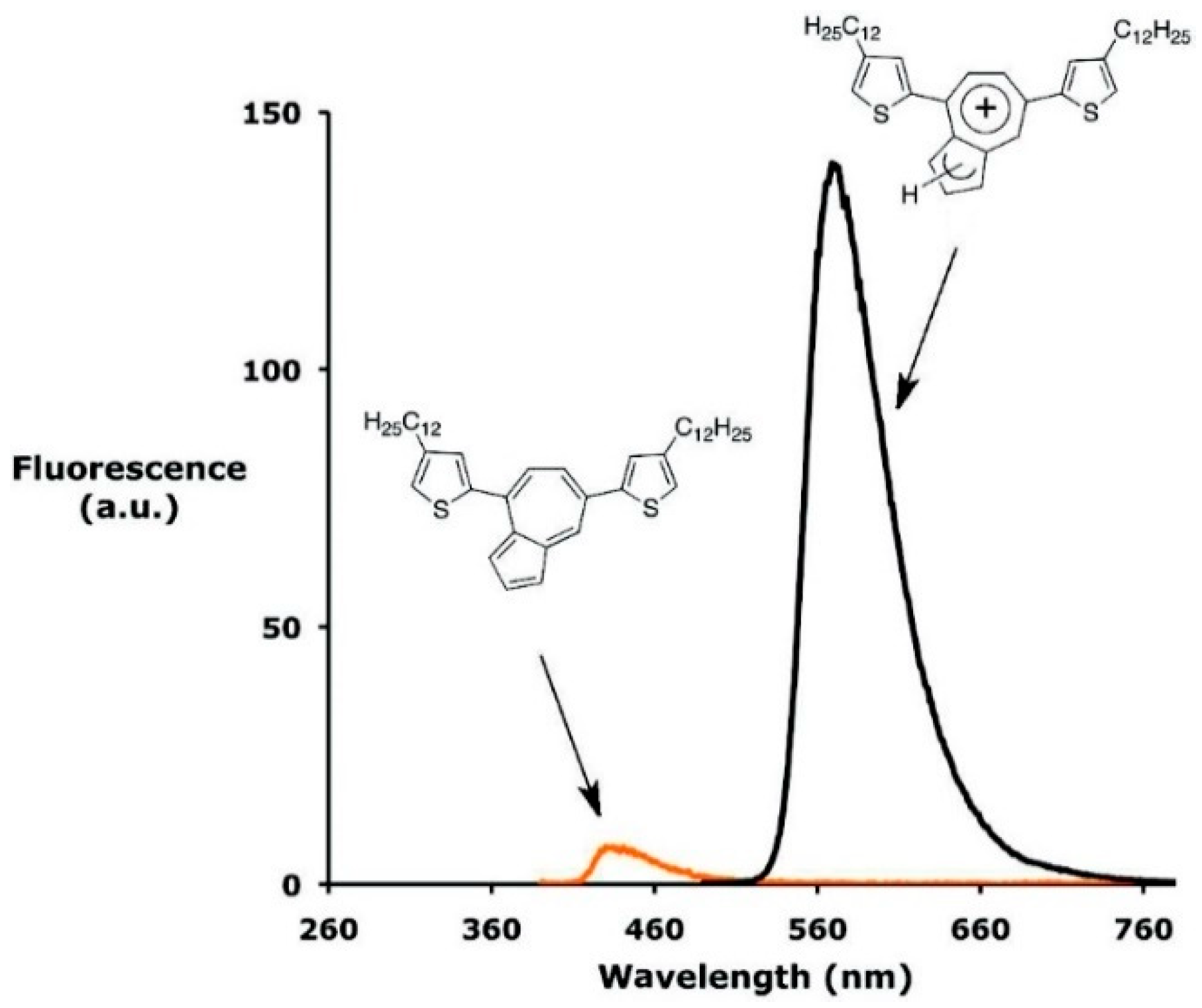
41] studied 5,5′-(azulene-4,7-diyl)bis(3-dodecylthiophene),
7, and 2,2′-(6-dodecylazulene-4,7-diyl)dithiophene,
8 (
Scheme 6). The two compounds can, in fact, undergo a reversible protonation/deprotonation process by treatment with trifluoroacetic acid (TFA) and triethylamine (NEt
3). The acidochromic switch can be easily followed by fluorescent spectroscopy (see
Figure 5) revealing a red shift and a remarkable increase in emission intensity upon protonation and the quenching of the signal by deprotonation with the base.
A similar behavior was reported by Koch et al. [
42] for a series of 2-alkynyl azulenes,
9–
20 (
Scheme 7), characterized by a weak fluorescence that can be enhanced and red-shifted by treatment with acids.
Shoij et al. [
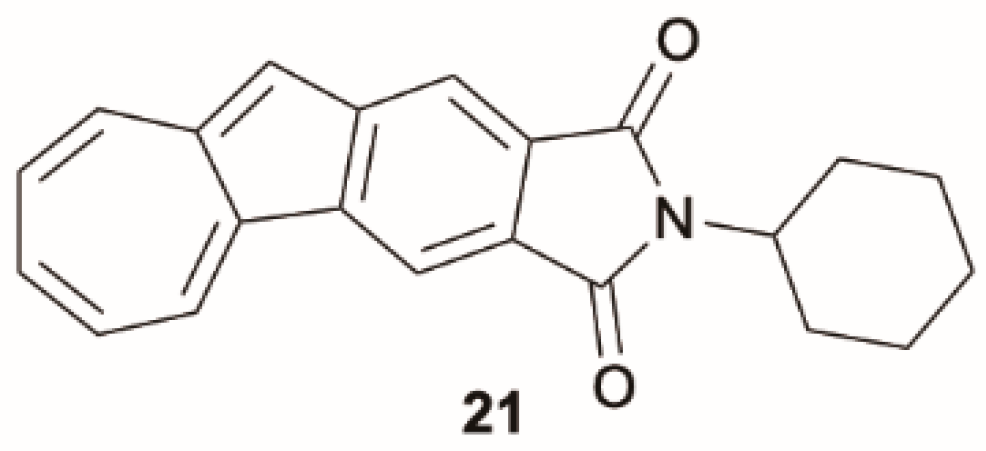
43] reported on a family of azulene-fused phthalimides, among which derivative
21 (
Scheme 8) displays the typical azulene fluorescent anti-Kasha feature. Compound
21 and a series of 2-arylazulenes reported by the same authors [
44] show the typical intensification of the S
1 emission through protonation, leading to suppression of anti-Kasha behavior.
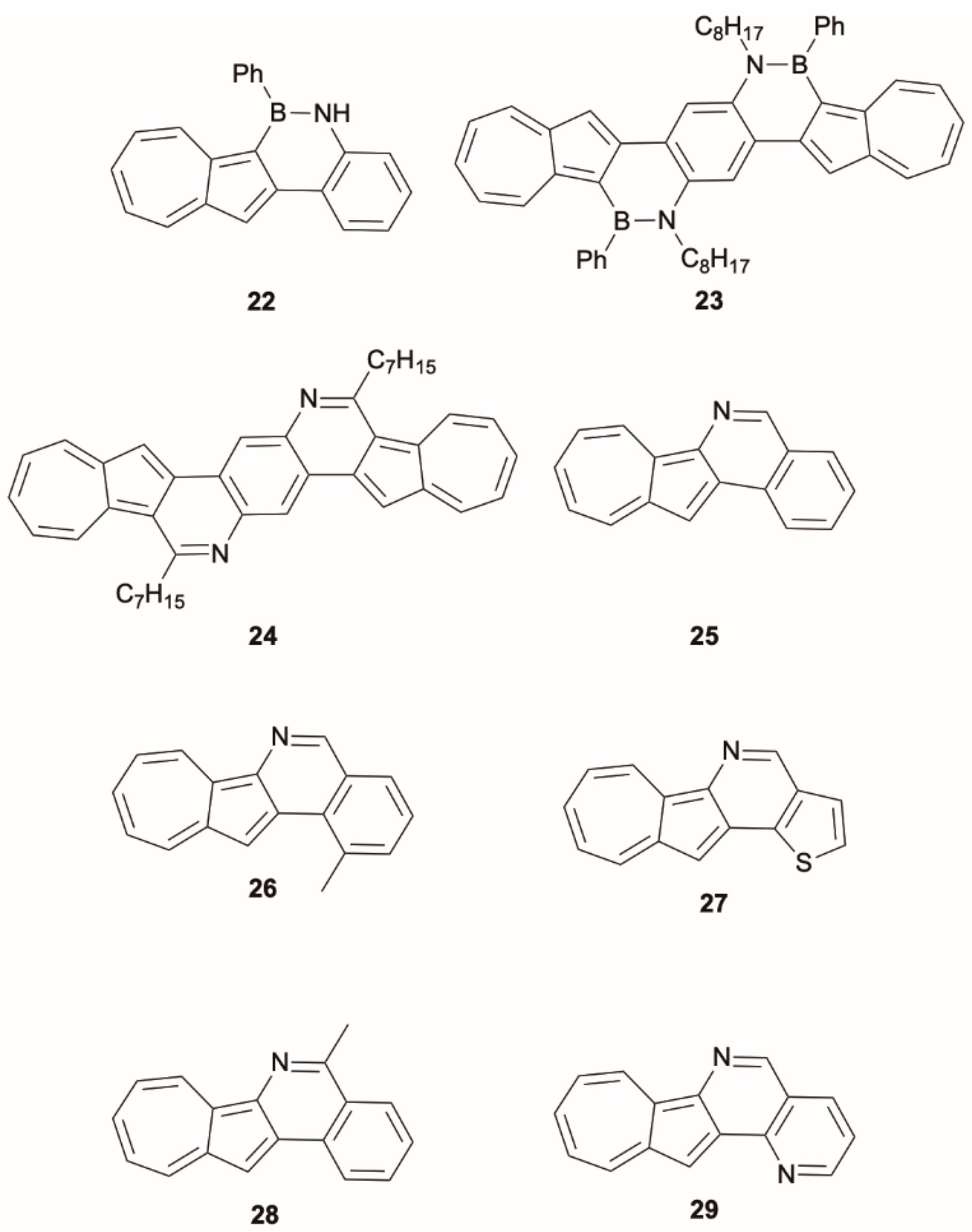
In the search of new polycyclic heteroaromatics for potential optoelectronic applications, new examples of azulene derivatives showing anti-Kasha behavior were reported by Xin et al. [
45], who prepared the first two azulene-based BN-heteroaromatics,
22 (
Scheme 9) and
23, by nitrogen aromatic borylation. Their analysis was supported by TDDFT calculations at the CAM-B3LYP/Def2-TZVP level of theory. The same research group [
46] described the reductive cyclization of 1-nitroazulene precursors to produce a series of azulene-pyridine-fused heteroaromatics,
24–
29, displaying photophysical features typical of azulene derivatives with a weak emission band originating, based on CAM-B3LYP/6-311G(d,p) TDDFT calculations, from S
2 or higher excited states and no detectable emission from S
1.
3. Cyclic Triimidazoles
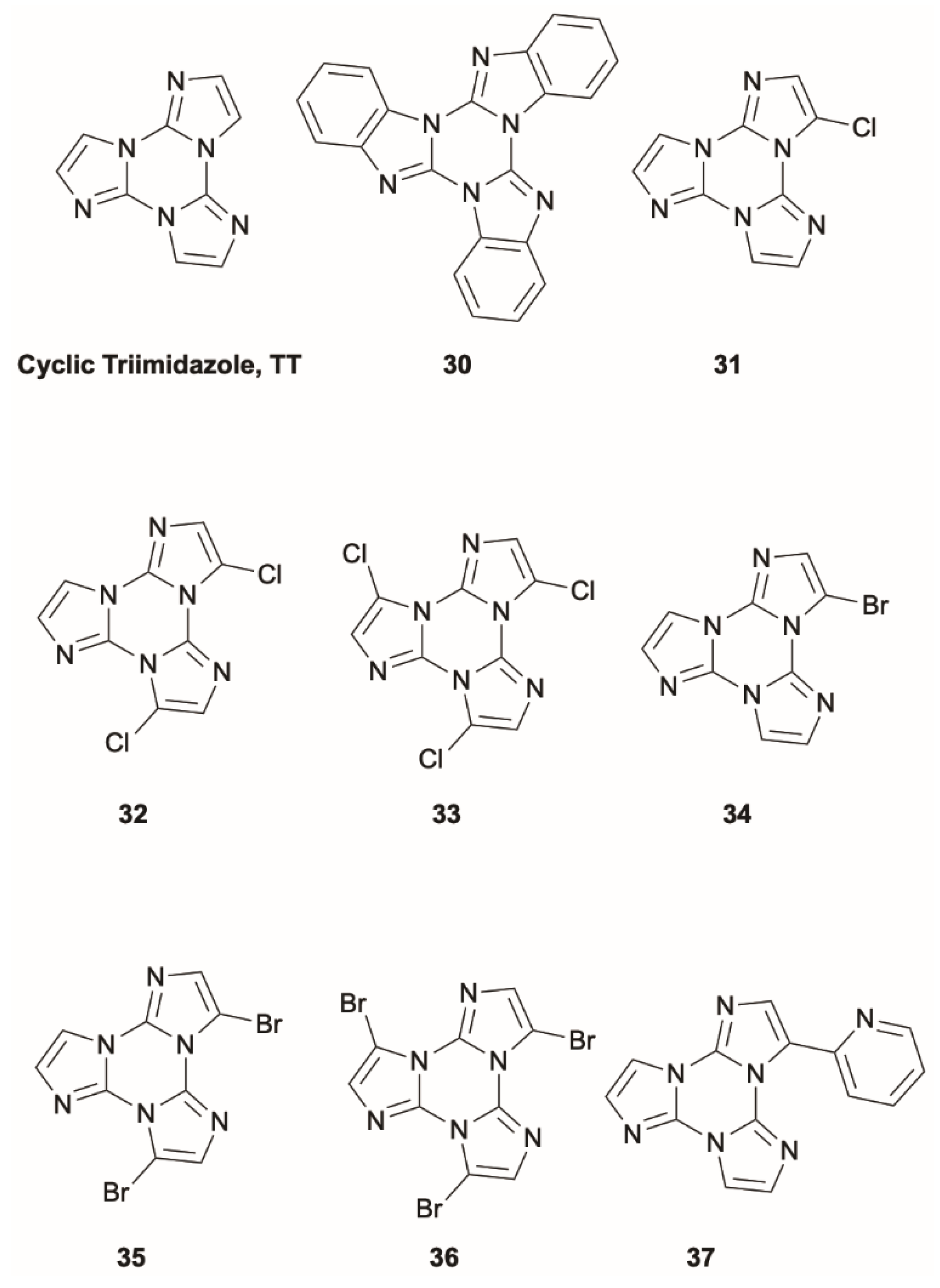
Lucenti et al. [
47] investigated an extensive series of “cyclic triimidazole”,
TT (
Scheme 10), derivatives (where
TT is triimidazo[1,2-
a:1′,2′-
c:1″,2″-
e][1,3,5]triazine) characterized, mainly in the solid state, by an intricated multiemissive behavior of molecular and aggregation-induced origin, including RTP associated with π–π interactions. Within this rich photophysics, for some members of this family, a fluorescent or phosphorescent anti-Kasha behavior was recognized.
In particular, benzo[4,5]imidazo[1,2-
a]benzo[4,5]imidazo[1,2-
c]benzo[4,5]imidazo[1,2-
e][1,3,5]triazine,
30 (
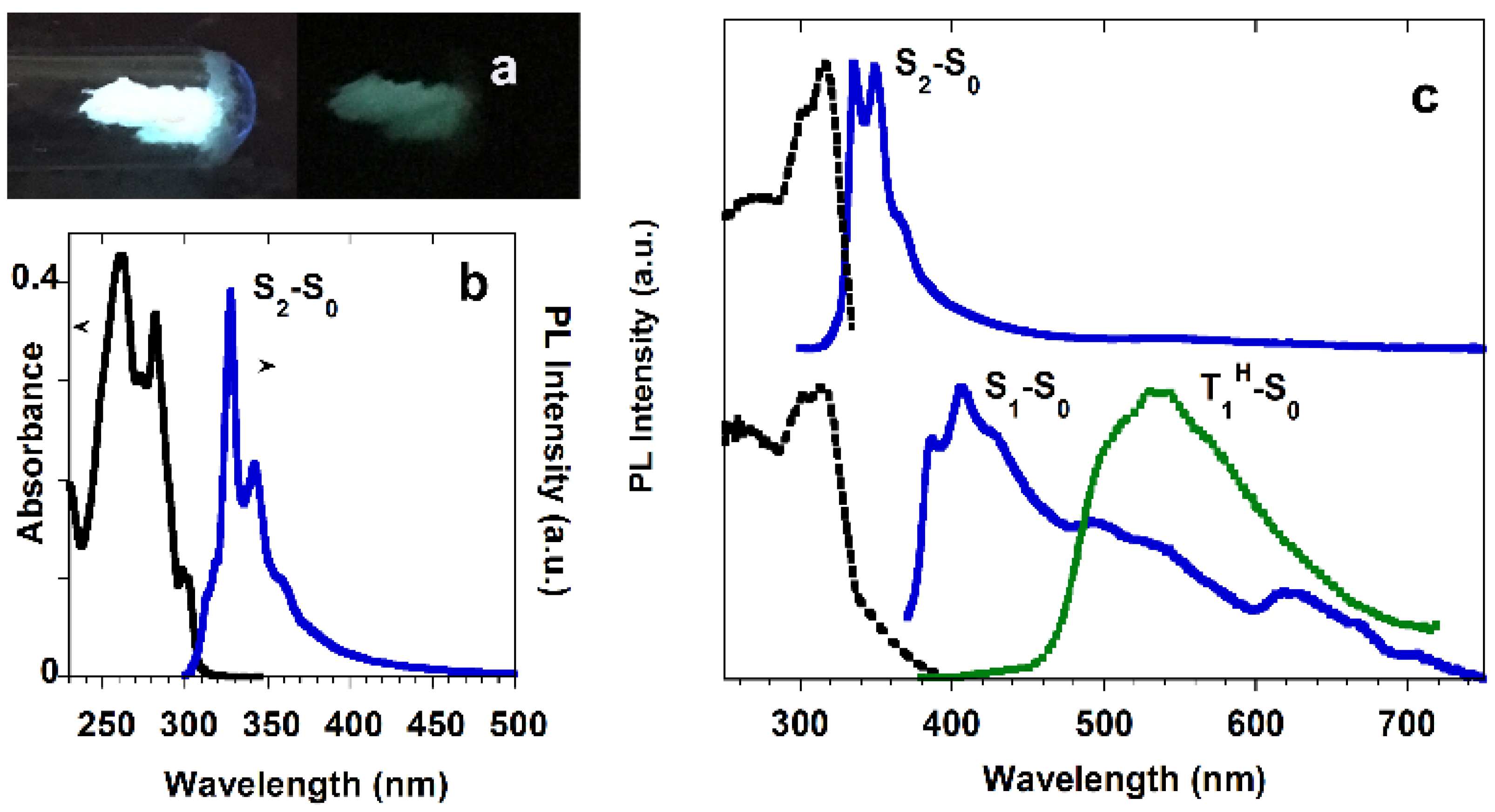
Scheme 10), displays a single fluorescent emission at about 330 nm (Φ = 17%) in diluted DCM solution at RT, which, based on TDDFT calculations at the ωB97X/6-311++G(d,p) level of theory, was associated with deactivation from an S
2 excited state [
48]. This attribution was supported by a calculated substantial S
2-S
1 energy gap (0.35 eV) and an S
0-S
1 transition with zero oscillator strength owing to the high symmetry (ideally
C3h) of the molecular π-electron system. Intriguingly, solid
30 shows both phosphorescence and dual fluorescence (Φ = 18%) at RT, which can be selectively activated by varying the excitation wavelength (see
Figure 6). At 260 nm excitation wavelength, a near-UV fluorescence emission at 350 nm of S
2-S
0 origin, similar to that of the chromophore in dilute solution, is observed. By exciting at 370 nm, a structured blue fluorescence at 407 nm superimposed onto the phosphorescent emission at 530 nm is produced. The authors explained the presence of this second fluorescence of S
1-S
0 nature, selectively activated only by populating the S
1 state, by partial loss of the molecular symmetry of the chromophore owing to interchromophoric interactions in the solid state.
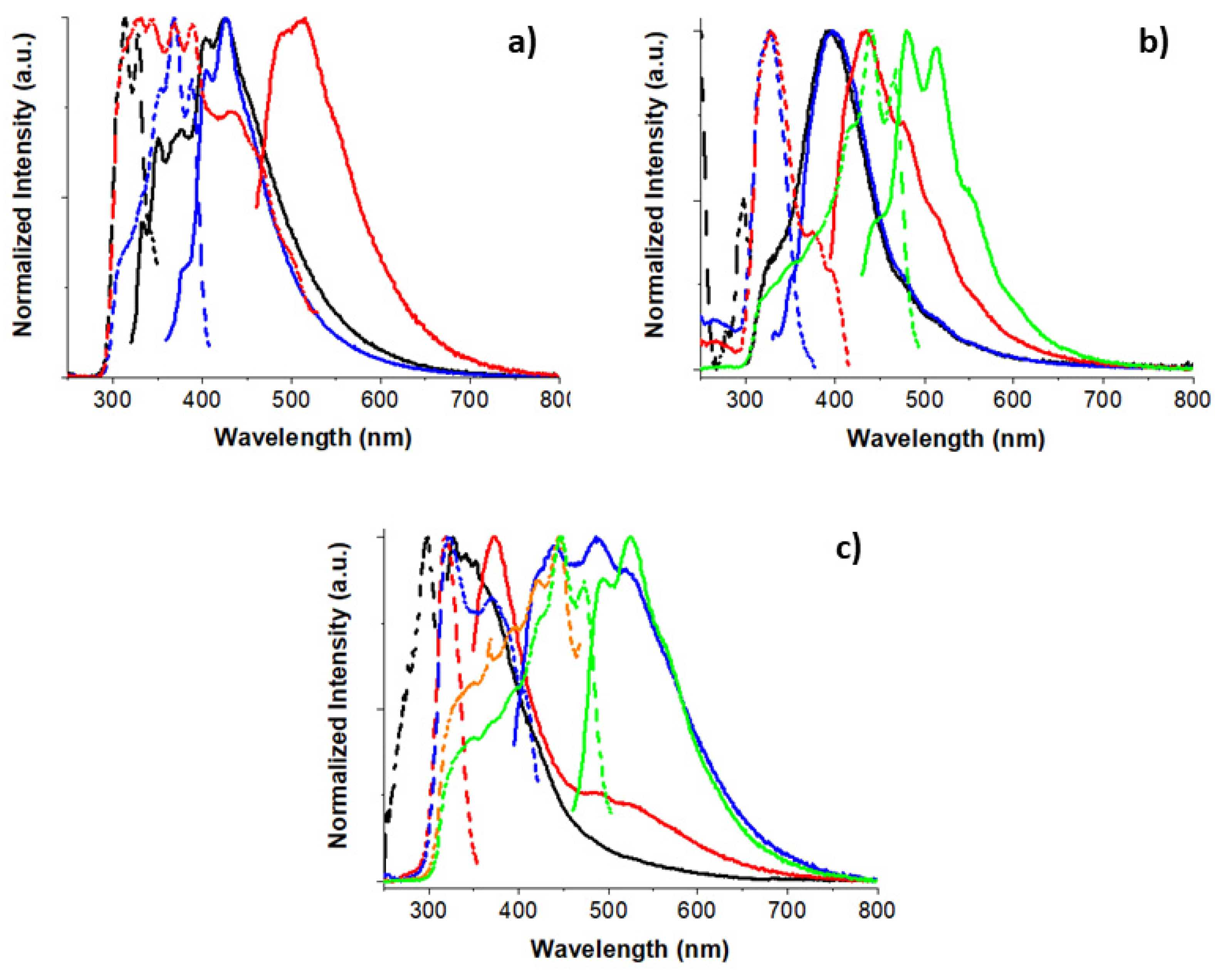
Similarly, mono-, di- and trihalogenated
TT compounds [
48,
49,
50],
31–
36 (
Scheme 10), display dual fluorescence in the solid state due to a large S
n-S
1 energy splitting and a low or zero oscillator strength of the S
0-S
1 transition. In particular,
31–
33 (Φ = 24%, 12% and 9%, respectively) are characterized by multiple long-lived emissive components (due to molecular, T
1 and aggregated forms) in addition to dual fluorescence (one at about 340 nm for the three compounds and the other at about 350, 400 and 370 nm for
31,
32 and
33, respectively; see
Figure 7), better resolved at low temperature [
49]. The two fluorescences, based on analysis of excitation spectra showing a first absorption at 320–330 nm and a second absorption at 300 nm, were assigned to deactivation from S
1 and S
n (
n > 1), respectively, with an experimental S
2-S
1 energy gap in the 0.27–0.38 eV range. This anti-Kasha origin of the dual fluorescence was supported by ωB97X/6-311++G(d,p) DFT/TDDFT calculations which provided an S
1 state of low oscillator strength (
f = 0.013, 0.009 and 0 for
31,
32 and
33, respectively) due to symmetry reasons and high-energy singlet levels with large oscillator strength (S
3 and S
4,
f > 0.4).
Analysis of the emissive properties of
31–
33 in solution was precluded by the very low quantum efficiency of the compounds at low concentration. On the other side, DCM solutions of
34–
36 [
48,
50] display a weak but recognizable fluorescence with maxima at 328, 380 and 370 nm, respectively (Φ equal to 3% for
34 and almost vanishing for
35 and
36), which was ascribed, on the basis of ωB97X/6-311++G(d,p) DFT/TDDFT calculations, to a high-energy S
n state (
n = 3; 4, 5; 6, 7 for
34–
36, respectively). Again, the absence of the S
1-S
0 radiative deactivation channel was explained by the low
f of the S
0-S
1 transition (0.024, 0.016 and 0 for
34–
36, respectively) associated with the high symmetry of the
TT π-electron system, only partially disrupted in
34 and
35, and to the sizable S
n-S
1 energy gap (calculated as 0.43, 0.44 and 0.48 eV for
34–
36, respectively).
Interestingly, unlike S
1 and S
n having (π, π*) character, the intermediate S
m levels (S
2, S
2,3 and S
2–4 for
34–
36, respectively) correspond to forbidden transitions (
f = 0) with partial (π, σ*) character, where the involved σ orbitals are mainly delocalized on Br atom(s) and C–Br bond(s). Going to the solid state (Φ < 0.1, 14 and <0.1 for
34–
36, respectively) (see
Figure 8 for
34), molecular and intermolecular-induced phosphorescences are observed in the spectra of the three compounds. Moreover, while an S
m-S
0 emission is visible for
34–
36 (at 326, 345, 365, 382; 395, 419, 443; 394, 418, 444 nm, respectively), the S
1-S
0 one appears only for
34 and
36 (with maxima at 426 and 415 nm, respectively). This latter emission, associated with the distorting packing forces which reduce the symmetry of the molecular π-electron system, is the only emission observed by exciting at high energy (E > S
m) while dual fluorescence is produced by exciting to S
m levels. A possible interpretation was provided by ωB97X/6-311++G(d,p) DFT/TDDFT calculations on the π–π dimer of
36, which revealed a manifold of S
m levels of (π, π*), (π, σ*) and mixed (π, π*)/(π, σ*) character where the latter possess the strongest oscillator strength. It is therefore expected that excitation to S
m results in both radiative deactivation (from (π, π*)/(π, σ*) levels) to S
0 and IC (from (π, π*) levels) to S
1, while excitation to (π, π*) S
n leads to IC to S
1. In the case of
35, no mixed S
m (π, π*)/(π, σ*) states came out from calculations on its π–π dimer, and the (π, σ*) excitations have
f < 0.01, suggesting that S
m levels have predominant (π, π*) character with
f greater than that of S
1. Therefore, irrespective of excitation energy, these (π, π*) S
m levels are responsible for the observed single fluorescence.
3-(Pyridin-2-yl)triimidazo[1,2-
a:1′,2′-
c:1″,2″-
e][1,3,5]triazine,
37 (
Scheme 10), is the only member of the
TT family showing anti-Kasha behavior from an excited triplet level [
51]. This compound crystallizes in three phases characterized by a highly intricated photophysics comprising multiple fluorescent and phosphorescent bands (Φ = 50%). The dual fluorescence observed at about 350 and 450 nm was associated with two different molecular conformations; therefore, it does not represent an exception to the Kasha rule. On the other side, two (at about 370 and 400 nm) of the four observed phosphorescences were ascribed to molecular origin and interpreted, on the basis of ωB97X/6-311++G(d,p) DFT/TDDFT calculations, as derived from triplet levels of different character. In particular, the high energy contribution resulted from the calculated T
6 level having (σ/π, π*) character, while the low energy contribution derived from the calculated T
1 with (π, π*) symmetry.
5. Cyanines
The peculiar electronic structure of cyanines, having an odd number of conjugated p
z orbitals and an even number of π electrons, results in the stabilization of the S
1 state with respect to S
2. Therefore, frequently, these dyes are characterized by an intense S
0-S
1 transition in the visible/near-IR region and much less intense S
0-S
n (
n > 1) transitions in the visible/near-UV region. Another relevant aspect of cyanine dyes is their large two-photon absorption cross-sections for near-IR wavelengths. These peculiar properties of cyanine dyes have attracted much interest in many applications such as two-photon photosensitizer or antenna systems. In addition, the large S
2-S
1 energy gaps (0.6–1.0 eV) associated with low S
2-S
1 IC rates represent an interesting feature for possible anti-Kasha behavior. In fact, some cyanine dyes exhibit dual fluorescence when excited directly to S
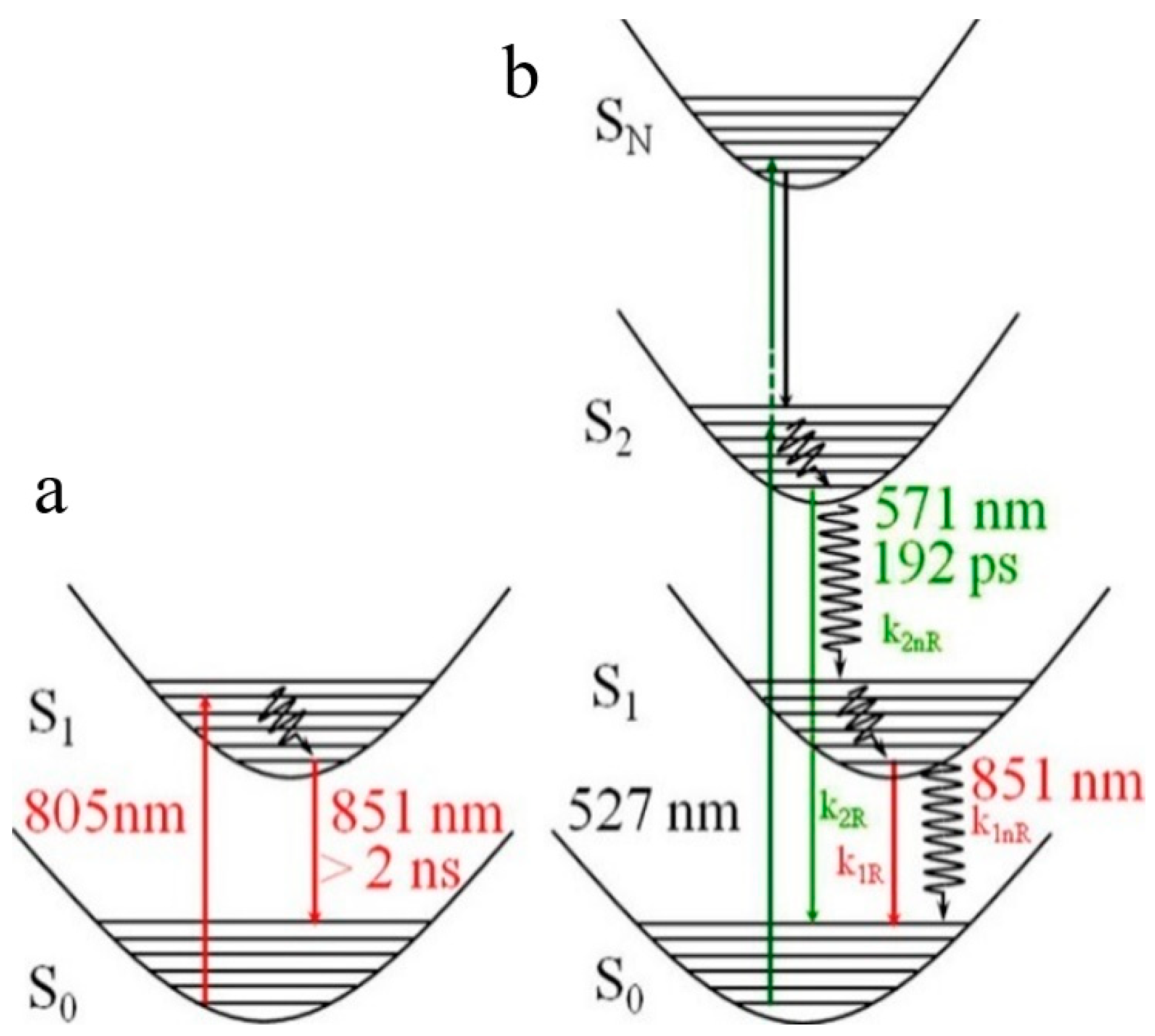
2, as reported by Das et al. [
55] in 2013. In particular, cyanine IR125 (
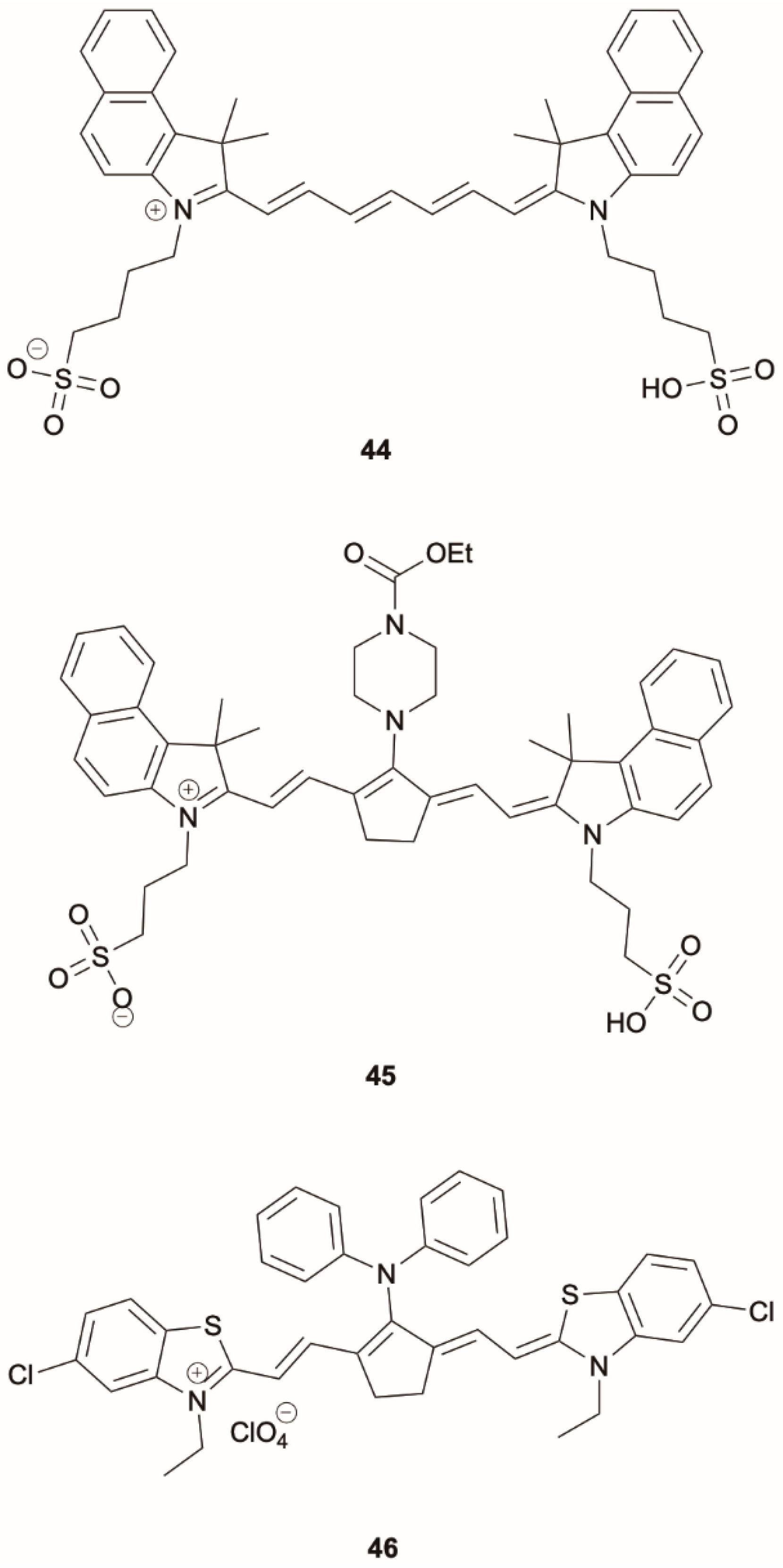
44) (
Scheme 14) in DMSO produces a single fluorescence at 851 nm when excited at 805 nm, but a second fluorescence at 571 nm (of S
2-S
0 origin) is activated together with the one at 851 nm (of S
1-S
0 nature) upon excitation at 527 nm. According to steady-state and time-resolved experiments, dual fluorescence was schematized as reported in
Figure 12. By high-energy excitation, direct fluorescence from S
2 is produced together with IC to S
1, from which fluorescence is also observed. By low-energy excitation, only S
1 is populated, therefore resulting in single fluorescence.
Later on, Das et al. [
56] reported two near-infrared (NIR) tricarbocyanine dyes, IR144 and IR140 (
45 and
46, respectively) (
Scheme 14), displaying two distinct fluorescent emissions in different alcoholic solutions (MeOH, EtOH, PrOH and BuOH): one in the visible region (500–550 nm), assigned to an S
2-S
0 transition, and the other in the NIR (850–900 nm), assigned to an S
1-S
0 transition. For both
45 and
46, a variation of the S
2 emission intensity and lifetimes by varying the alcohol chain length was observed. Differently from
46,
45 displays an S
2 excited state dynamics which depends on the solvent polarizability as predicted by the energy gap law. By increasing the polarizability of the alcohol series (from MeOH to BuOH), S
2 emission lifetimes of
45 can be extended from a few picoseconds to quite larger values (83.46 ps in BuOH), opening up a new area for cell imaging and tissue engineering in different alcohols.
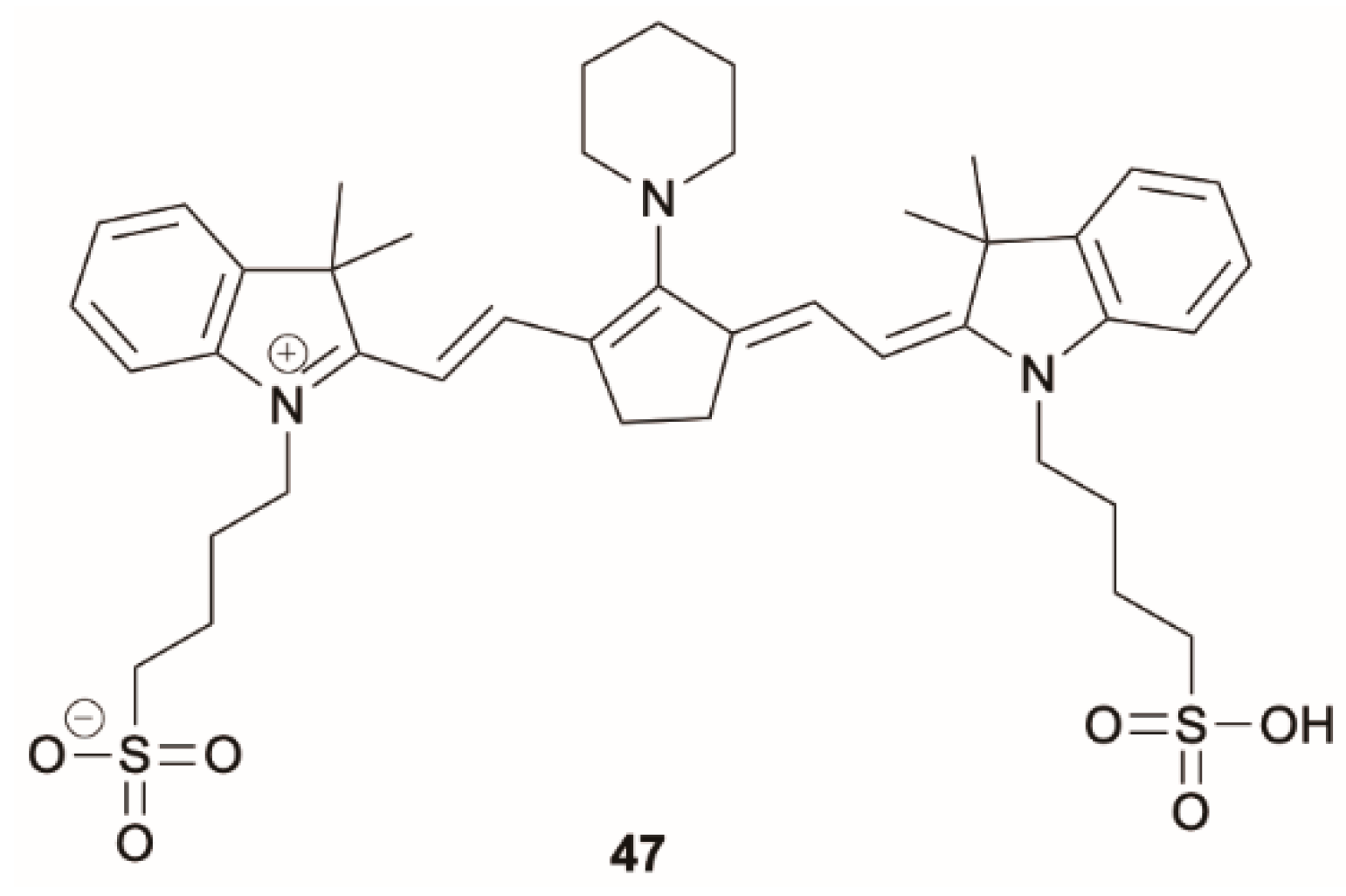
Nairat et al. [
57] proposed an original way to modulate the Kasha rule through photonic control in cyanine dyes by using shaped pulses for direct S
2 excitation. The authors tracked the S
2 and S
1 fluorescence yield for different cyanine dyes, i.e., IR144 (
45) and mPi-IR806 (
47) (
Scheme 15), in solution as a function of the femtosecond pulse produced by a linear chirp device. These findings offer a photonic method to control S
2 population, allowing the exploration of photochemical processes initiated from higher excited states.
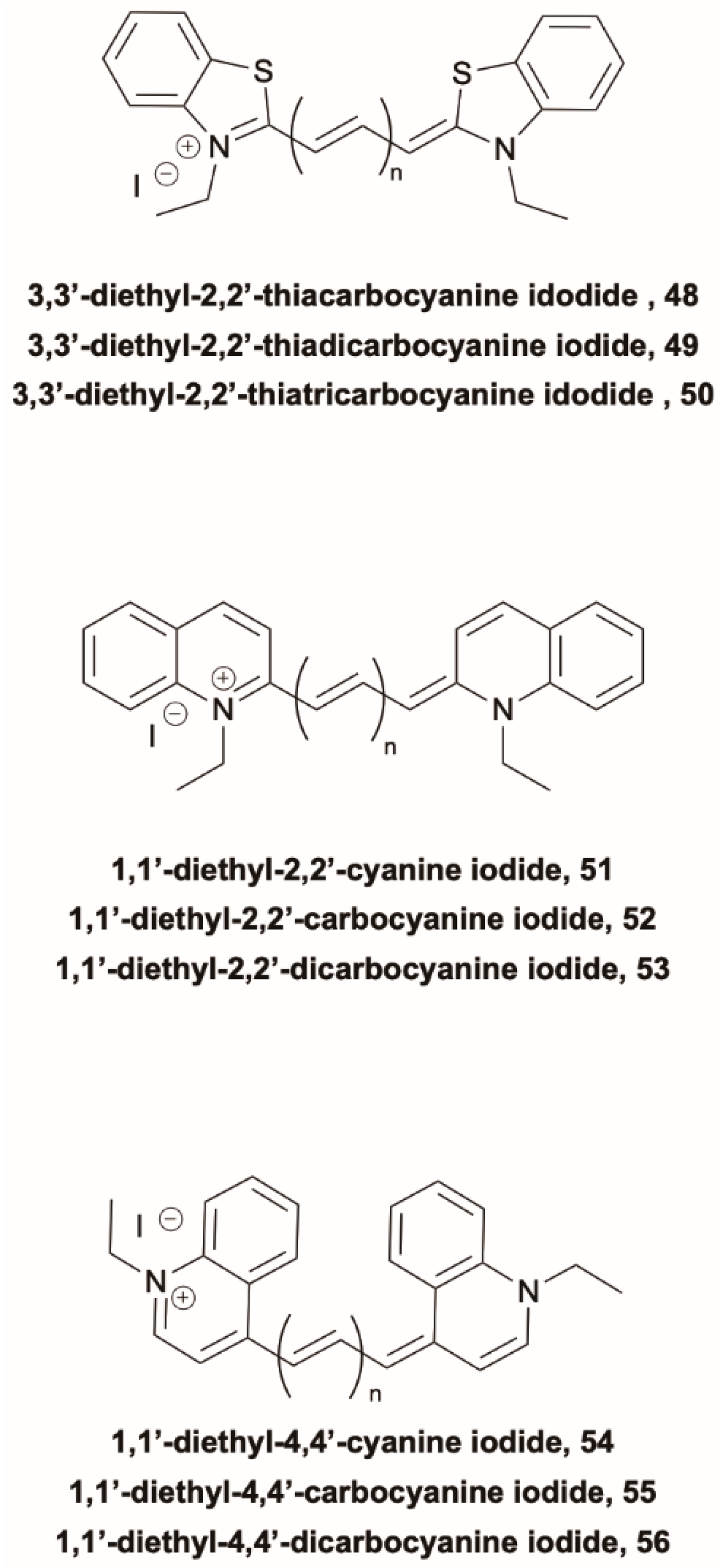
Guarin et al. [
58] reported direct measurements of the S
2 lifetime for three cyanine families with benzothiazolyl (
48–
50) (
Scheme 16), 2-quinolyl (
51–
53) (
Scheme 16) and 4-quinolyl (
54–
56) (
Scheme 16) end groups and different –(C=C)
n– skeleton sizes (
n = 1, 2 and 3 for each family) in order to investigate the structural features able to slow down or inhibit S
2-S
1 IC. Clear emission peaks in the region between the first and second absorption transitions, ascribable to emission from S
2, were detected for the longer cyanines in ethanol solution (at 429 and 494 nm for
49 and
50, respectively; at about 460 and 490 nm for
52 and
53, respectively; at 460 and 523 nm for
55 and
56, respectively), while the shortest ones displayed weak emission from S
2 only in more concentrated solutions. The dynamics of the upper states was followed by femtosecond fluorescence upconversion and by time-resolving spontaneous emission. Moreover, the time evolution of the S
1 emission allowed the monitoring of the population growth of S
1 through IC from S
2. The authors observed that S
2 lifetime can vary by more than two orders of magnitude depending on the conjugation length and end groups. For the shortest cyanines,
51 and
54, ultrafast (<0.4 ps) S
2 lifetimes were measured, with a correspondingly fast S
1 population. These rapid dynamics were associated with the nonplanar ground state geometry due to steric hindrance between the end groups. For all the other cyanines (
48–
50,
52,
53,
55 and
56), having planar structures, the S
2 lifetimes were found to closely follow the energy gap law, so that the cyanine with the largest gap,
50, displays the slowest (17.3 ps in ethanol) S
2 decay time among all the analyzed compounds.
Similarly, Kumari and Gupta [
59] showed that indocyanine green ICG,
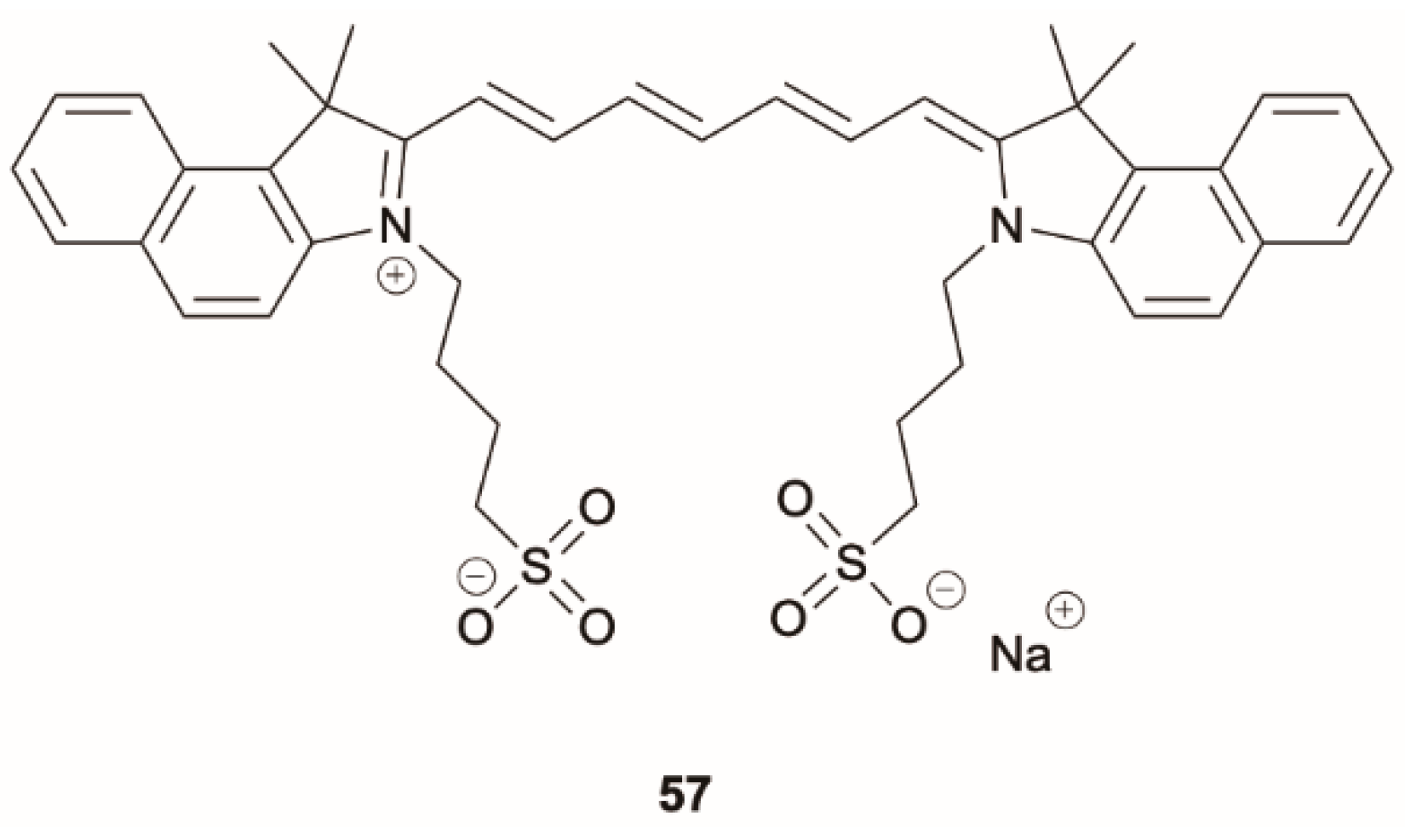
57 (
Scheme 17), an exogenous contrast agent used for clinical applications in the NIR region for its 810 nm emission from S
1, displays direct fluorescence at 572 nm from S
2 when excited at 393 nm. S
2 level can be populated also by two-photon excitation with a 790 nm wavelength femtosecond laser, in the well-known “tissue-optical window”, providing a two-photon contrast agent for biomedical imaging applications. The authors demonstrated the use of
57 as exogenous contrast agent for HeLa cell imaging, exploiting its direct emission from the S
2 state.
6. Thiophenes
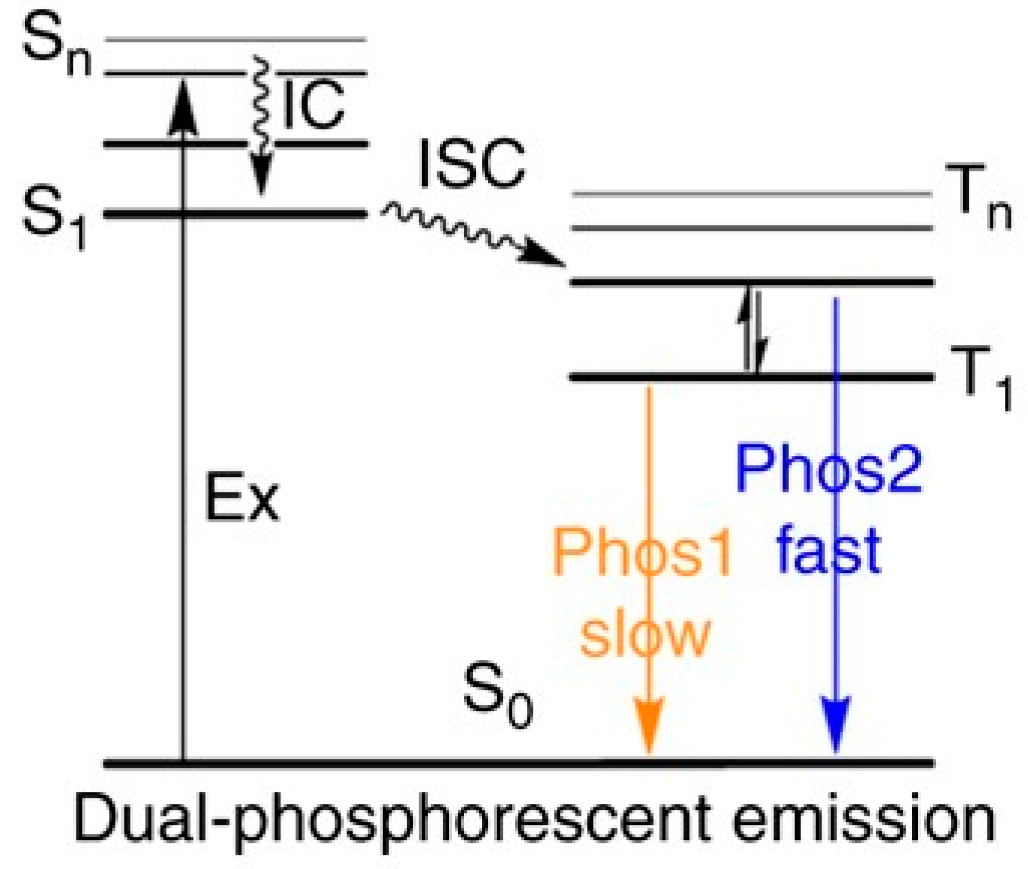
Thiophene-based compounds are potential candidates to display anti-Kasha phosphorescence from T
2 since thienyl substituents have been reported, through a combined spectroscopic and B3LYP/6-31G(d) DFT and TDDFT investigation, to favor S
1 to energy-matched T
2 ISC [
60].
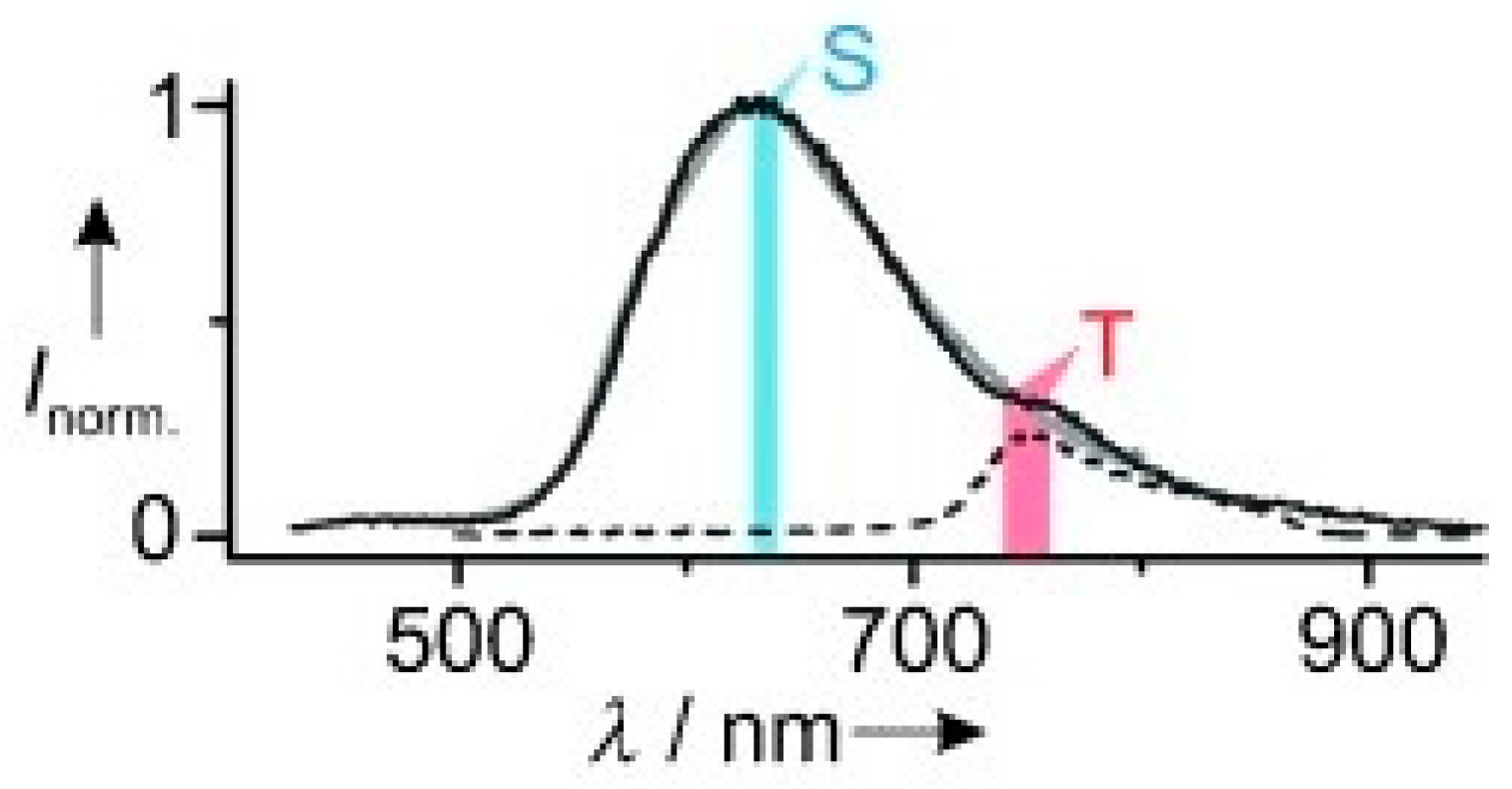
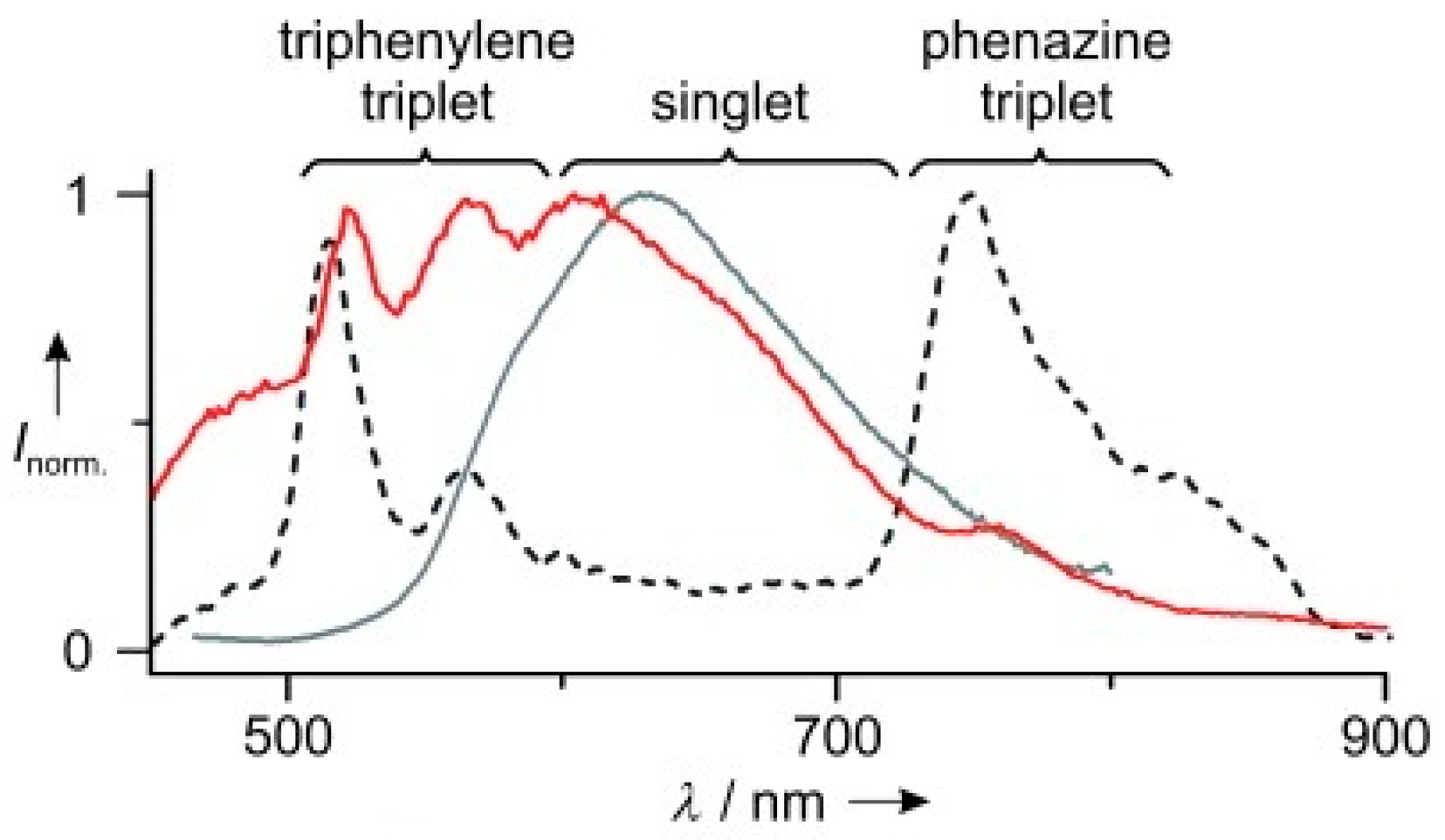
Chaudhuri et al. [
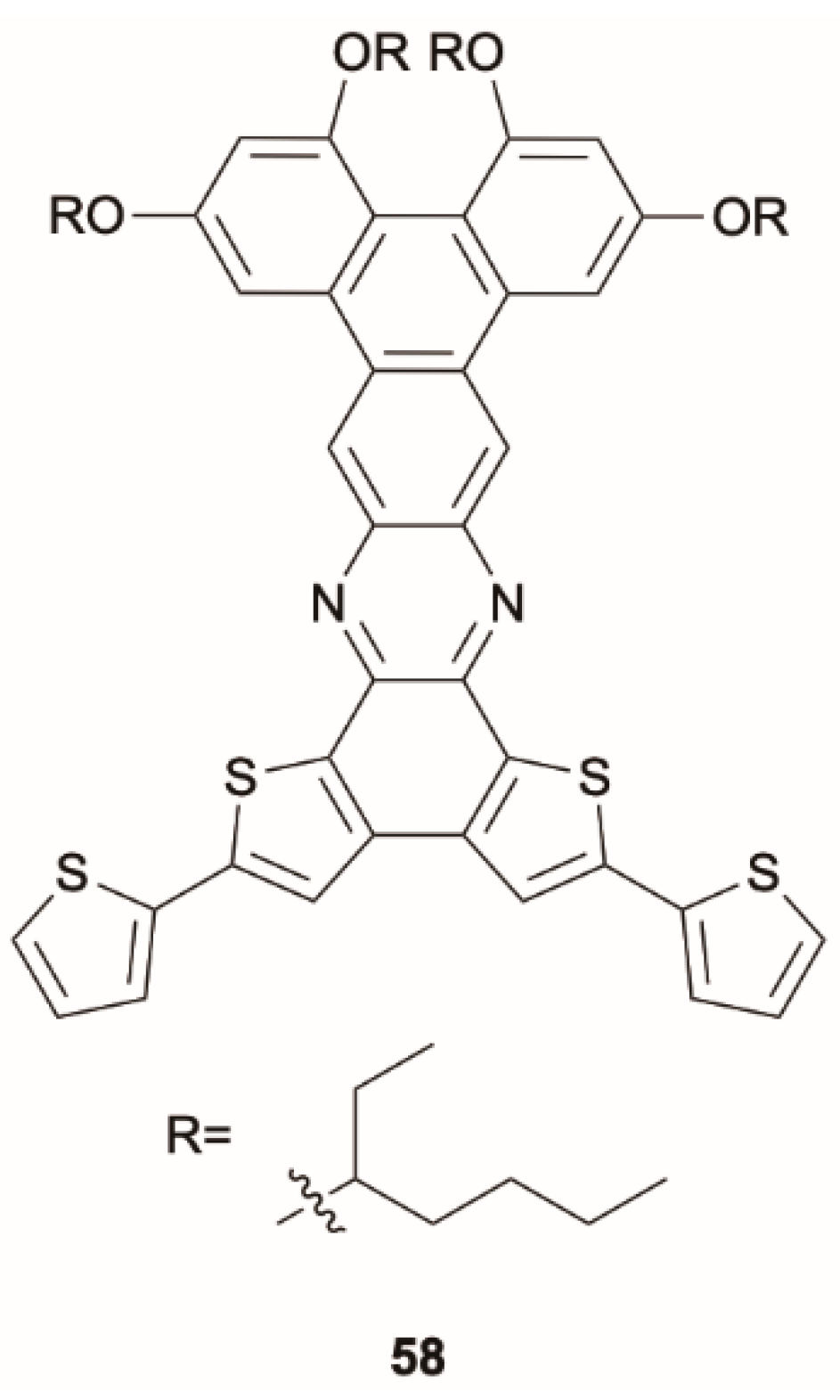
61] prepared and studied compound
58 (
Scheme 18), which has a thiophene-decorated phenazine linked to a triphenylene block. Compound
58 displays in solution one fluorescent emission at 630 nm corresponding to deactivation from S
1. Concomitant phosphorescent emission at 760 nm at room temperature becomes visible only in polystyrene matrix with exclusion of oxygen (see
Figure 13). Intriguingly, room temperature EL spectra of
58 in poly(9-vinylcarbazole) matrix display fluorescence/phosphorescence dual EL (see
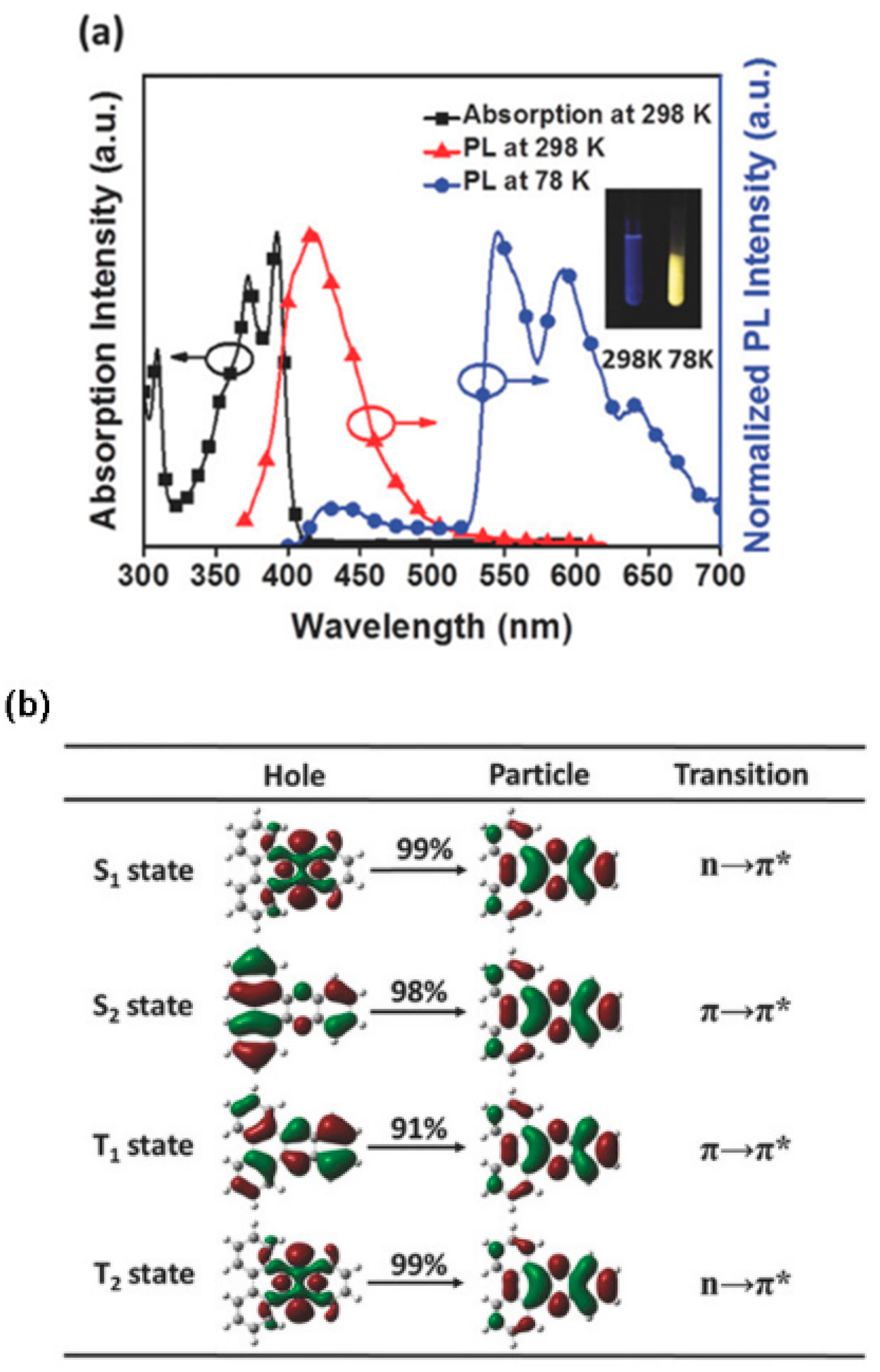
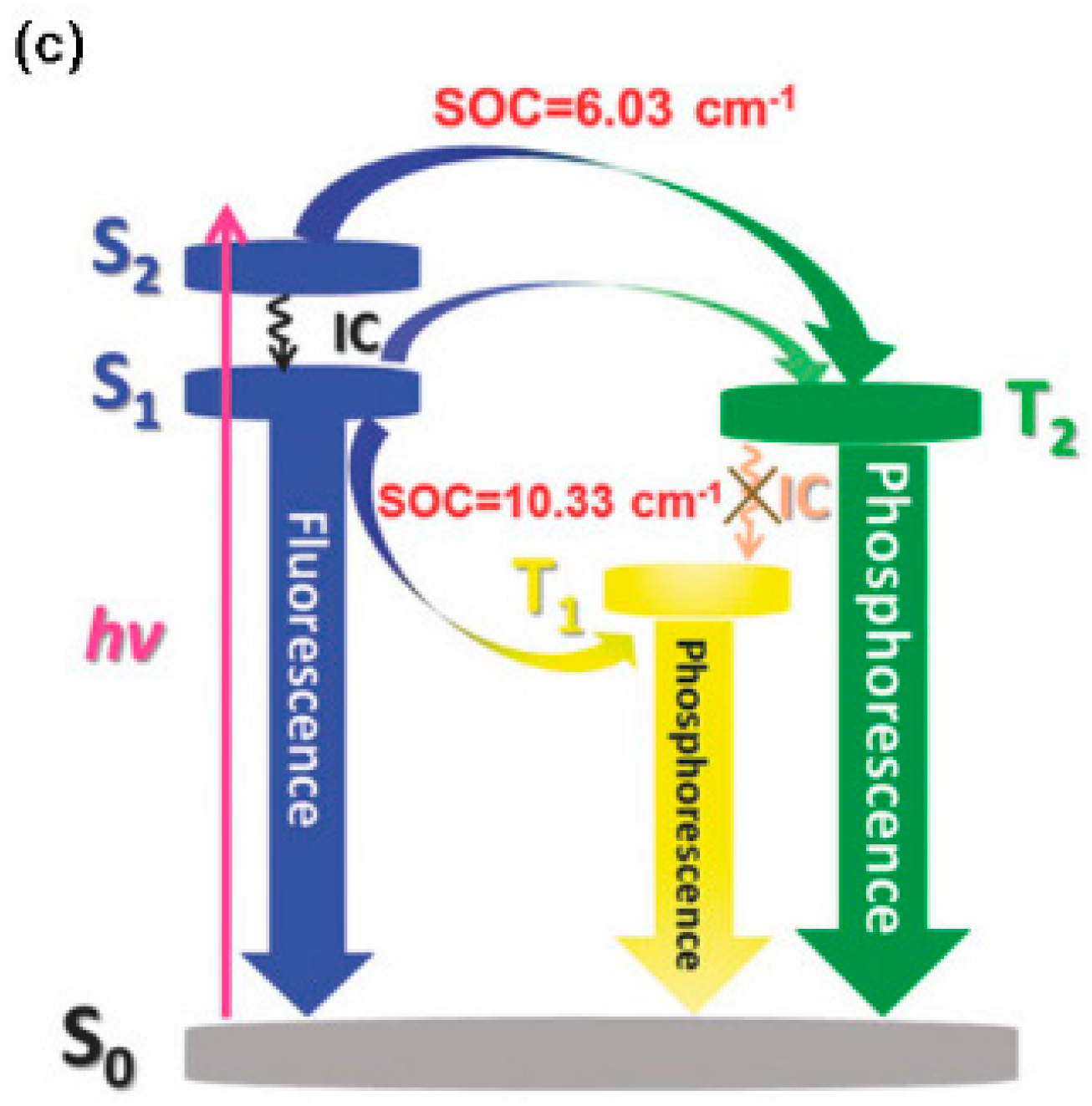
Figure 14). Moreover, EL and gated spectra of diluted polystyrene films, collected at 4 and 25 K, respectively, show an additional peak at 530 nm from a high-energy triplet. Based on PBE0/6-31G(d) geometry optimization and PBE0/6-311G(d) TDDFT calculations on the analog compound with the long alkyl chains (R) replaced by methyl groups, the two phosphorescences were assigned to triplet states localized on the triphenylene (the high-energy emission) and the phenazine moieties (the low-energy one). At RT, the high-energy triplet emission is quenched by vibrations that favor IC from the “hot” triphenylene triplet to the lower-energy phenazine one. It was therefore suggested that anti-Kasha dual phosphorescence may favor the realization of room-temperature electrophosphorescent devices.
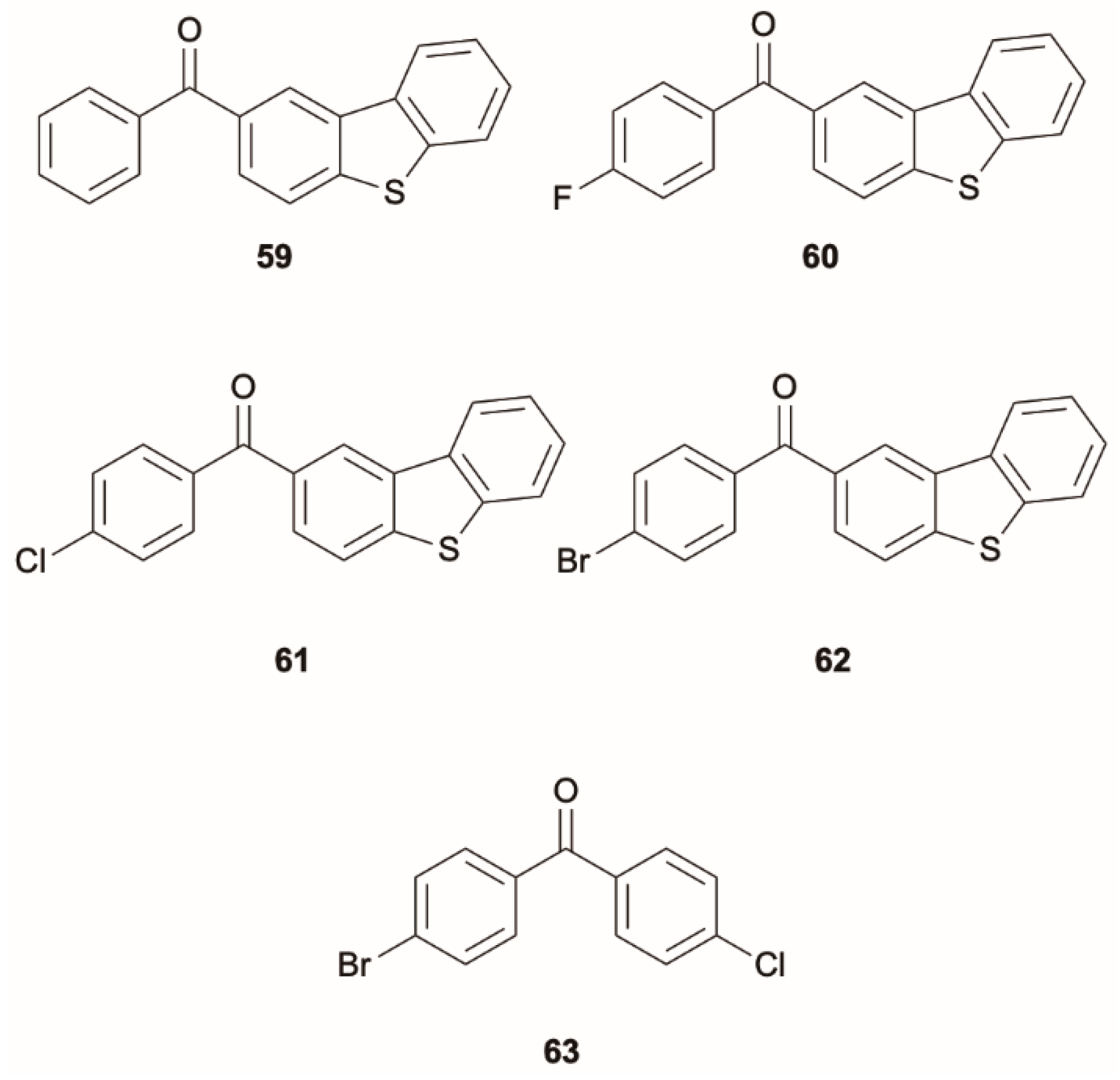
He et al. [
62] proposed a design strategy to develop pure-phosphorescent single-molecule white light emitters by incorporating benzophenone derivatives into the dibenzothiophene unit. By Friedel–Crafts acylation, five compounds, namely dibenzo[
b,
d]thiophen-2-yl(phenyl)methanone (
59), dibenzo[
b,
d]thiophen-2-yl(4-fluorophenyl)methanone (
60), dibenzo[
b,
d]thiophen-2-yl(4-chlorophenyl)methanone (
61), dibenzo[
b,
d]thiophen-2-yl(4-bromophenyl)methanone (
62) and 4-bromo-4′-chlorobenzophenone (
63) (
Scheme 19) were synthesized, for which phosphorescence was expected to be triggered by the presence of both the carbonyl group and the heavy atom. Moreover, the dibenzothiophene moiety should favor dual phosphorescence by providing various triplet excited states with different molecular orbital configurations. The derivatives display CIE behavior showing weak luminescence in solution and intense emission as crystals at ambient conditions arising from two phosphorescent contributions: a fast one at 470 nm (τ = 0.06–0.71 ms), which has been attributed to a radiative decay from T
2, and a long-lived one at about 570 nm (τ = 104–123 ms) due to emission from T
1 (see
Figure 15). The assignment of the fast decay to a phosphorescence rather than a TADF was supported by low-temperature measurements, revealing an increase in lifetime with a decrease in temperature. Moreover, quantum mechanics/molecular mechanics (QM/MM) studies on selected compounds, where the QM region was treated at B3LYP/6-31G(d) level for both DFT and TDDFT calculations, provided two lowest excited states (T
1 and T
2) below S
1, with large S
1-T
1 separation (in the 0.72–0.88 eV range), suggesting that TADF is less likely to occur at RT. The key point for successfully obtaining dual phosphorescence relies on the phenyl group shared between benzophenone and dibenzothiophene moieties providing, respectively, (
n, π*) and (π, π*) character to the transitions. As a result, although both T
1 and T
2 states have mixed (
n, π*) and (π, π*) configuration, T
2 is mainly an (
n, π*) transition while T
1 possesses more (π, π*) character. According to the El-Sayed rule, a larger SOC is expected for T
2-S
0 than for T
1-S
0. This leads to a short lifetime for T
2 and a long lifetime for T
1. The anti-Kasha emission might then occur after the thermal population of T
2 from T
1 due to a small ΔE(T
1-T
2) (0.19 and 0.27 eV from experiment and theory, respectively, for
61), comparable to RT thermal energy.
Later on, Paul et al. [
63] applied computational techniques to re-examine the previously reported anti-Kasha behavior of
61 [
62]. According to the new calculations, performed by DFT and TDDFT approaches at the Grimme’s dispersion corrected B3LYP/6-311G(d,p) level of theory, the ΔE(T
1-T
2), 0.48 eV, is too large to obtain a sufficient Boltzmann population of T
2 from T
1. Therefore, they proposed that T
2 is instead populated by S
1-T
2 ISC. These two levels are in fact separated by only 0.09 eV and are characterized by strong Duschinsky mixing [
64] between their normal modes.
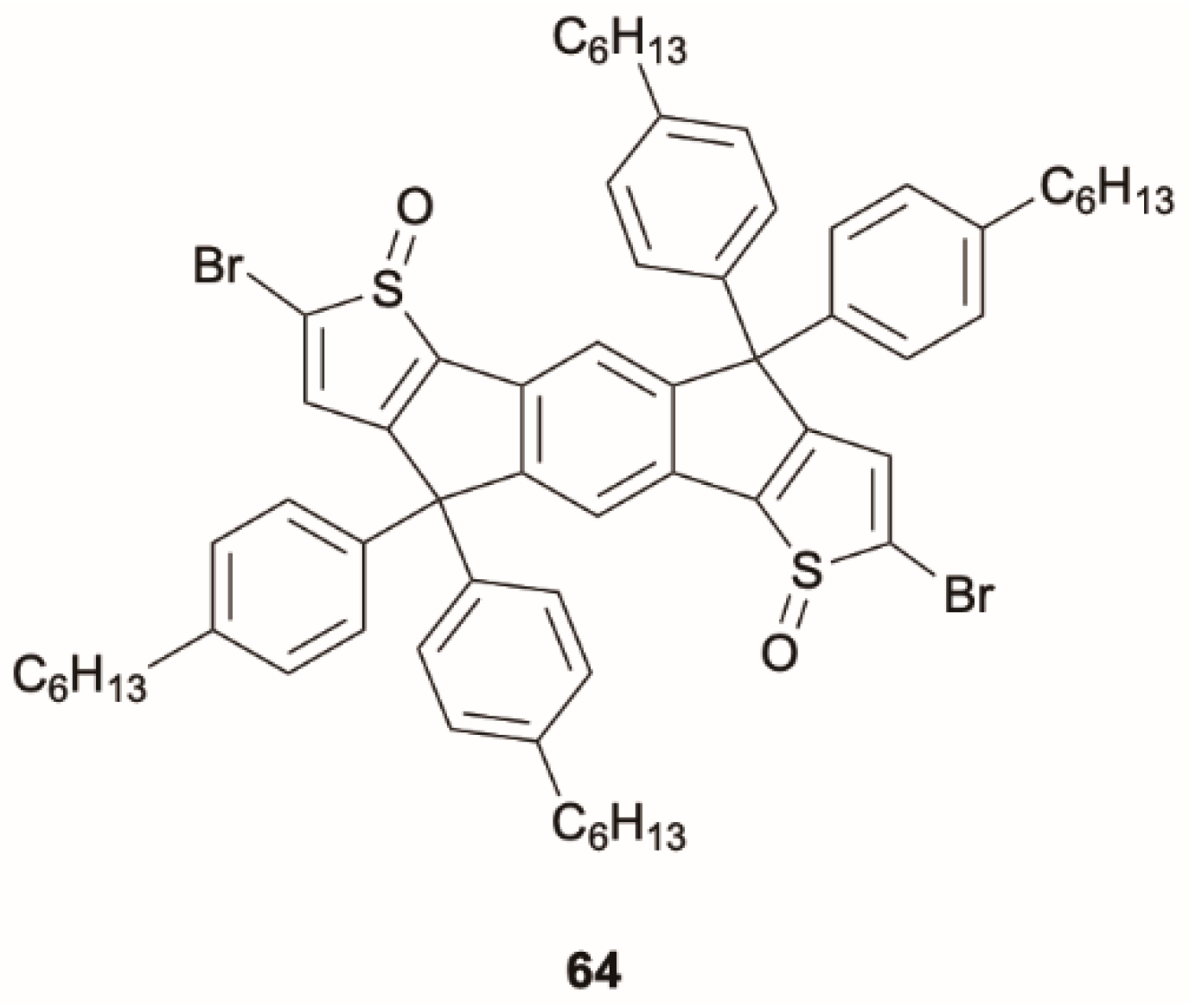
Cao et al. [
65] prepared and investigated a series of highly planar indaceno[1,2-
b:5,6-
b′]dithiophene 1,6-dioxide derivatives containing out-of-plane side chains aimed at preventing quenching of the emission due to strong intermolecular π–π interactions. Among the studied compounds, film of
64 (
Scheme 20) displays anti-Kasha phosphorescence.
Diluted chloroform solution of
64 displays, when excited at 380 nm, a vibrationally resolved emission originating from a locally excited state at 400–500 nm and an additional broad band of ICT character at 500–700 nm. The film of
64 shows excitation-dependent photoluminescence. In particular, upon excitation at 340 nm, a fluorescent band at 570 nm appears in the spectrum. While, at 253 nm excitation, dual emission comprising a phosphorescence at 400 nm (τ = 0.102 ms) and a fluorescence at 570 nm (τ = 0.812 ns) is observed. The phosphorescence was attributed to anti-Kasha deactivation originating from a high-energy triplet level. In agreement, DFT-TDDFT calculations on a model molecule where hexyl was replaced by methyl, performed at B3LYP/6-31G(d) level for S
0 and S
1 and at M06-2X/6-31G(d) level for higher singlet states and for triplet states, revealed small S
2–3-T
3–4 energy gaps and a large T
3-T
2 one. This justifies an easy ISC to populate high triplet levels from which phosphorescence is observed due to inhibited IC. Therefore, it was suggested that upon excitation to S
1, only the corresponding fluorescence at 570 nm is observed. However, by populating upper excited states (S
n), relaxed S
1 (by IC) and T
n (by ISC) are obtained, from which fluorescence and anti-Kasha phosphorescence are respectively produced (see
Figure 16).
8. Miscellaneous
Wilson et al. [
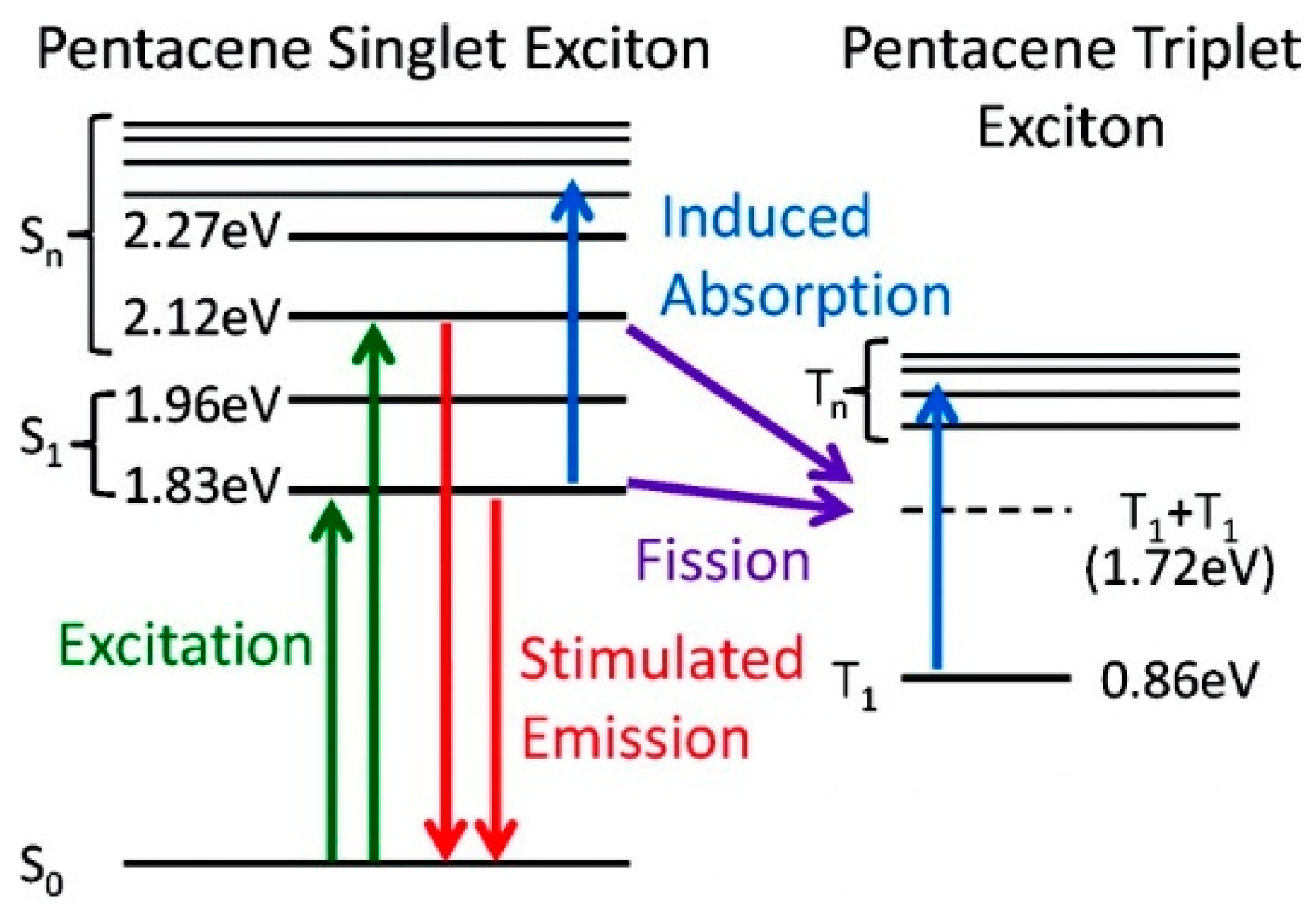
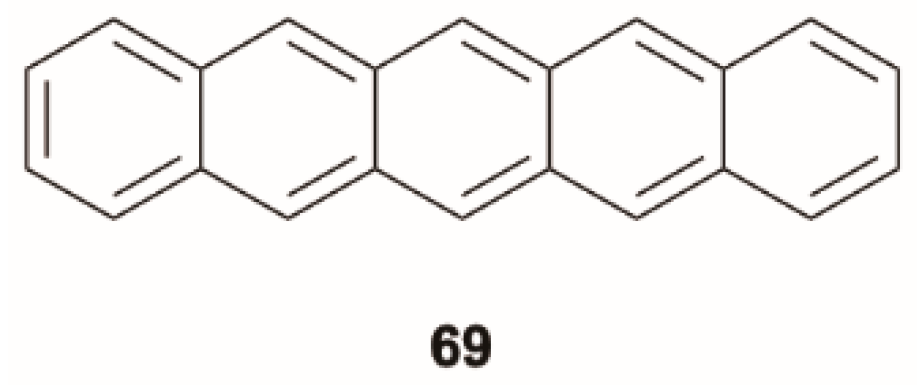
69] probed the process of exciton fission in polycrystalline thin films of pentacene,
69 (
Scheme 23), by ultrafast TA spectroscopy. The authors monitored the singlet population through the detection of stimulated emissions (SEs), which were found to be excitation-dependent. The anti-Kasha blue-shifted SE obtained by excitation of higher-lying transitions was attributed to light emitted by the “hot” exciton before its relaxation to S
1 (as suggested by the red arrows in
Figure 17). The dynamics of triplet generation indicated that the singlet fission process does not require first relaxation to S
1 but can proceed directly from higher-lying singlet states on a similarly rapid (~80 fs) time scale. These observations imply that pentacene S
1 state is short-lived, even on the time scale of excitonic cooling, in agreement with the very low luminescence efficiency of pentacene crystals.
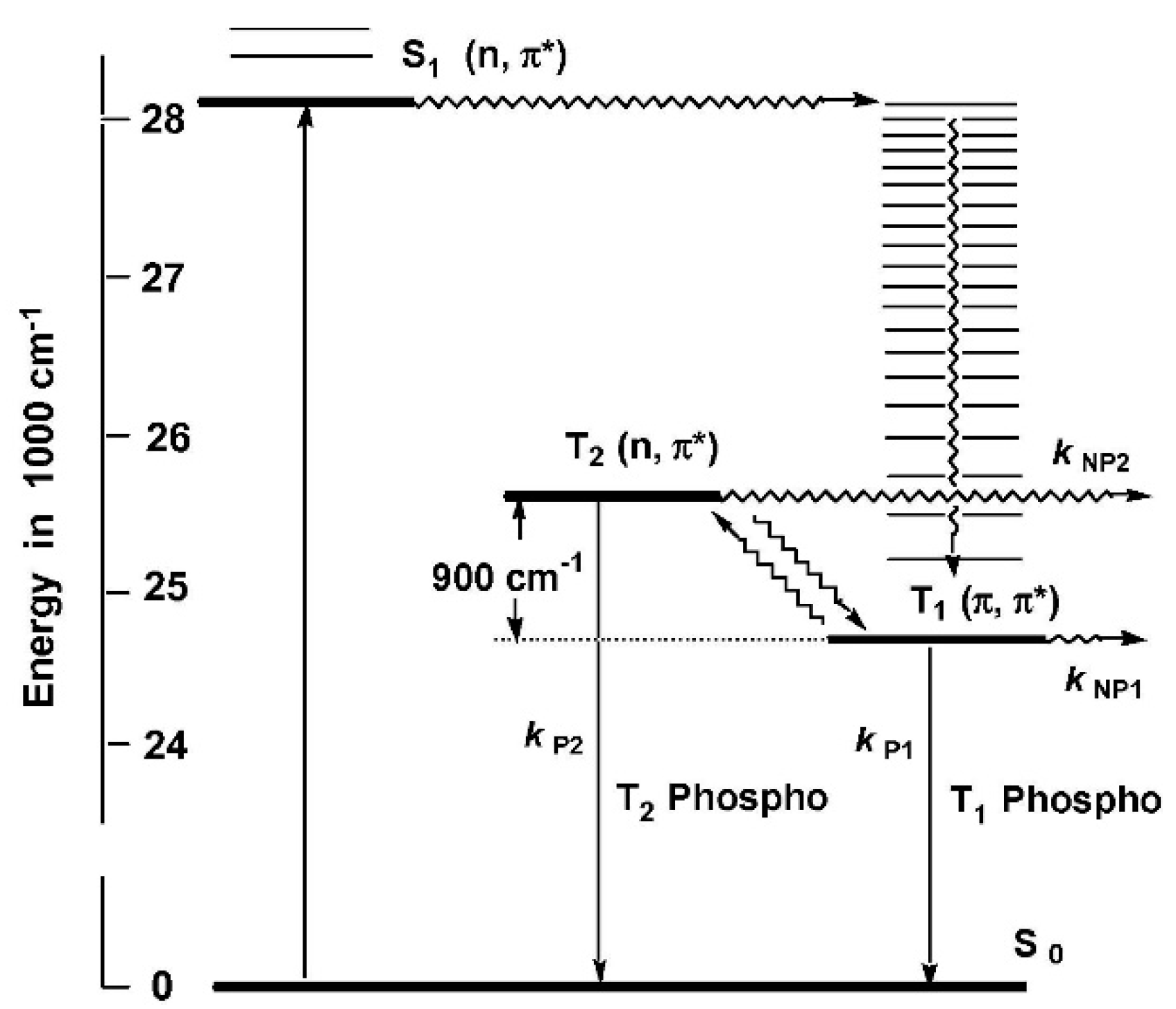
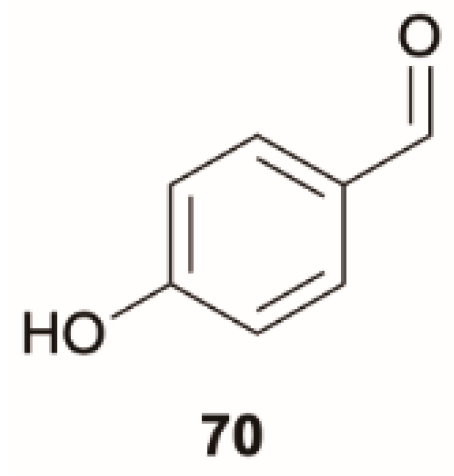
Some derivatives of benzaldehyde [
9] were reported to emit from T
2 due to its closeness to T
1 and to the higher S
0-T
2 oscillator strength with respect to the S
0-T
1 one. However, for hydroxybenzaldehyde,
70 (
Scheme 24), despite the S
1 (
n, π*) > T
2 (
n, π*) > T
1 (π, π*) excited-state ordering being favorable to observing phosphorescence from T
2, only phosphorescence from T
1 was observed in a rigid matrix at 77 K [
9]. In 2014, Itoh [
70], speculating that anti-Kasha emission from T
2 could be missed due to its weakness, performed a detailed steady-state and time-resolved investigation of
70 in a 1,4-dichlorobenzene matrix in the −196 to −20 °C interval. At 77 K the only phosphorescent emission at 405 nm was assigned to the T
1 (π, π*) state. At −70 °C an additional phosphorescent contribution at 391 nm of T
2 (
n, π*) origin, with intensity increasing with temperature up to −20 °C, was observed. A ΔE(T
1-T
2) equal to 0.11 eV was estimated from both steady-state and time-resolved experiments which allowed the drawing of the mechanism reported in
Figure 18 in which the thermal population of T
2 from T
1 justifies the observed emissive behavior.
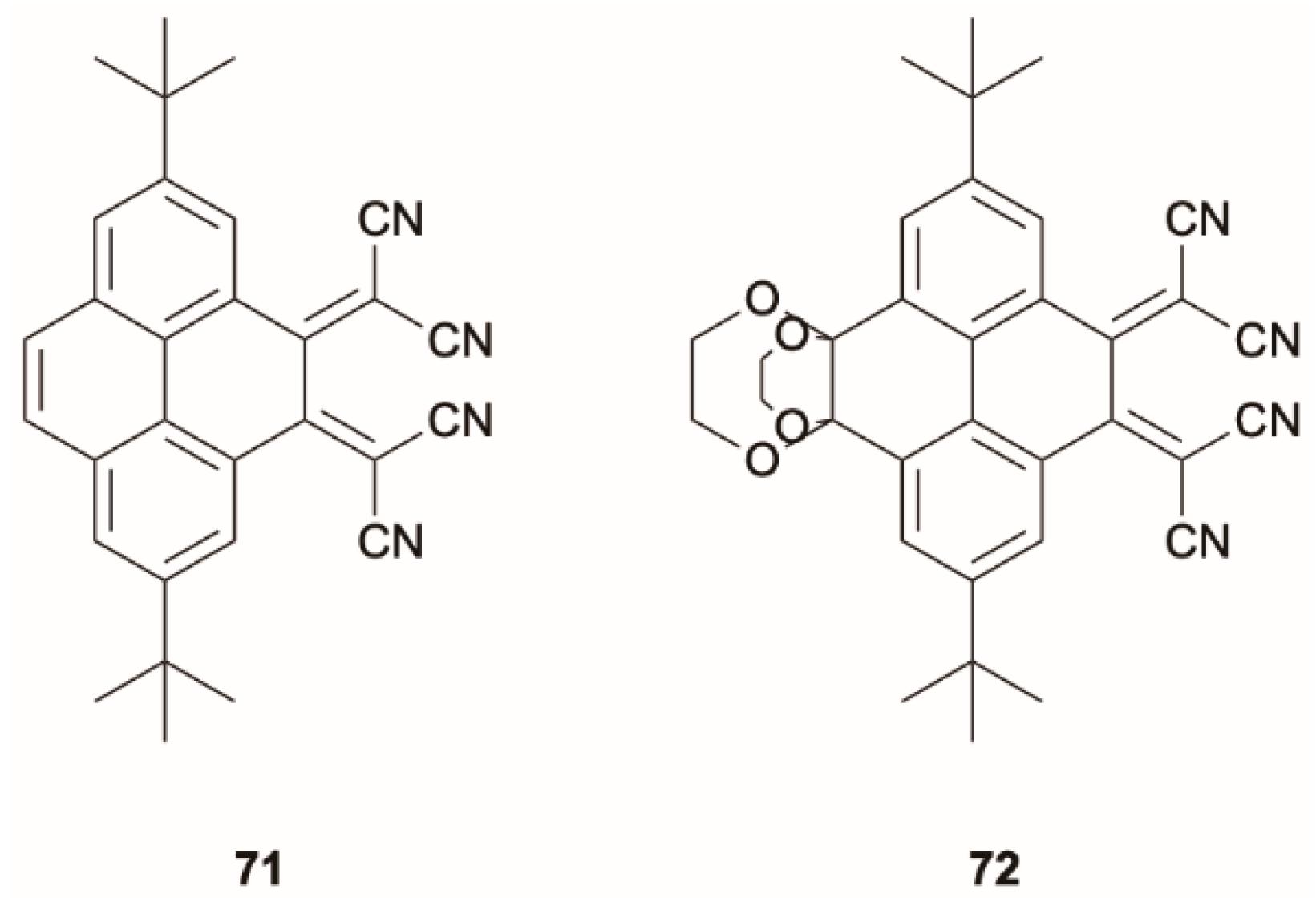
While pyrene, previously reported as an exception to Kasha rule [
9], was recently demonstrated [
14] to emit uniquely from S
1, the introduction of dicyano methane substituents on the pyrene scaffold to generate compounds
71 and
72 (
Scheme 25) promotes, in DCM solution, an intense ICT emission that originates from S
2, while no radiative deactivation is obtained upon S
0-S
1 excitation [
71] (see
Figure 19). Violation of the Kasha rule was attributed to the large S
1-S
2 energy gap (0.98 and 0.94 eV in
71 and
72, respectively, as determined by DFT and TDDFT calculations at B3LYP/6-31G(d,p) level) favoring emission from S
2 over S
2-S
1 IC as a consequence of the reduced overlap between vibrational levels of S
1 and S
2.
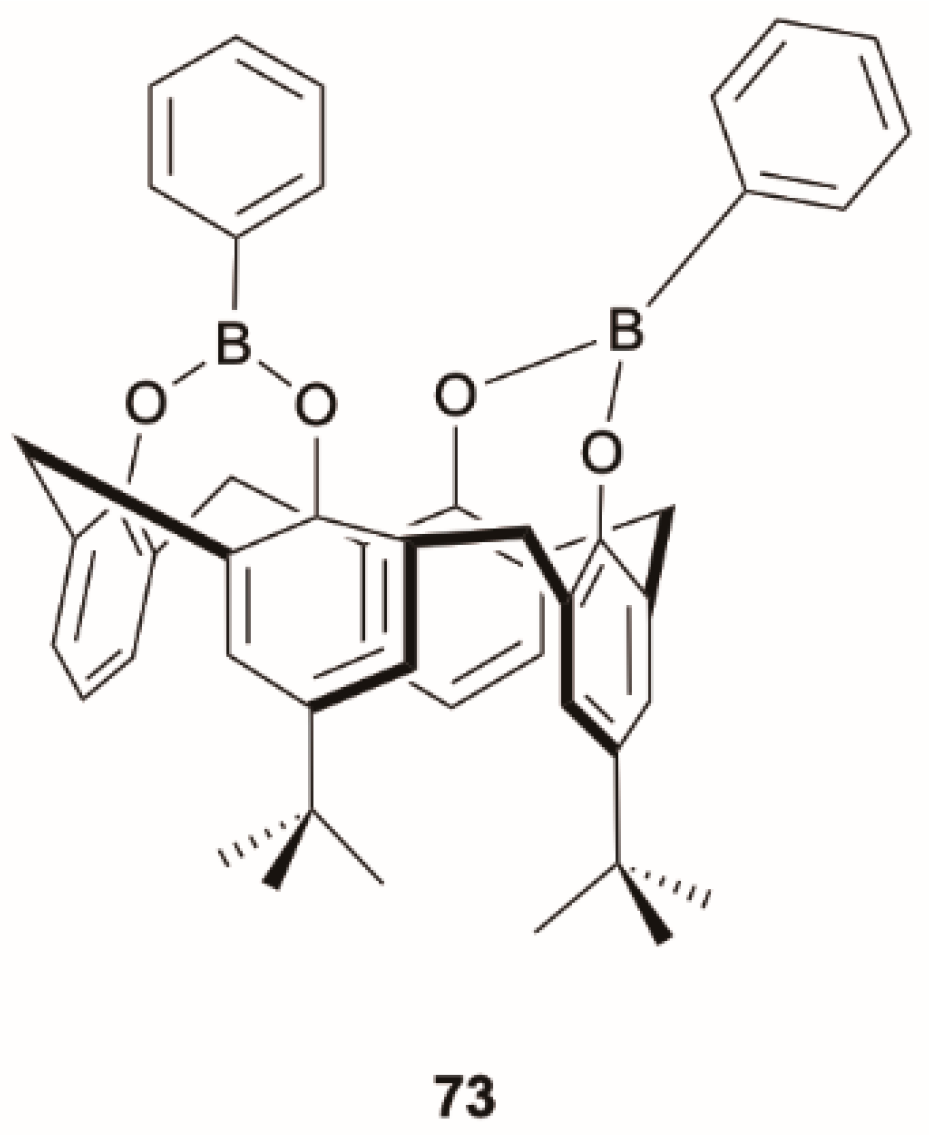
Bis(bora)calix[4]arene,
73 (
Scheme 26), was reported by Arimori et al. [
72] as a fluorescent sensor selective for fluoride due to a decrease in UV absorption and fluorescence upon F
− binding. In 2016, Jin et al. [
73] performed a thorough DFT/TDDFT study to interpret this behavior. Calculations were performed by using a GGA functional, PBE1W, which provided a better agreement with experimental data, and the 6-311G(d) basis set. The prompt fluorescence of
73 was assigned to emission from S
2 owing to a sizable ΔE(S
1-S
2) gap (0.48 eV) and very low S
0-S
1 oscillator strength. The quenching of fluorescence observed for the fluoride-binding complex
73-F was explained by a theoretically predicted fast IC to a dark S
1 state.
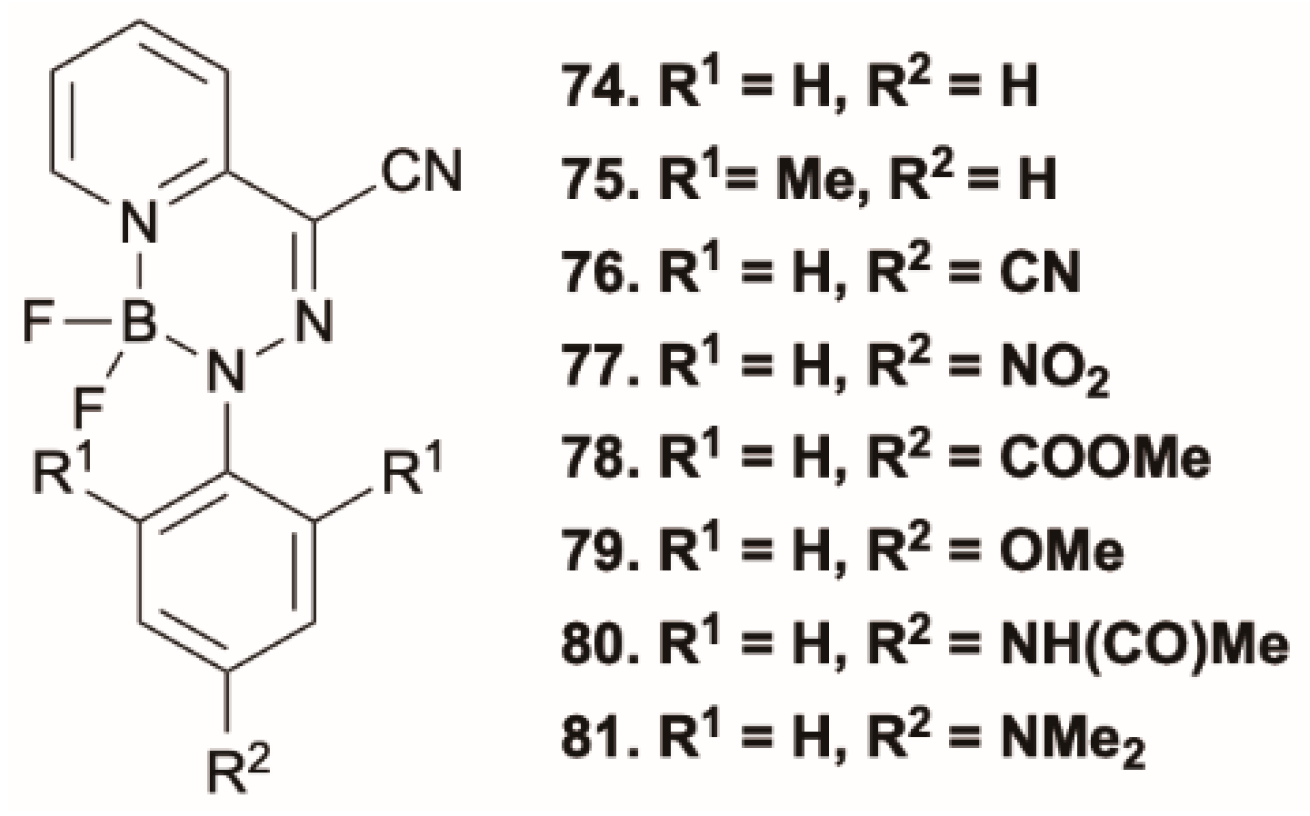
Qian et al. [
74] reported on a series of boron difluorohydrazone derivatives,
74–
81 (
Scheme 27), showing enhanced emission in the solid state and in highly viscous environments and a quenched emission in nonviscous solvents. Based on DFT/TDDFT calculations at the PBE/TZVP level, the authors attributed this behavior to the presence, in these molecular rotors, of an S
1 dark state and of a higher excited state, S
n (with
n > 1, varying within the series of compounds), having high oscillator strength. In nonviscous solvents, the free intramolecular rotation in the excited state leads to a conformation having a low S
n-S
1 energy gap, which allows rapid S
n-S
1 IC and then very weak emission or nonradiative decay to S
0. Highly viscous solvents, on the other hand, inhibit the formation of the nonradiative excited-state structure, decreasing the rate of S
n-S
1 IC and then resulting in anti-Kasha emission from S
n. Such behavior was supported by a strict correlation between the viscosity sensitivity of the fluorescence and the computed barrier to rotation in the excited state: the higher the barrier, the greater the viscosity sensitivity. Moreover, the anti-Kasha emission was experimentally confirmed in
81 by the observation of different absorbance and circular dichroism band shapes at 77 K, a feature that can be explained by the existence of two or more distinct electronic transitions. Furthermore, a weak emission emerged when excitation was shifted towards lower energies, indicating that the molecule is relaxing from the dark state S
1.
Subsequently, Zhou et al. [
75], contesting the use of the pure functional PBE adopted by Qian et al. [
74] for
74–
81, performed a new TDDFT study on the same series of compounds using the B3LYP and CAM-B3LYP functionals, besides complete active space self-consistent field (CASSCF) calculations. It was reported that S
1 for the same series of compounds
74–
81 is not a dark but a bright state, which calls into question the anti-Kasha origin of the enhanced emission of these compounds in viscous environments. The authors proposed an alternative origin, based on restriction, in viscous solvents, of flip-flop motion promoting fast S
1-S
0 IC. Confutation of the previously proposed anti-Kasha behavior of these compounds, however, still deserves experimental validation.
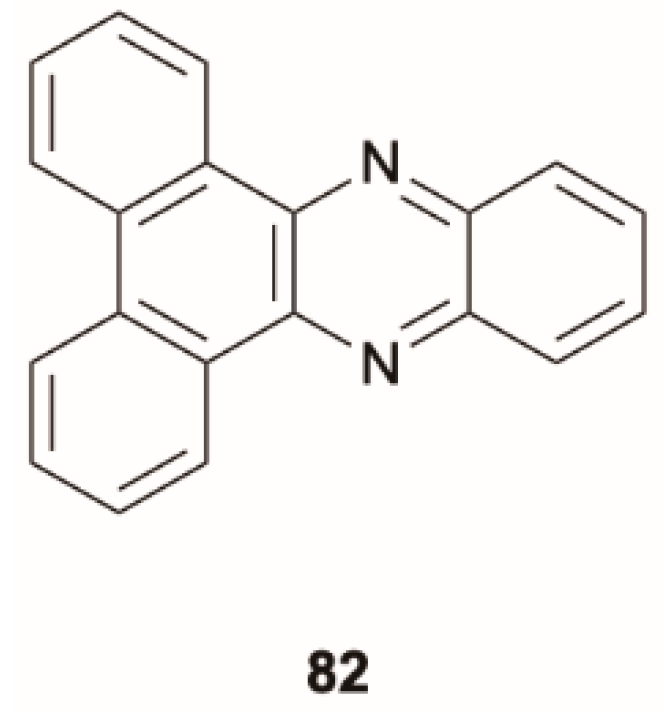
Zhou et al. [
76] reported single-molecule white light emission from dibenzo[
a,
c]phenazine,
82 (
Scheme 28). In solution,
82 displays steady-state PL dominated by a structured phosphorescence with a lifetime of 2.7 μs, accompanied by a fluorescent emission. This latter was associated to an S
1 state with (
n, π*) character while the phosphorescence was assigned to a (π, π*) T
1 level, with S
1-T
1 ISC facilitated by El-Sayed considerations (see
Figure 20), as confirmed by the very short fluorescence lifetime of
82 (0.07 ns) [
77]. Powders of
82 display multiple emissions comprising one S
1-S
0 fluorescence and two RTPs, reaching Commission Internationale de l’Éclairage (CIE) coordinates of (0.28, 0.33) and Φ of 1%. From steady-state measurements at variable temperature and time-resolved studies, the authors demonstrate that the two RTPs are relatively independent of each other and excluded a T
1-T
n thermally activated reversed IC. With the support of TDDFT calculations, performed at the B3LYP/6-31G(d,p) level of theory, the high-energy RTP was assigned to a T
2 state of (
n, π*) character. T
2-T
1 IC competes with T
2 radiative deactivation, leading to the dual phosphorescent emission. The excitation wavelength dependency of the RTPs demonstrated that the high-energy one could be efficiently generated through multiple alternative ISC channels (S
2-T
2 and S
1-T
2, see
Figure 20c).
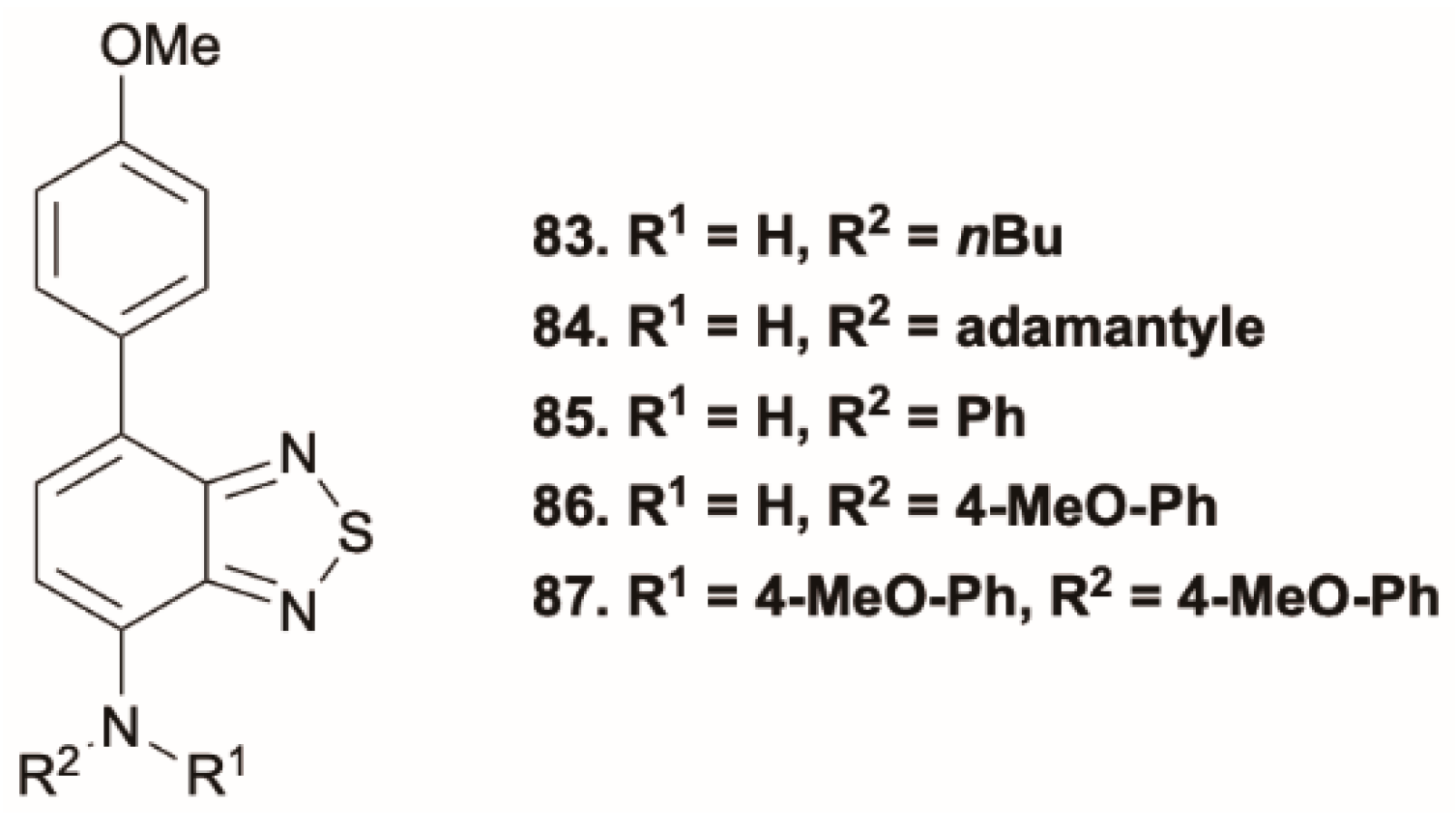
Peng et al. [
78] investigated the photophysical properties in different states of a series of amino benzothiadiazoles. Anti-Kasha dual emission was observed in solution for
83–
87 (
Scheme 29) and was correlated to the strength of the electronic communication between the amino group and the benzothiadiazole moiety. In fact, by increasing the CT character of the molecule, the molecular rigidity is increased and S
2-S
1 IC is inhibited. For
83 and
84, having the strongest CT character and the highest planarity among the members of the family, excitation-dependent dual emission in the visible region was observed both in toluene and acetonitrile solutions. Compounds
85 and
86, characterized by reduced electron-donating ability from the amino group, display dual emission only in acetonitrile where the ICT is enhanced. For
87, dual emission appears in both solvents but is hardly visible in acetonitrile.
Sun et al. [
79] and Liu et al. [
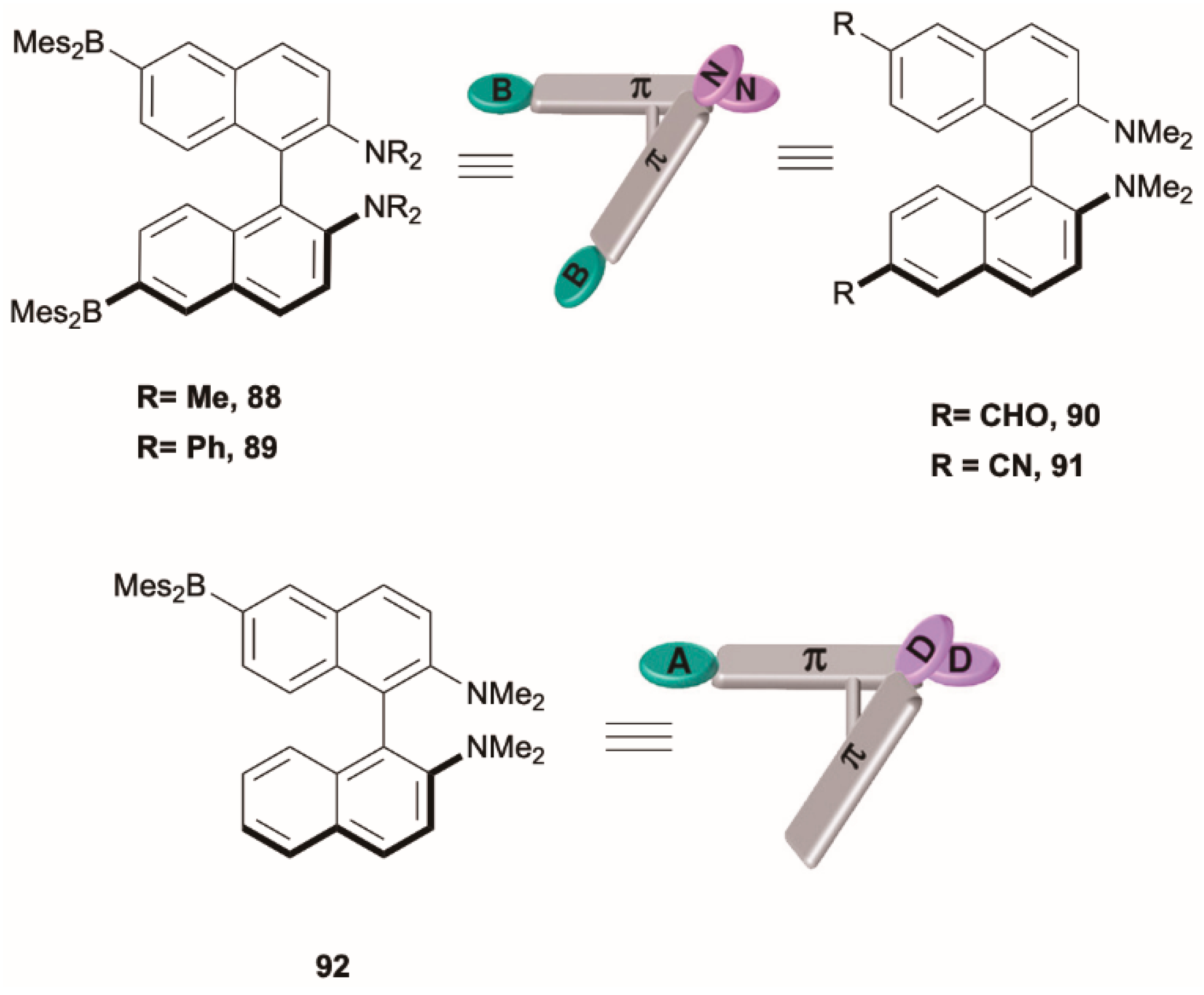
80] revealed a family of highly twisted
o,
o′-substituted binaphthyls,
88–
92, featuring a D–π–A structure with ICT character within each aryl group (
Figure 21). These compounds, unlike the “normal” D–π–A molecules, consist of two independent D–π–A subunits, each of them bearing either dimesitylboryl, Mes
2B (
88,
89,
92) or CHO (
90) or CN (
91) groups as strong electron acceptors, and NR
2 groups, with R = Me (
88,
90–
92) or Ph (
89) as donors. All these compounds revealed temperature-dependent dual fluorescence and switchable circularly polarized luminescence (CPL). Comparison between
88 and
89 [
79] indicates that, in the presence of the less bulky NMe
2 group (
88), the two fluorescence bands are well separated in polar solvents and are very sensitive to temperature, enabling the use of
88 as a ratiometric fluorescence thermometer. On the other hand, for
89, the dual emission was observed only in the CPL spectra, probably due to the overlap of the two fluorescences. Combining experimental and theoretical results, the latter obtained by DFT and TDDFT calculations at the PBE0/6-31G(d) level of theory, the dual fluorescence of
88 was assigned to deactivation from S
1 and S
2. The computed excitation energies of these two states were very similar (ΔE = 0.07 eV), justifying a thermal equilibrium population between them which explains the observed temperature-dependent dual fluorescence. Moreover, the optimized geometries of S
1 and S
2 reveal different features, S
1 having mainly inter-subunit CT character and S
2 having mixed intra-subunit CT and LE character. Calculations on
89 provided essentially identical features; owing to its higher structural rigidity with respect to
88, only small changes are observed in the optimized S
1 and S
2 geometries with respect to the ground state. This explains the observed overlap of the S
1-S
0 and S
2-S
0 deactivations.
From comparative analysis of the effect of the A group in
88,
90 and
91 [
80], it appeared that the acceptor groups greatly affect the relative intensity and the energy separation of the two emissions. The reference compound
92, lacking one electron-accepting group, was found to possess less sensitive temperature-dependent dual fluorescence, indicating that the symmetric structure of
88–
91 is very important for realizing highly sensitive temperature-dependent dual fluorescence.
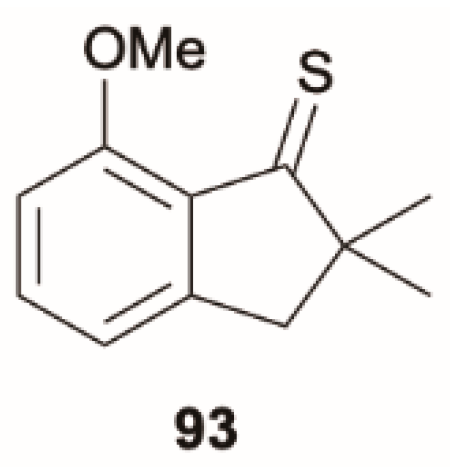
Liu et al. [
81] reported a study on intramolecular O–H
…S H-bond formation in a thione derivative which displays dual RTP due to a remarkable photoinduced intramolecular H-bond on/off switching reactions. The corresponding methylated compound,
93 (
Scheme 30), which lacks the dual RTP, represents instead an example of anti-Kasha fluorescence (see
Figure 22). In cyclohexane at RT,
93 displays dual emission comprising one fluorescence (at 380 nm) and an RTP (at 690 nm). Through a deep photophysical analysis, integrated with DFT and TDDFT calculations at the B3LYP/6-311++G(3df,3pd) level of theory, the authors assigned the first excited state, S
1, to an optically forbidden (
n, π*) transition and the higher-lying S
2 state to an allowed (π, π*) one. The calculated ΔE(S
2-S
1) energy gap, amounting to about 1.37 eV, justifies an anomalously slow S
2-S
1 IC, allowing the radiative S
2-S
0 transition to become competitive.
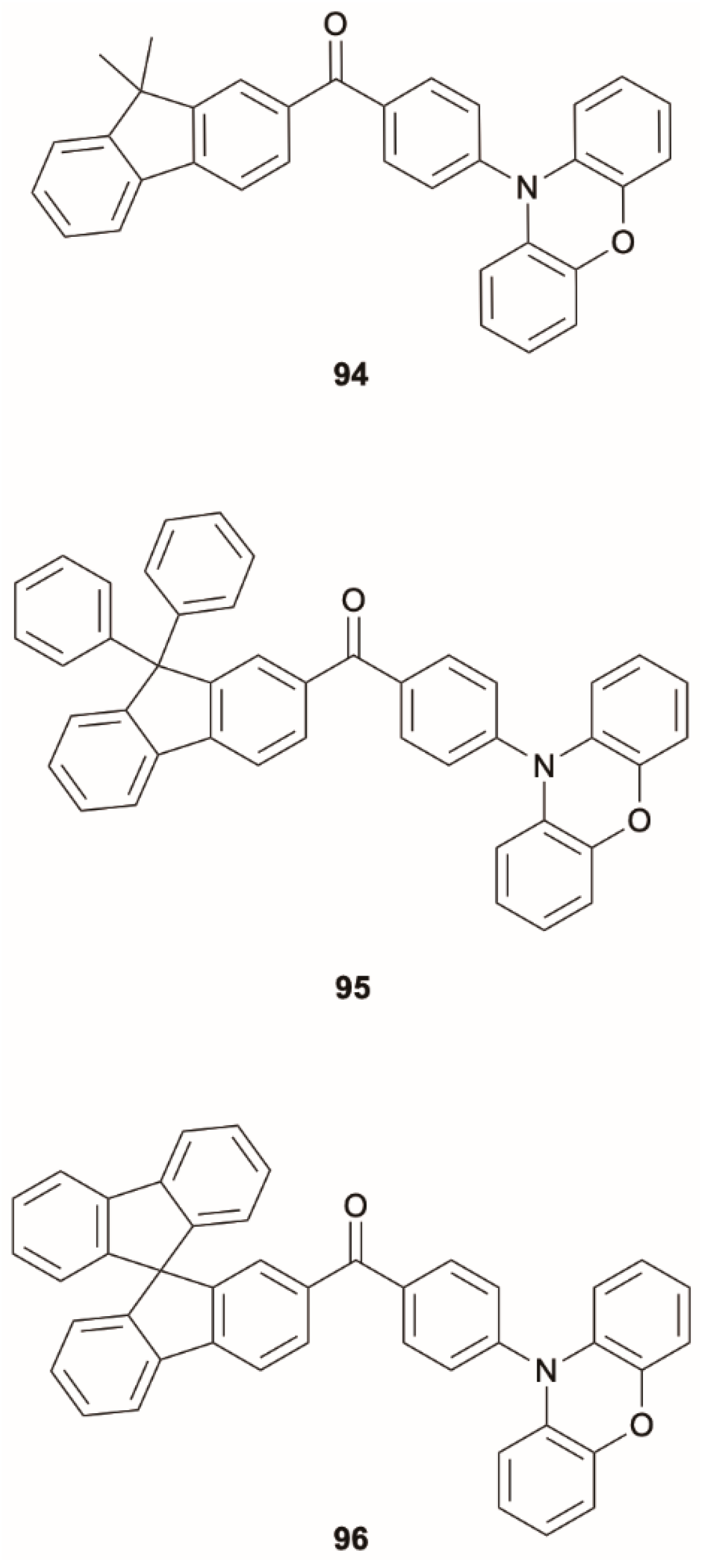
Guo et al. [
82] reported three donor-acceptor (D–A) compounds,
94–
96 (
Scheme 31), consisting of electron-donating phenoxazine and fluorene derivatives coupled with an electron-withdrawing benzoyl core, that are weakly emissive in solution but display DF upon aggregation. DFT/TDDFT calculations using the PBE0 functional were employed to rationalize the emission mechanism of
94 as the representative of the family. The obtained oscillator strength for S
1 was almost zero (
f = 0.0006), indicating that S
1 is nonemissive. On the other hand, the optimization of the S
2 geometry provided a relatively large
f value (0.0523), suggesting that the efficient emission of
94 corresponds to an anti-Kasha radiative deactivation from S
2.
Additionally, natural transition orbital (NTO) analysis, both in THF and in solid state, has been performed. For S
2, a typical CT state was predicted, while a significant LE feature was found for T
3. Moreover, the small (0.22 eV) S
2-T
3 energy gap calculated in solid state makes an rISC process highly favorable; thus, in the solid state, T
3 can be converted to S
2, supporting the subsequent anti-Kasha delayed emission (see
Figure 23). The full emissive mechanism in the solid state can be therefore explained by a reduced IC and a promoted ISC, with respect to the solution, resulting in the DF.
Such aggregation-induced delayed fluorescence (AIDF) appears particularly useful for OLED applications since the use of fluorescent emission, with respect to phosphorescence, allows the problem of efficiency roll-off to be reduced, while the AIE property of the compounds allows the fabrication of efficient nondoped devices. Therefore, the EL behavior of the compounds was investigated in nondoped and doped OLEDs, and external quantum efficiency, ηext, of up to 14.3% was obtained in the device of 95. At 1000 cd m−2 the device kept excellent ηext of 14.1%, and the external quantum efficiency roll-off was as small as 1.4%, indicative of the superior efficiency stability of the device based on these AIDF materials.
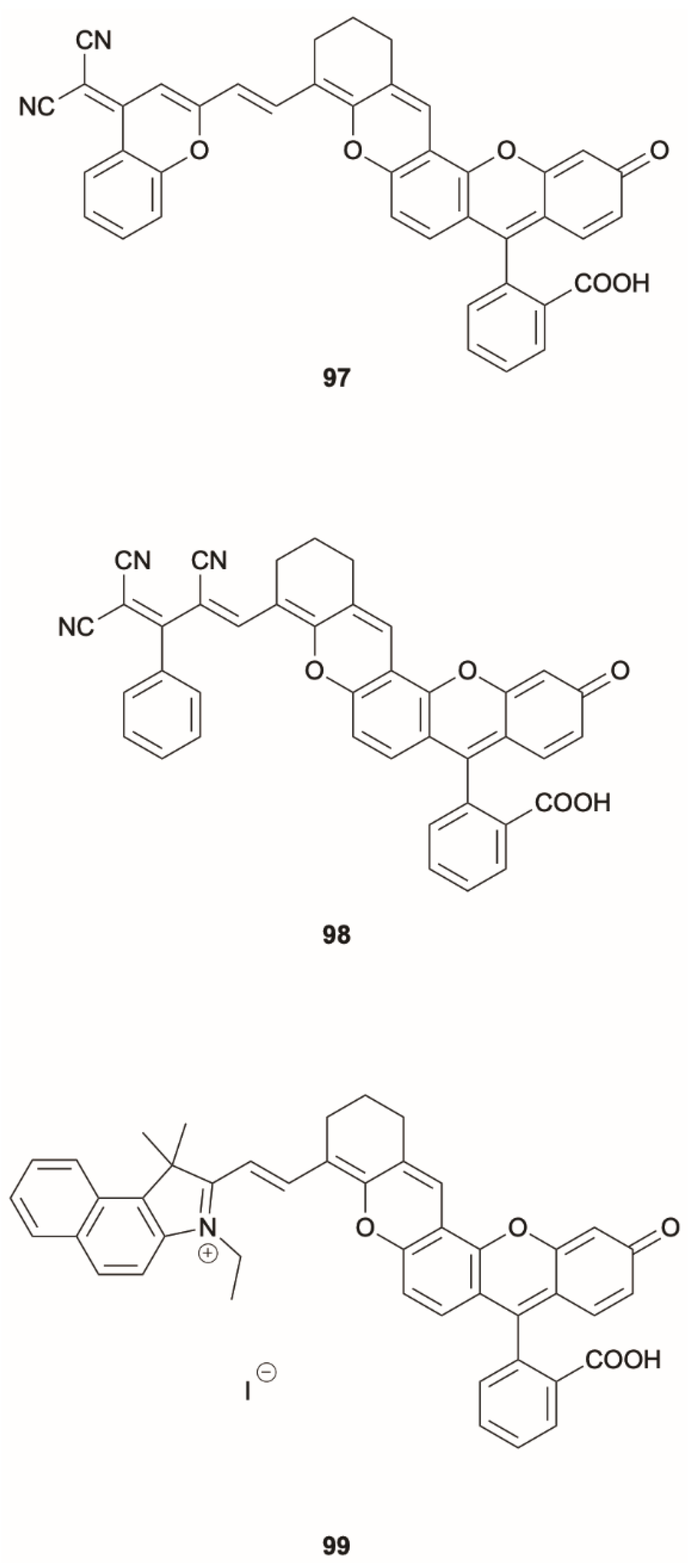
Shi et al. [
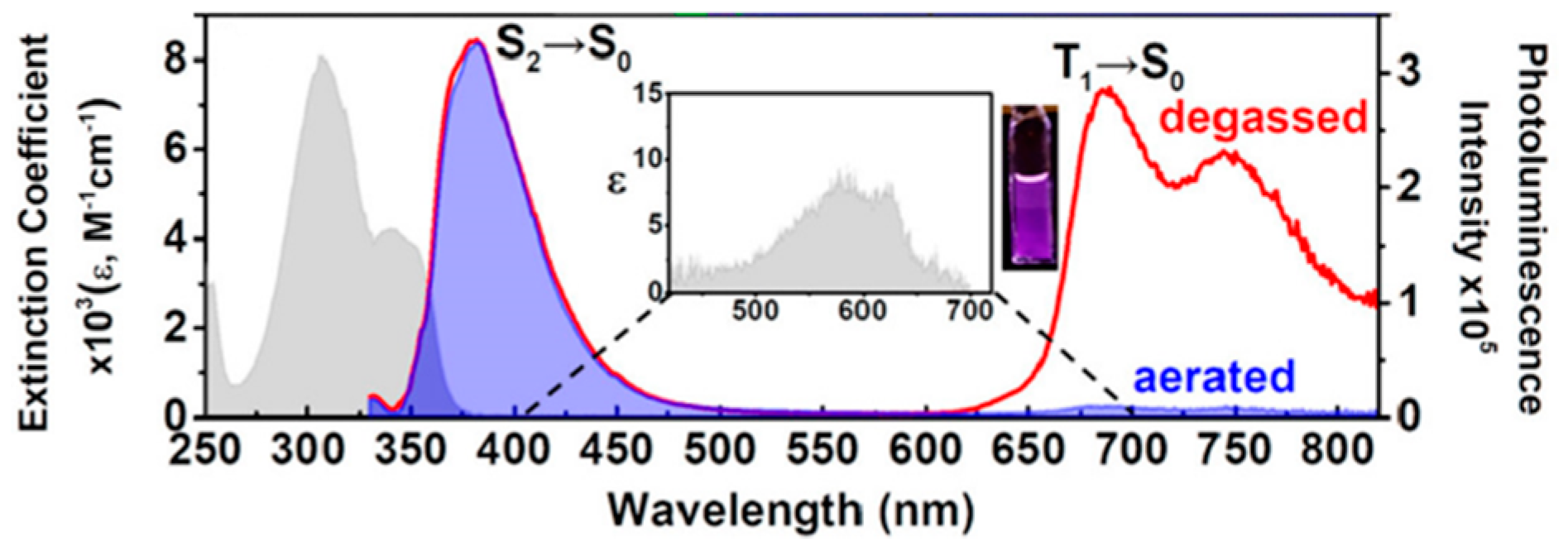
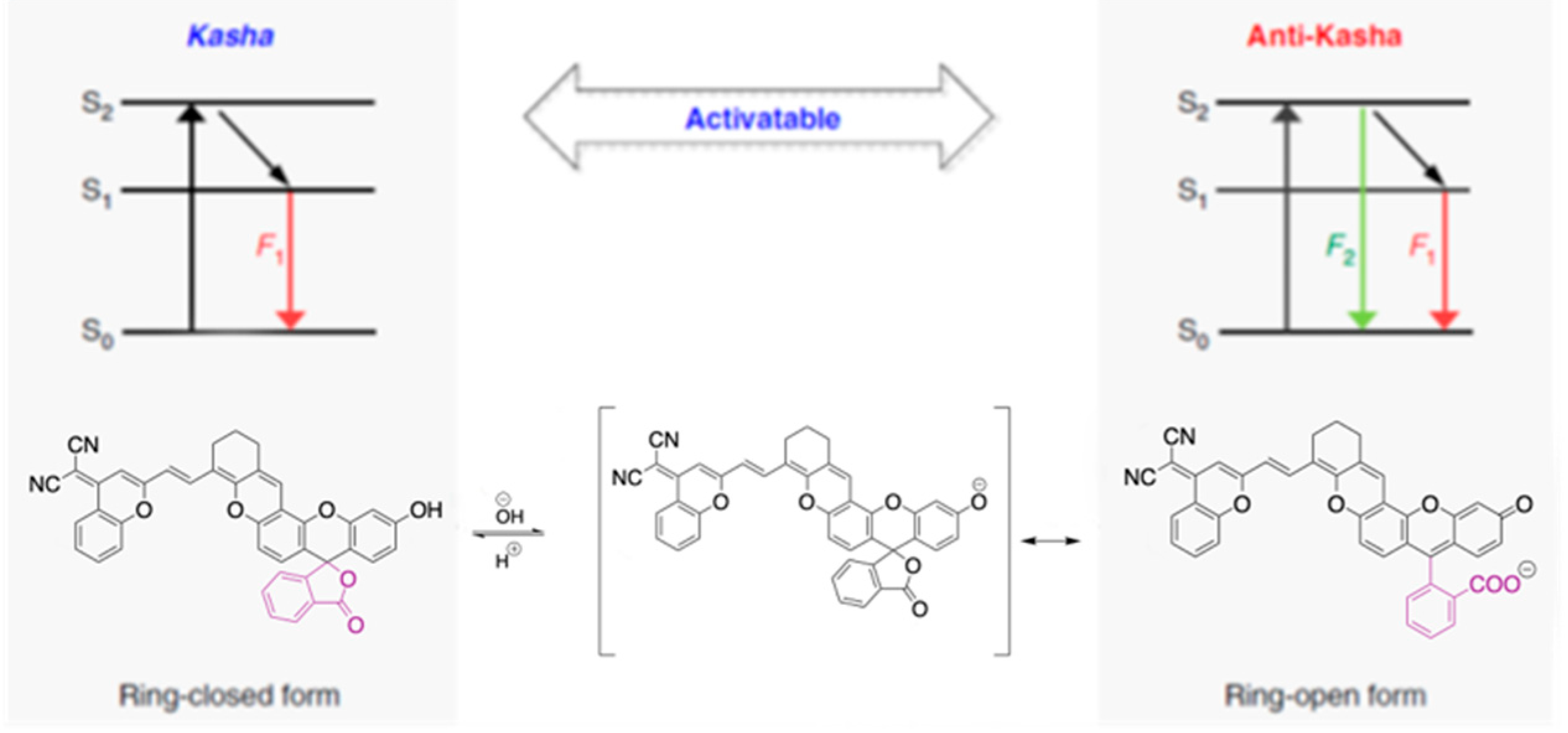
83] proposed a strategy to obtain ratiometric sensing for quantitative analysis of biomolecules in a dynamic cellular environment by using dyes able to switch from a close Kasha form to an open anti-Kasha one characterized by dual emission. In particular,
97 (
Scheme 32), where fluorescein is covalently linked to a chromene unit, displays in solution two fluorescences at 520 and 700 nm, assigned, through DFT-TDDFT studies at M06-2X/Def2-SVP level, to emissions from S
2 and S
1, respectively. According to the quantum chemical calculations, a ΔE(S
2-S
1) equal to 0.74 eV was determined. Such a high energy gap, together with the different S
2 and S
1 orbital localization, the former mainly on the fluorescein fragment and the latter on the chromene moiety, justify the slow S
2-S
1 IC resulting in the observed dual fluorescence. For the corresponding closed form of
97, only Kasha emission from S
1 was recorded (see
Figure 24).
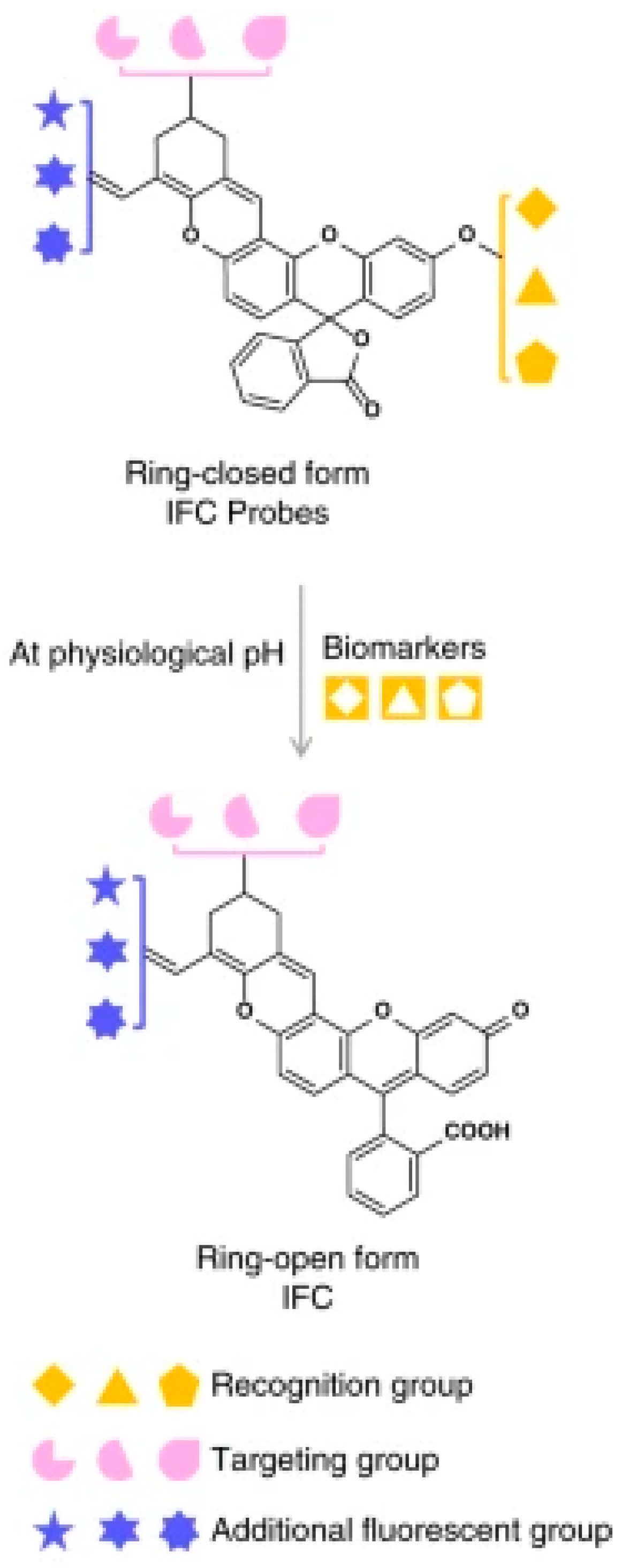
Starting from
97, similar compounds (
98,
99) (
Scheme 32) were prepared and studied. Through D–π–A structural modifications, the ΔE(S
2-S
1) gap can be modulated to provide diverse anti-Kasha/Kasha chromophores exhibiting an invariant S
1-S
0 NIR Kasha emission and an anti-Kasha visible S
2-S
0 one. In the physiological pH range, these chromophores remain in their open form, emitting from both S
2 and S
1. A quantitative sensing platform can be realized by replacing the hydroxy group with different biorecognition units (
Figure 25) When the probes in their closed form (with only the NIR emission from S
1) encounter the corresponding analyte, the hydroxyl group is restored, turning the probes to their open form at the desirable physiological pH, with both S
2 and S
1 emissions. The invariant NIR Kasha emission from the S
1 state acts as internal reference while the green emission from the anti-Kasha-active S
2 state linearly increases with the concentration of targeted analyte. Using this strategy, the authors demonstrated the ratiometric quantification of cysteine and glutathione in living cells and animals.
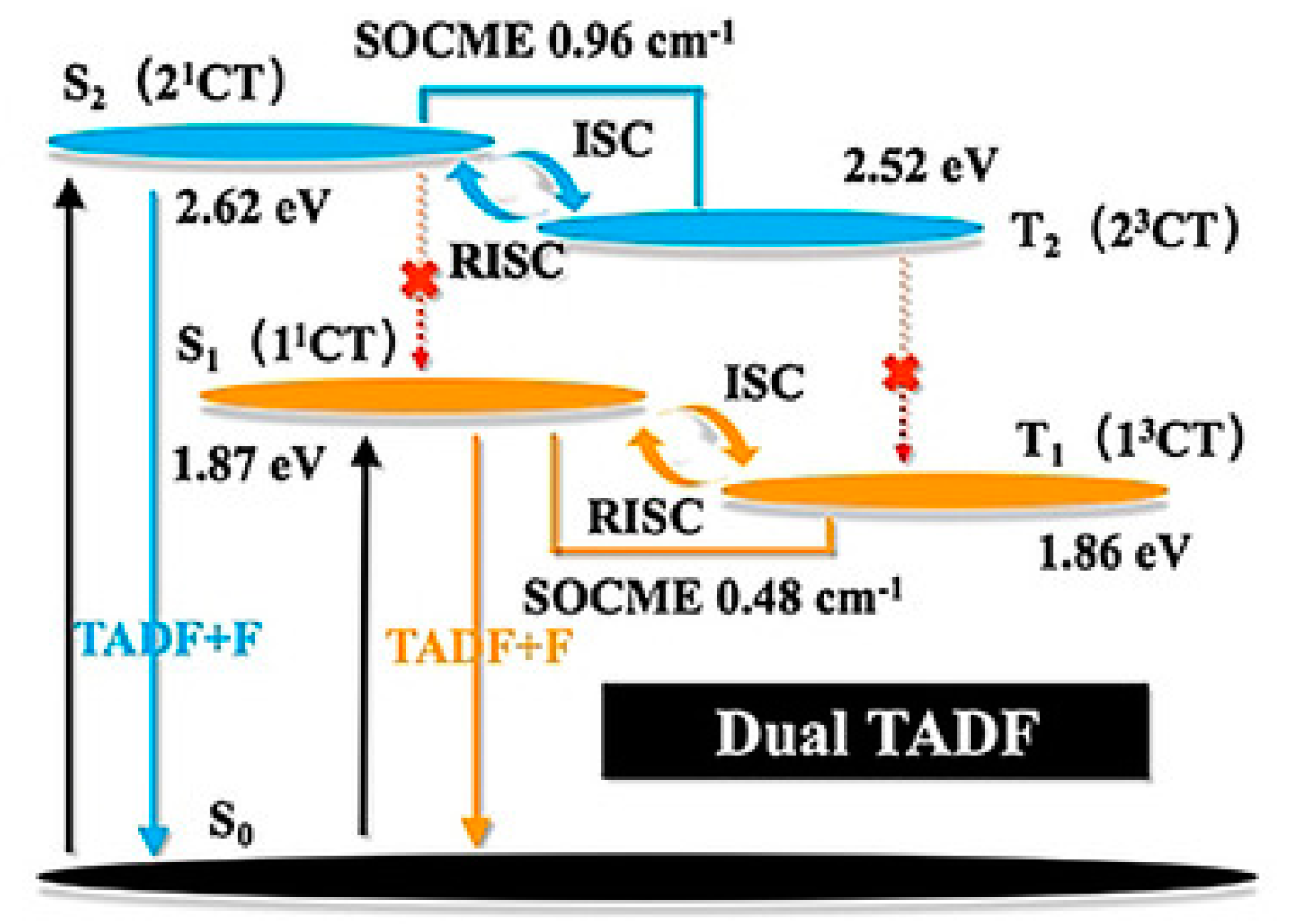
Luo et al. [
84] reported dual TADF emission in a single luminophore arising from anti-Kasha/Kasha pathways from different excited electronic states (see
Figure 26). The authors designed the unsymmetrical D–A–D′ molecular structure
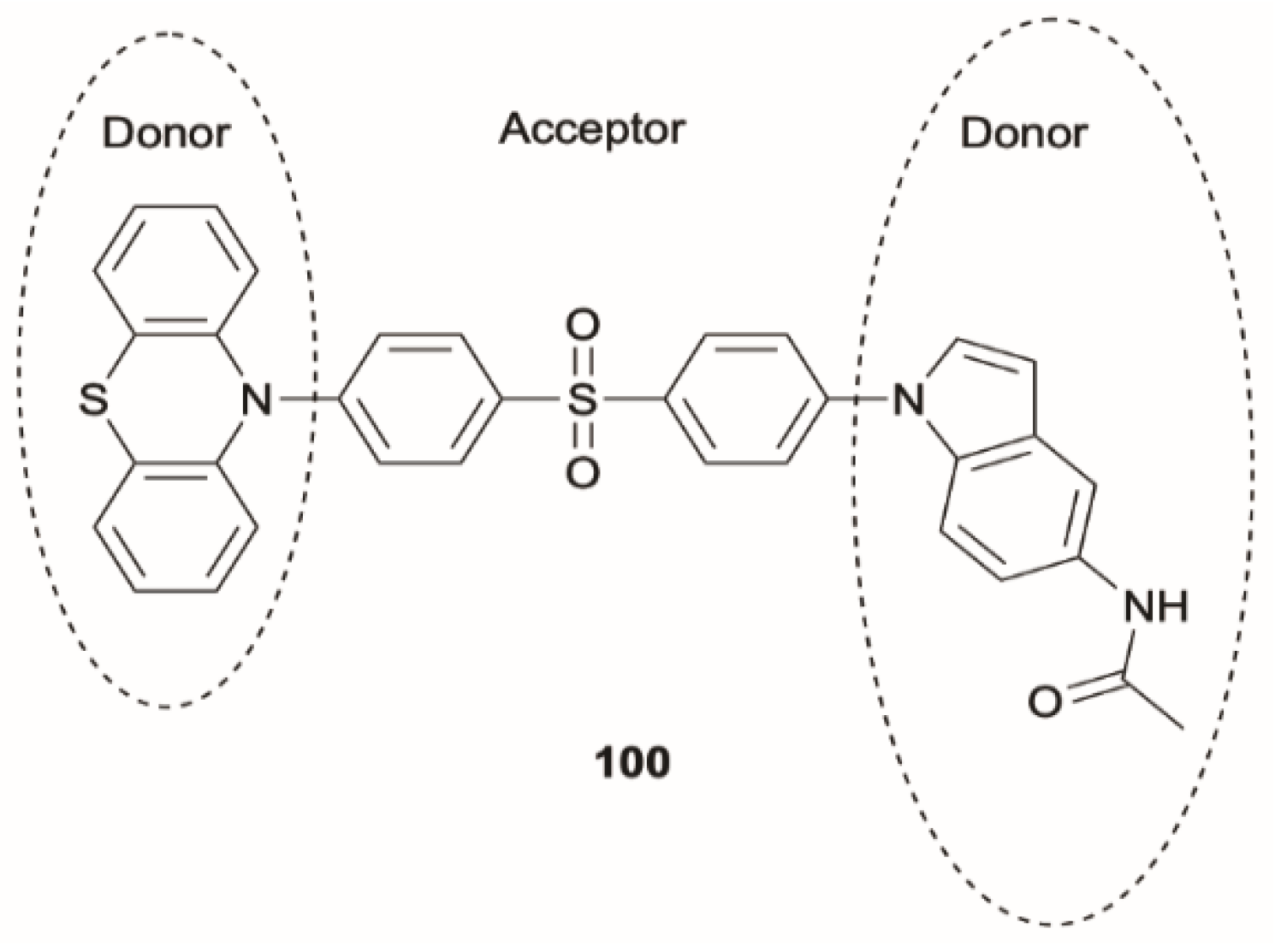
100 (
Scheme 33) (where D = phenothiazine, D′ = N-(1H-indole-5-yl) acetamide and A = diphenylsulfone) which possesses two CT states localized on the D–A and D′–A subunits of the molecule, as elucidated by means of B3LYP/6-31G(d) DFT and TDDFT calculations. For each CT singlet-triplet pair, the calculated energy splitting was found to be less than 0.1 eV, allowing, both in diluted solution and in the aggregated state (THF/water, solvent/nonsolvent mixture), very fast ISC and rISC rates that produce two independent DFs with different colors and lifetimes. Due to its favorable features,
100 was successfully employed in time-resolved fluorescence cell imaging. The main advantage of this probe was its capability to provide complementary dual-channel lifetime mapping that reduces time-resolved imaging distortions.
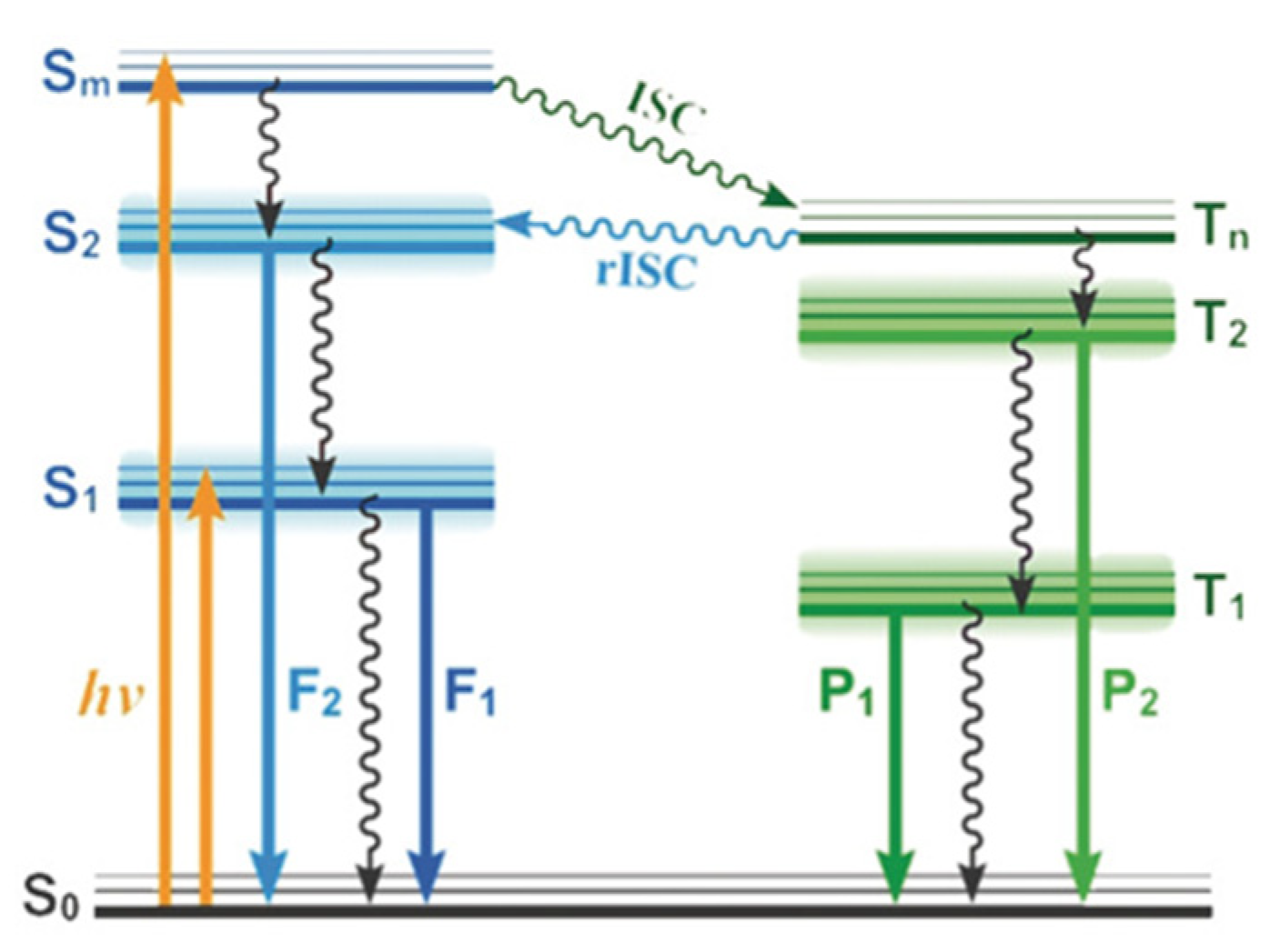
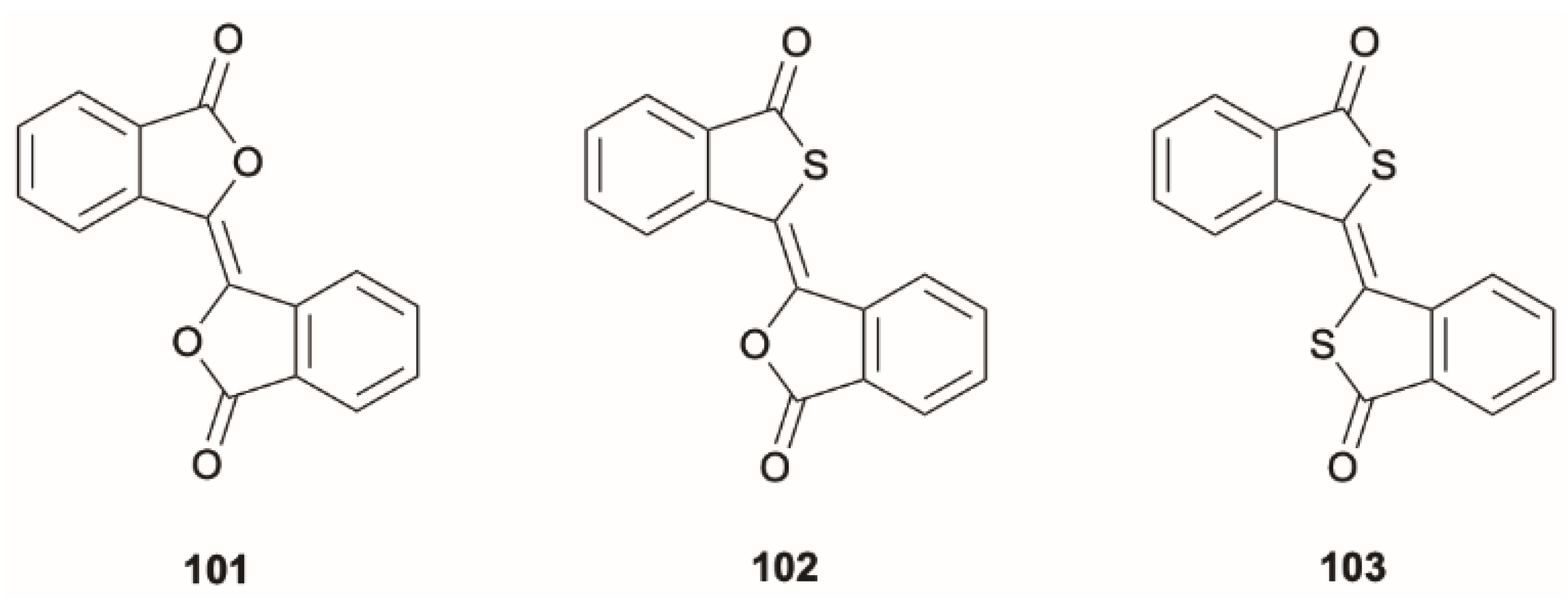
Wu et al. [
85] presented three heterocyclic 1,2-diphenyl ethylene derivatives,
101–
103 (
Scheme 34), that, at ambient conditions in the solid state, display excitation wavelength-dependent multiple emission, comprising prompt, delayed and RTP components originating from first and higher-energy excited singlet and triplet levels. The first and upper excited singlet and triplet states were responsible for the observed dual fluorescence and dual phosphorescence, as supported by QM/MM calculations with TDDFT excitation energies for the QM region computed at B3LYP/6-31G(d) level. In particular, prompt fluorescence from S
1 and prompt and TADF emission from S
2 were observed for all compounds, while dual T
2/T
1 RTP was recorded only for
101,
102 and
103 displaying single RTP from T
2 (see
Figure 27).
For 101, an S2 level of mainly (π, π*) character, energetically close to triplet states of mixed (n, π*)/(π, π*) nature, and an S1 and its close triplets of (π, π*) symmetry were calculated. For the other two compounds, an (n, π*) S2 level close to (π, π*) triplets together with an S1 and its adjacent triplets of (π, π*) symmetry were computed. Therefore, based on symmetry considerations and the small E(S2-Tn) (0.08, 0.04 and 0.02 eV for 101, 102 and 103, respectively), an easy DF from S2 and a negligible one from S1 were predicted. Moreover, the large ΔE(T2-T1) of 101, 1.40 eV, explains its observed dual RTP.
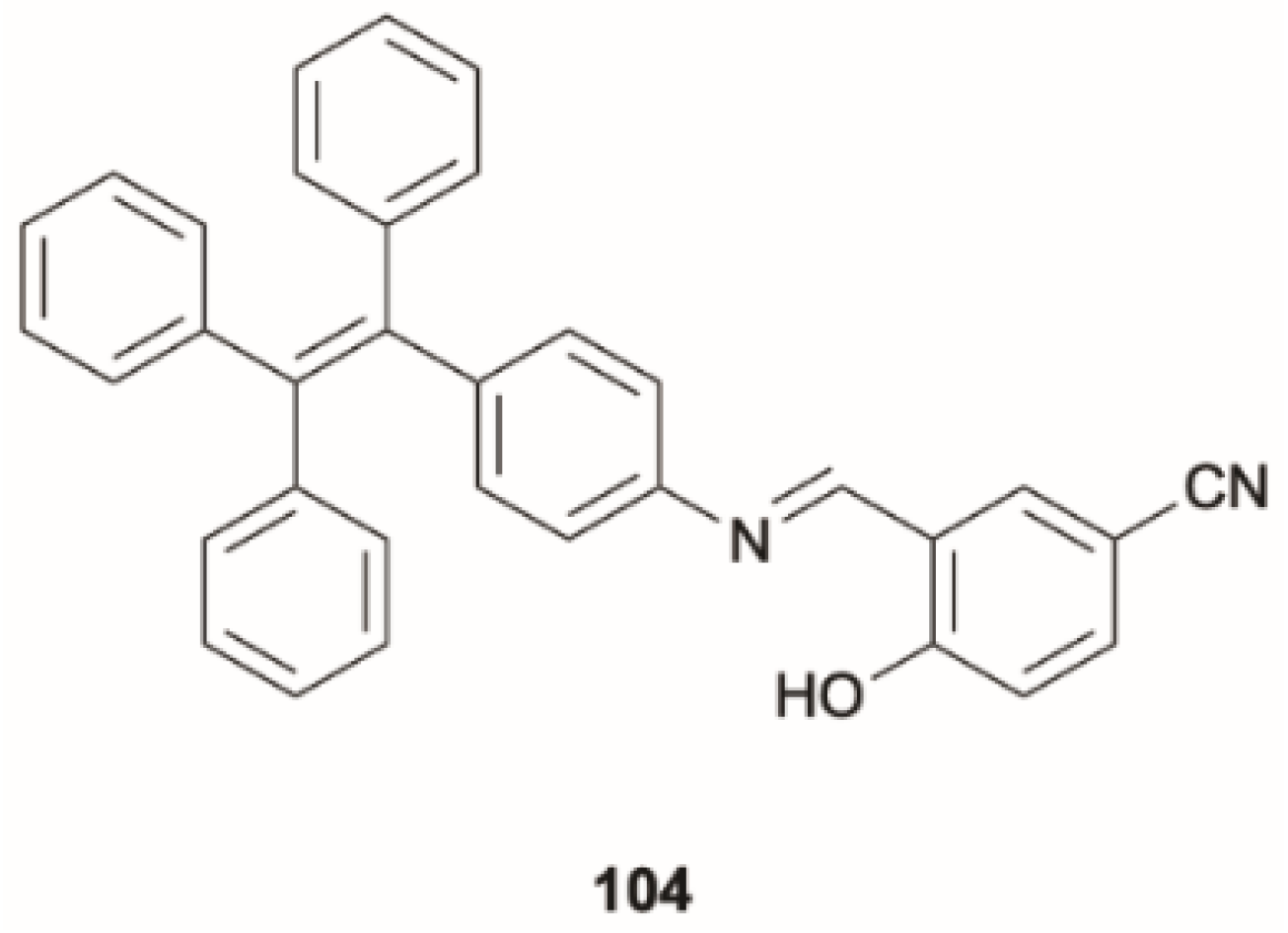
Sun et al. [
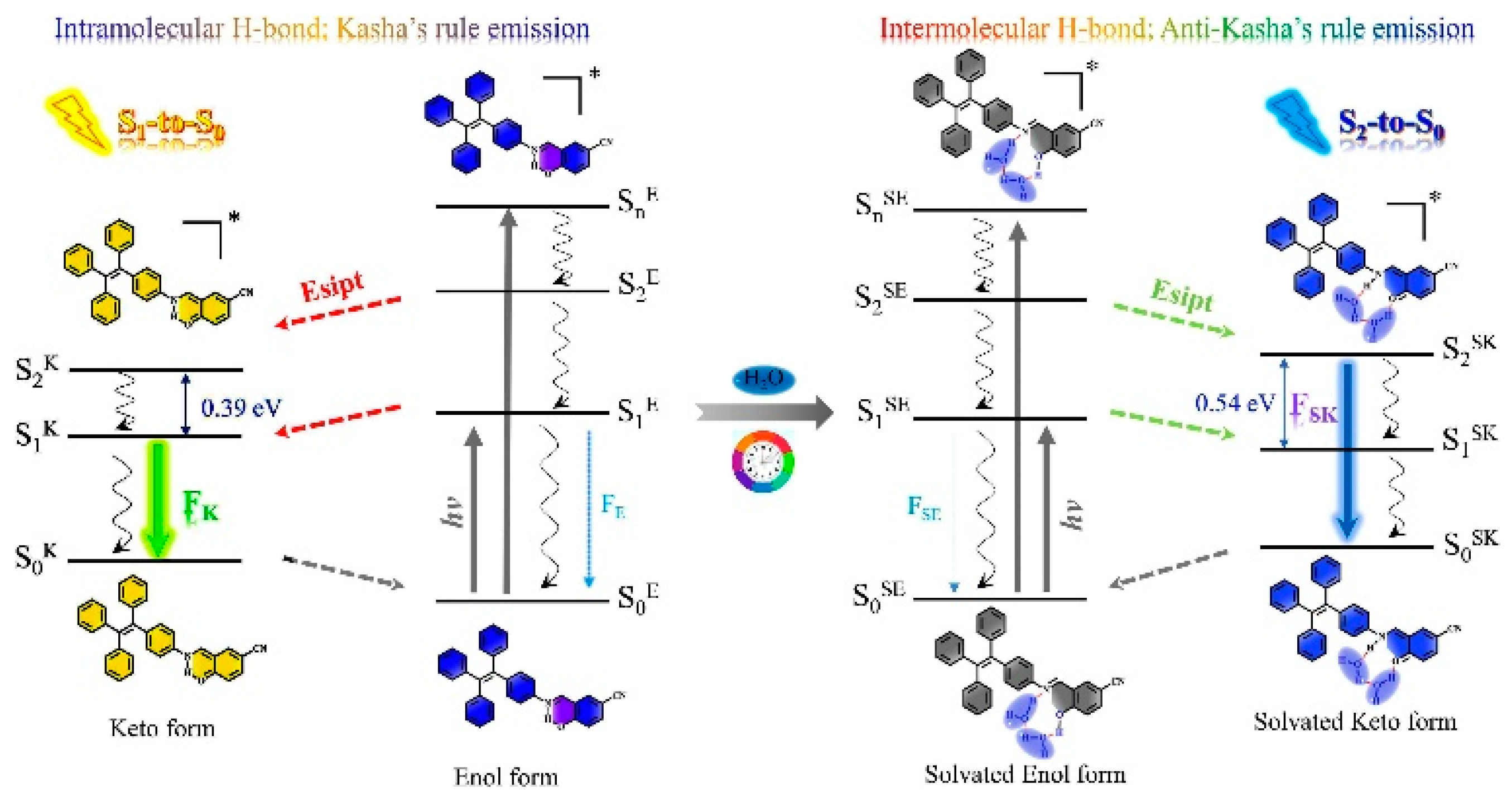
86] reported a tetraphenylethene-substituted Schiff base,
104 (
Scheme 35), which exhibits water-induced fluorescent switch from weak yellow-green Kasha emission to intense sky blue anti-Kasha one. According to TDDFT calculations at the B3LYP/6-31G(d) level of theory, in nonprotic solvents, an ESIPT process involving an intramolecular O-H
…N hydrogen bond is expected to lead to a ketone structure, responsible for the Kasha yellow emission (see
Figure 28). When water was added, an intramolecular hydrogen bond was broken in favor of an intermolecular one with a water molecule. ESIPT associated with this interaction leads to a solvated ketone structure whose 440 nm emission was assigned to radiative deactivation from S
2, and the S
2-S
1 IC was inhibited by the large ΔE(S
2-S
1), 0.54 eV. It should be underlined that this ESIPT process is correctly included in the present review, despite what was mentioned in the introduction, since it results in the formation of a new species from which anti-Kasha emission is observed. A practical use of
104 in rewritable papers and fluorescent sensors was developed with potential applications in information security protection, multilevel anticounterfeiting and data storage.
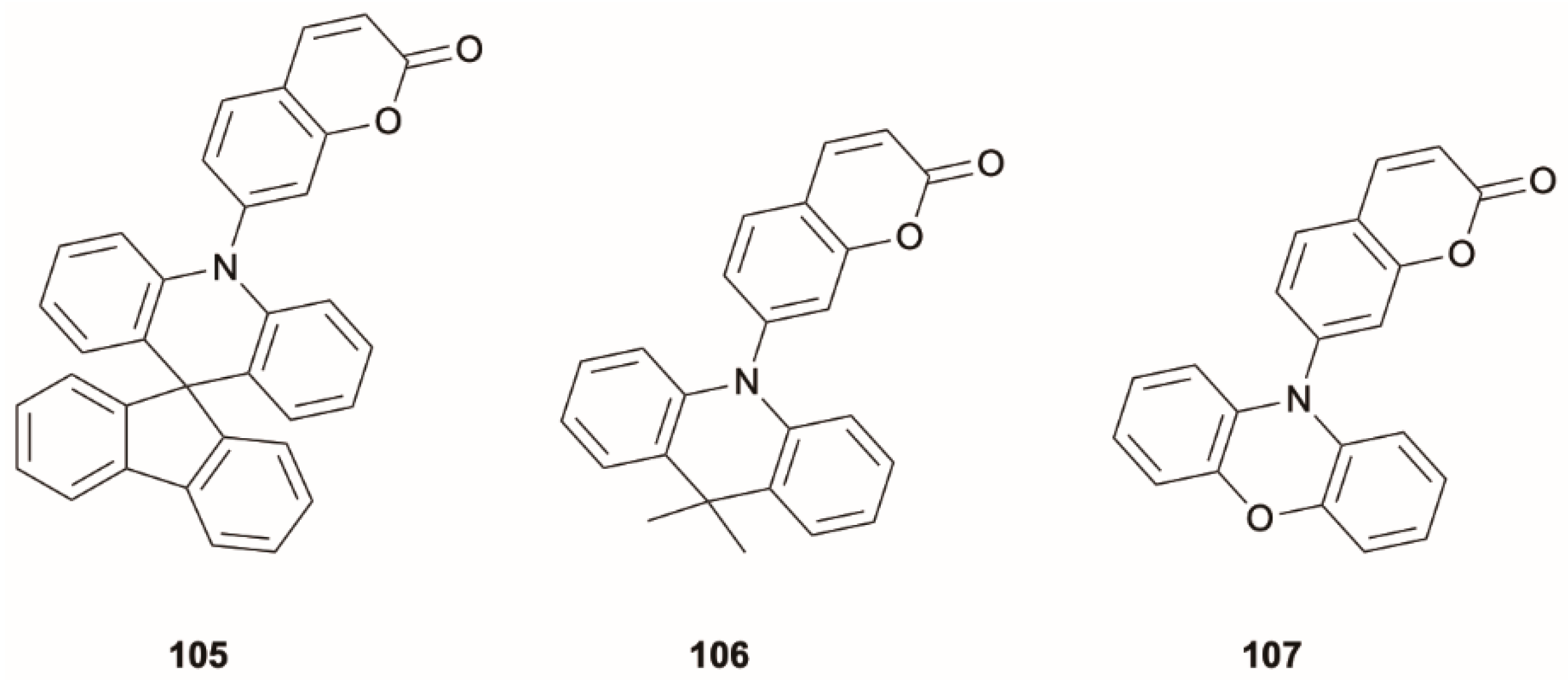
Due to the presence of (π, π*) and (
n, π*) levels, coumarin exhibits efficient El-Sayed-allowed ISC and therefore represents a useful scaffold to populate triplet levels in the absence of heavy atoms [
87]. Jhun et al. [
87] investigated a series of D-coumarin derivatives where D represents units with different oxidation potentials. Among the studied compounds, 10
H-spiro(acridine-9,9′-fluorene),
105; 9,9′-dimethyl-9,10-dihydroacridine,
106; and 10
H-phenoxazine,
107 (
Scheme 36), display anti-Kasha dual emission in toluene solution comprising a high-energy fluorescence at 420 nm of (π, π*) with partial CT character and a low-energy one in the 470–580 nm region of ICT character. Steady-state and time-resolved results revealed the TADF nature of the low-energy emission, which involves thermal repopulation of the
1ICT state from a close
3(
n, π*) one. This latter is populated through El-Sayed-allowed ISC from the
1(π, π*) level since both states are localized on the coumarin moiety (see
Figure 29). Intriguingly, such dual fluorescence is produced from (π, π*) and ICT transitions, both usually characterized by high oscillator strength. On the contrary, a single Kasha fluorescence was observed for the reference carbazole-coumarin compound, where the low (π, π*)-ICT energy gap (0.23 eV), smaller than those of the other investigated D-coumarin derivatives, allows a faster IC to the
1ICT state rather than ISC to the
3(
n, π*) one. It has to be noted that TDDFT calculations on this series of compounds, aimed at investigating the nature of the emissive state(s), revealed that the B3LYP functional provided results closer to the experimental values with respect to other tested (CAM-B3LYP and PBE0) functionals.
The dual fluorescence was successfully used by the authors for ratiometric temperature monitoring in the 120–300 K range and triplet quencher detection.
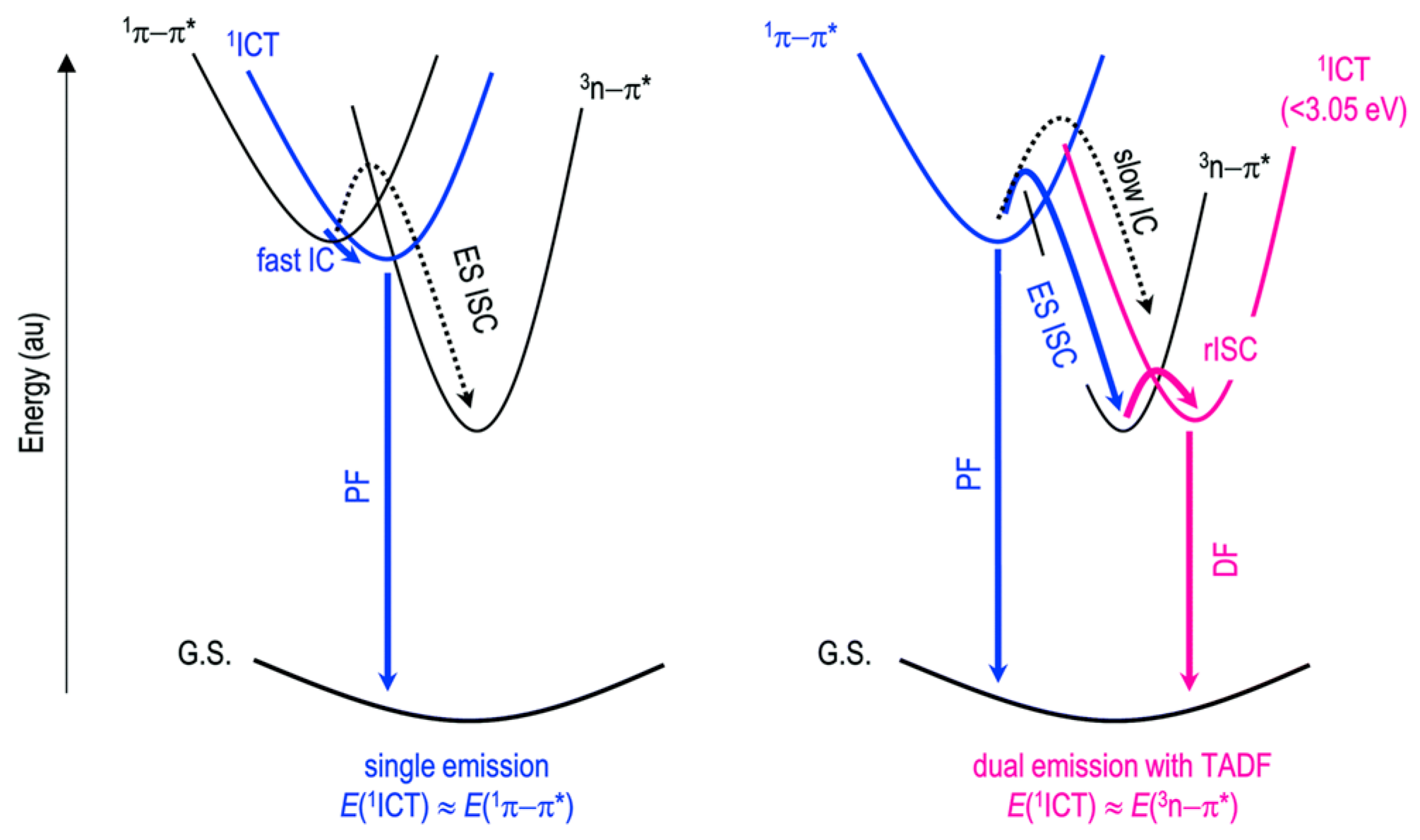
Imran et al. [
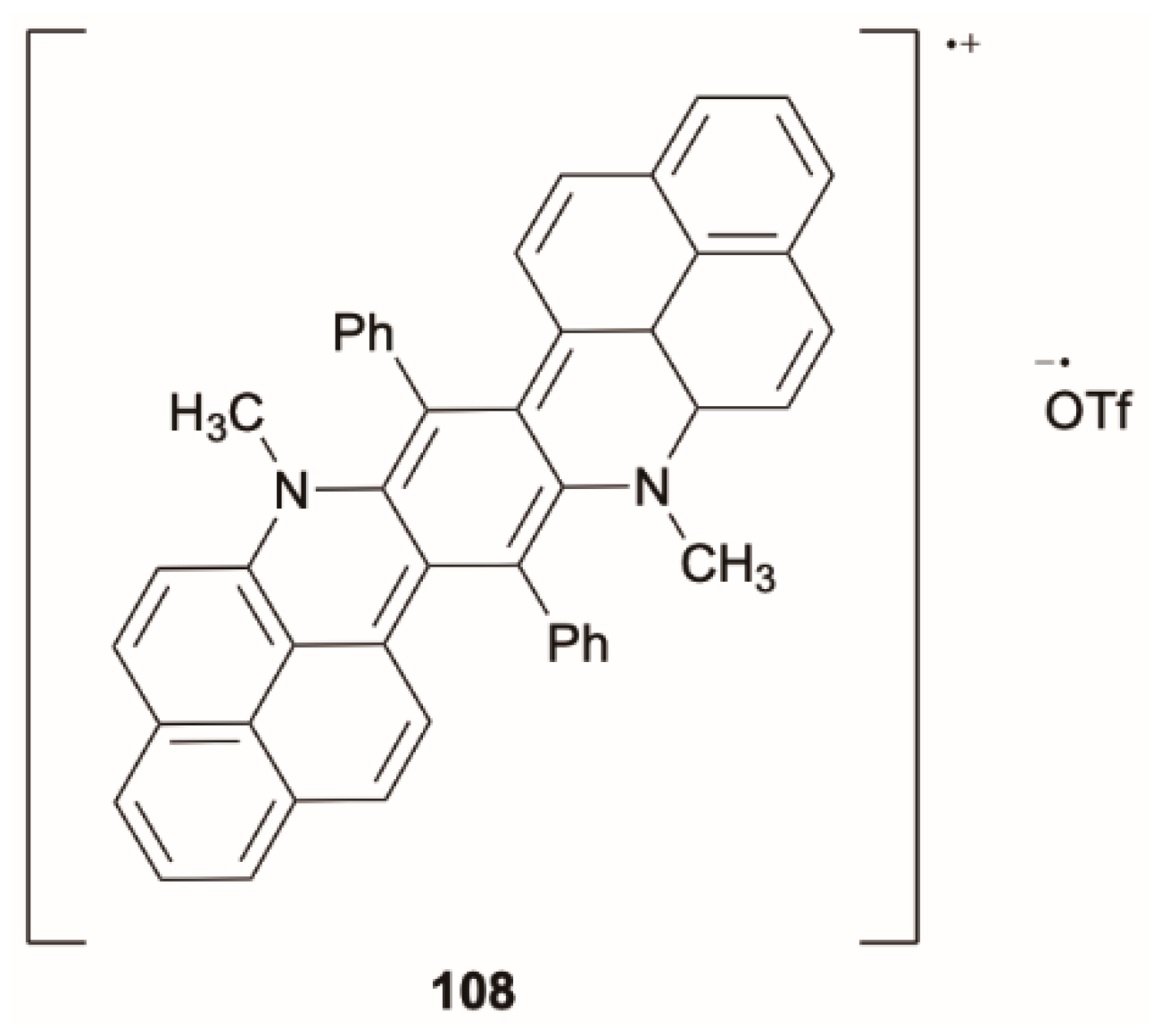
88] reported on the anti-Kasha behavior of radical cation
108 (
Scheme 37). When excited at 537 nm, DCM solutions of
108 display a red fluorescent emission centered at about 634 nm (Φ = 9.3%), which is intensified (Φ = 19.3%) when the compound is dispersed in PMMA matrix (1 wt%). The compound possesses one of the highest photostabilities among radical emitters with a half-life of 9.5 × 10
4 s under 350 nm irradiation. This property was ascribed to its cationic charge, which stabilizes ground and excited state molecular orbitals and inhibits their oxidation. DFT and configuration interaction singles calculations at the UB3LYP/6-31G+(d,p) level provided a low-energy excited doublet. According to the energy gap law, nonradiative deactivation was predicted from this state due to its closeness to the ground state. Therefore, the observed emission was associated with higher-energy calculated doublets, in violation of the Kasha rule.
Multiple anti-Kasha emissions from three singlet states of a single molecule were reported by Franca et al. [
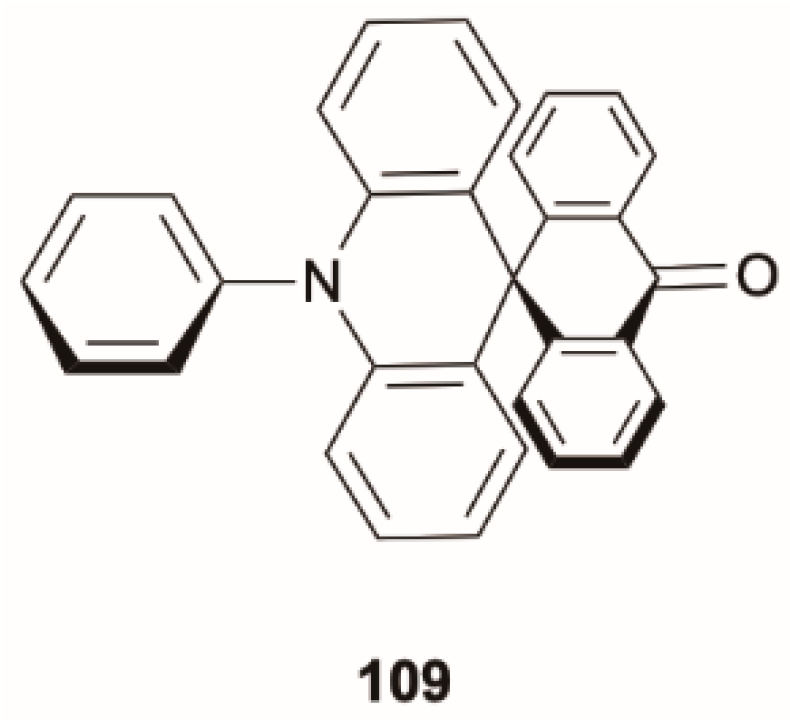
89] for a spiro compound, 10-phenyl-10
H,10′
H-spiro[acridine-9,9′-anthracen]-10′-one,
109 (
Scheme 38), characterized by rigidly connected orthogonal D–A units. Excitation of toluene solutions of
109 at high energy provides simultaneous emission from three states (see
Figure 30) which were described, based on previously reported calculations on this molecule [
90], performed at the DFT/MRCI-R/SV(p) level, as LE
D (π, π*) (
1B
1 in
Figure 30), LE
A (
n, π*) and CT (π, π*) states (in order of decreasing energy), the first two being locally excited states pertaining to the acridine (D) and the anthracenone (A) moieties, respectively. The lower-energy CT state, very close in energy to LE
A, provides both prompt and delayed fluorescence. The strongest allowed transition was ascribed to the high energy state, LE
D, located at about 0.6 eV above the other two. Emission from LE
D competes with both IC (to LE
A and CT lower-lying states) and ISC to a close triplet state which then decays to populate a lower triplet state yielding DF from rISC. Direct population of the weaker states produces both prompt (from LE
A and CT states) and delayed (from ISC between LE
A and
3LE
A followed by rISC to CT state) emissions.
The existence of the three emissions was unequivocally assessed by working in aerated/deaerated conditions and by varying both the excitation wavelength, resulting in a change in the relative intensities of the emissions, and the solvent. Strong solvatochromic relaxation was observed for the lowest-energy singlet CT state, while the close-lying LEA state was unaffected by polarity. The origin of such unprecedented rich photophysics of 109 was attributed to its rigid perpendicular geometry which effectively electronically decouples the D and A moieties. This ensures small ΔEST values and activates the TADF from a molecular CT state, in addition to the prompt emissions from the local LED and LEA states.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




































































