
A Landscape of Lignocellulosic Biopolymer Transformations into Valuable Molecules by Heterogeneous Catalysis in C’Durable Team at IRCELYON


Abstract
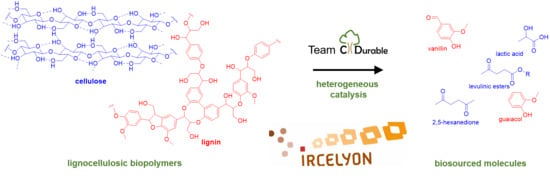
:
{kind=link}
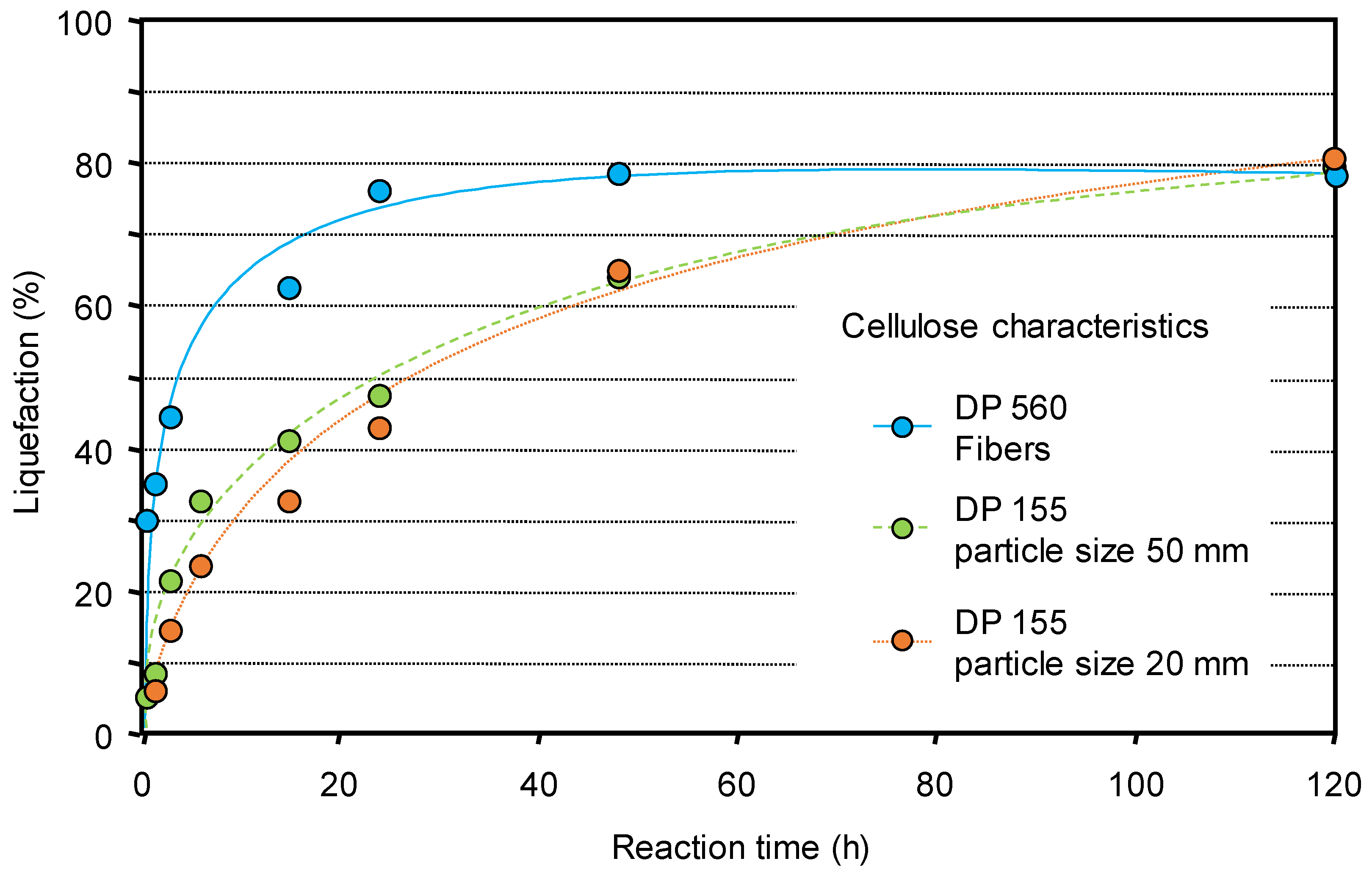{kind=link}
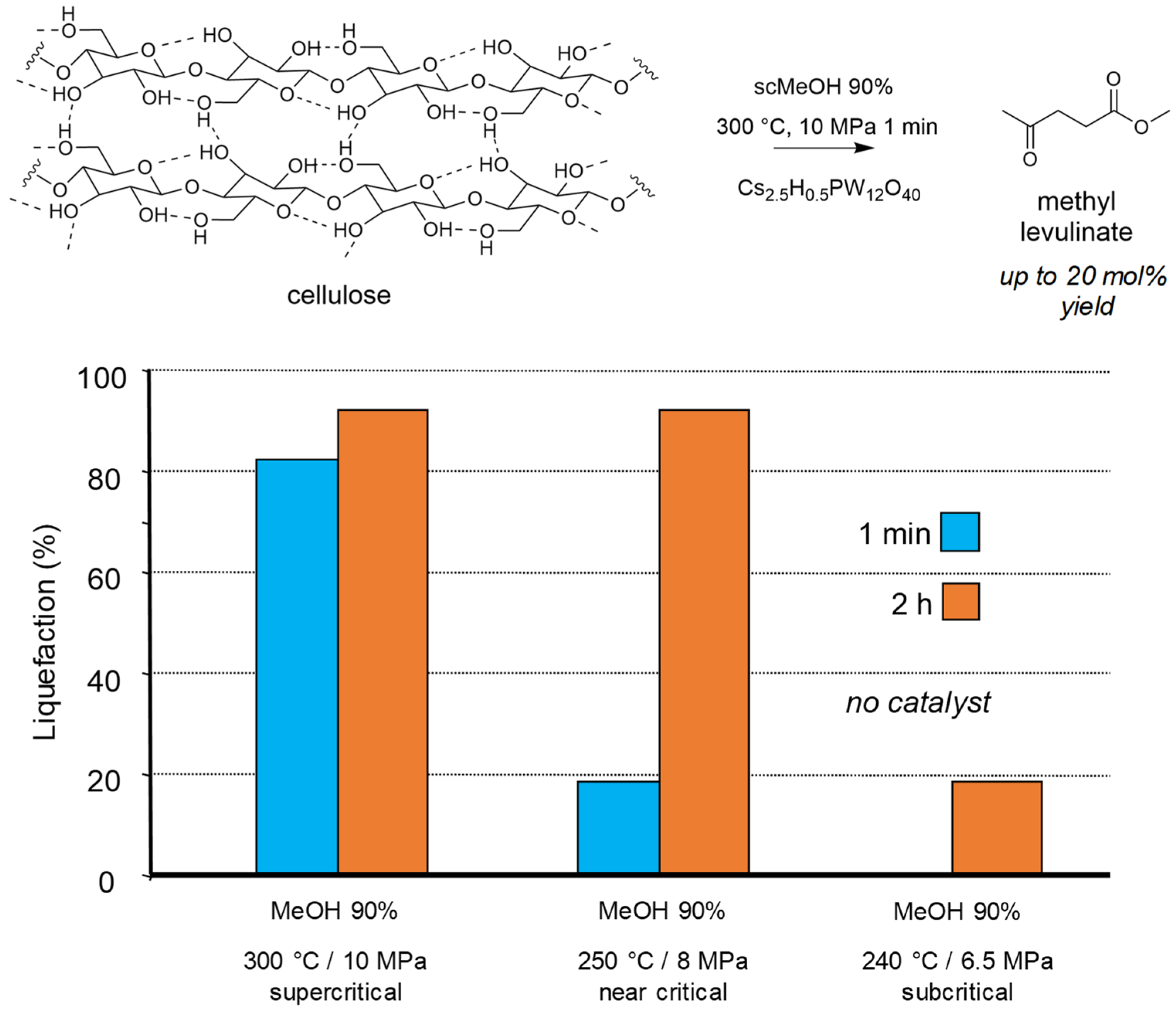{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Transformation of Cellulose
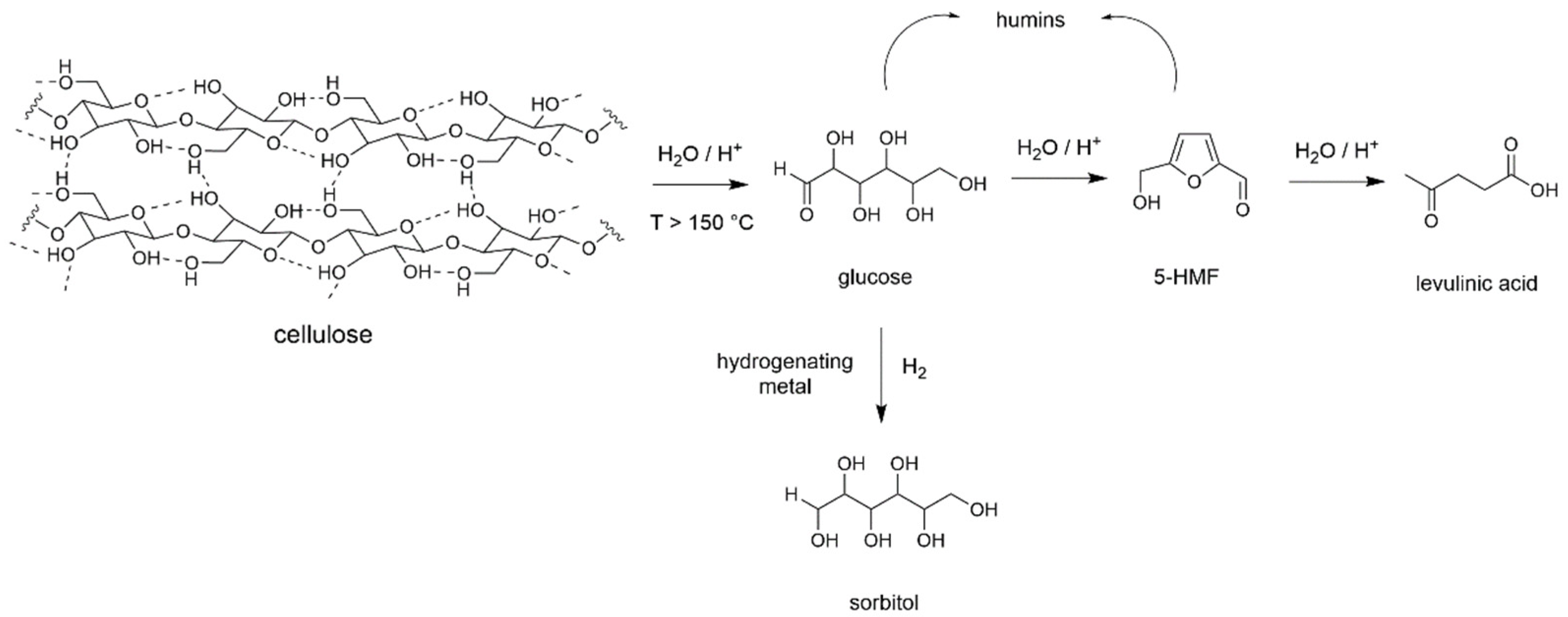
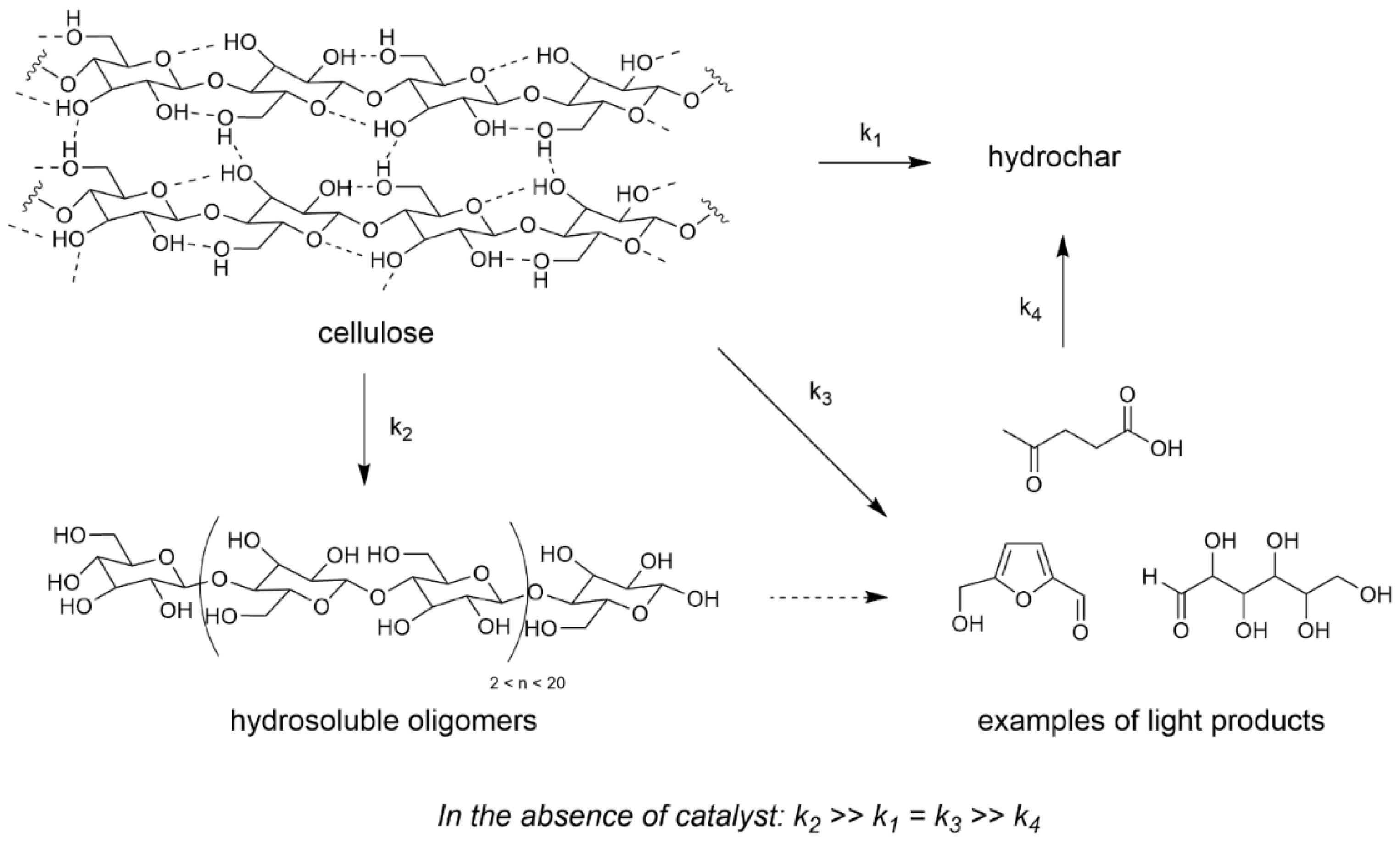
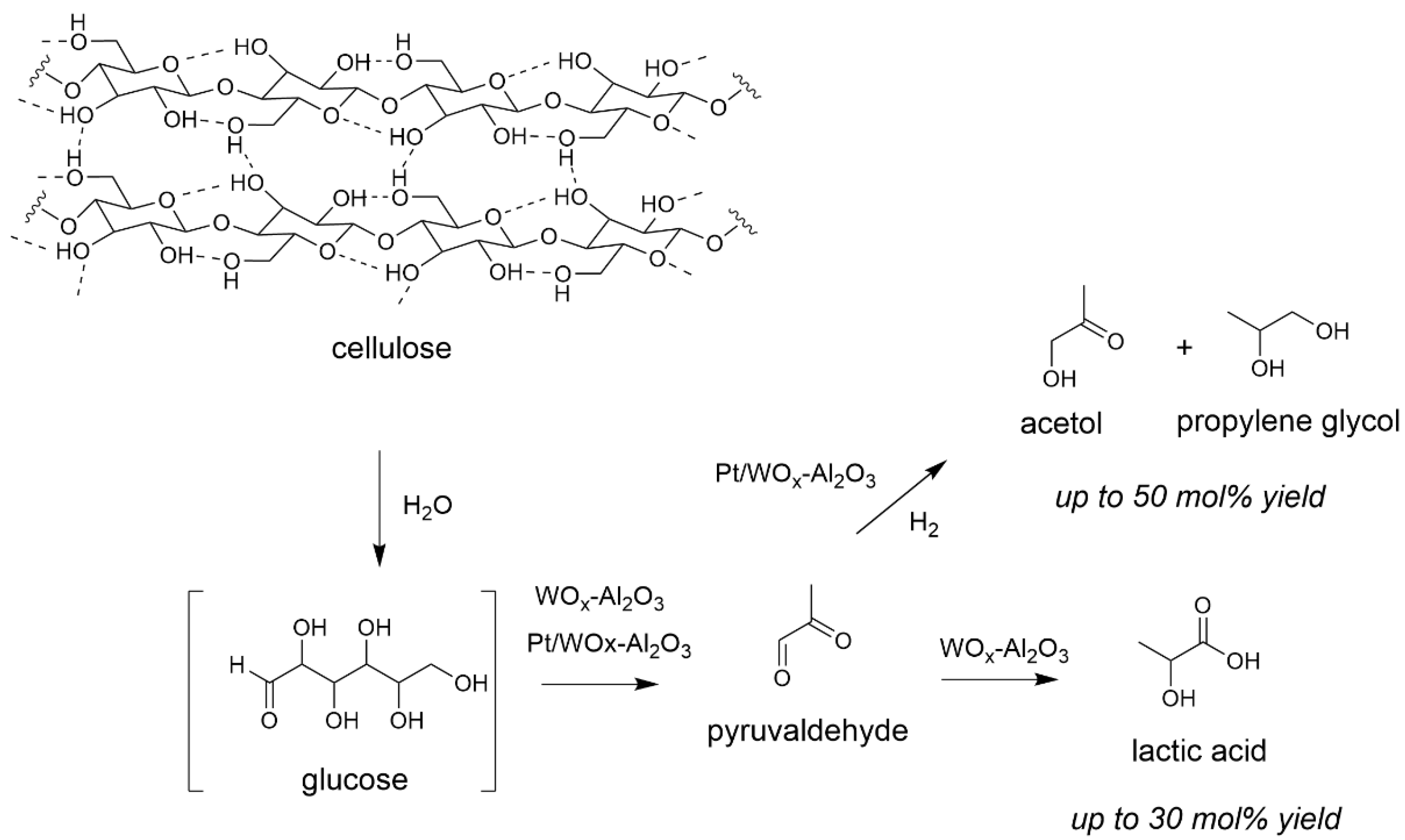
2.1. Transformation of Cellulose in Water
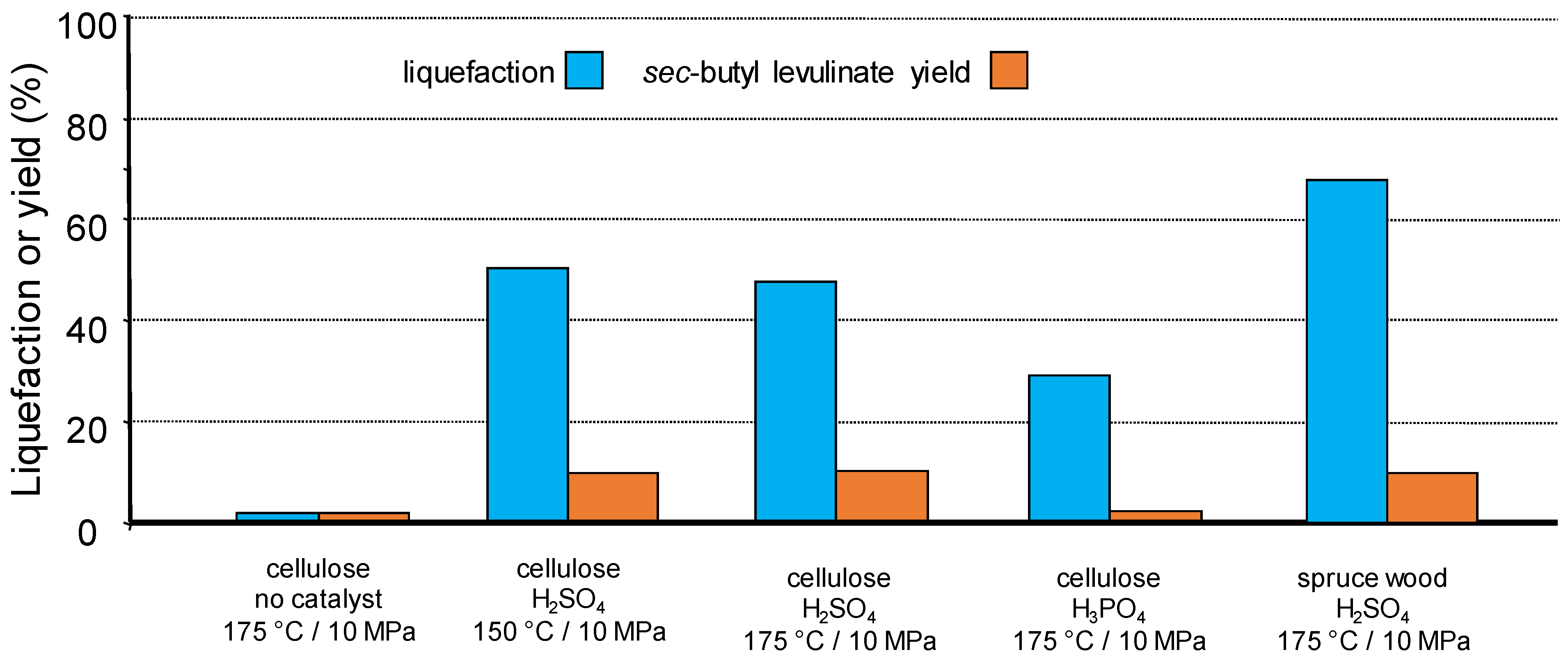
2.2. Transformation of Cellulose in Liquid and Supercritical Organic Solvents
3. Transformation of Lignin
3.1. Transformation of Lignin under Neutral Atmosphere
3.2. Transformation of Lignin under Oxidative Atmosphere
4. Transformation of Lignocellulose
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luterbacher, J.S.; Martin Alonso, D.; Dumesic, J.A. Targeted chemical upgrading of lignocellulosic biomass to platform molecules. Green Chem. 2014, 16, 4816–4838. [Google Scholar] [CrossRef] [Green Version]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, P.; Zhong, R.; Van den Bosch, S.; Coman, S.M.; Parvulescu, V.I.; Sels, B.F. Functionalised heterogeneous catalysts for sustainable biomass valorisation. Chem. Soc. Rev. 2018, 47, 8349–8402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, S.; Dutta, S.; Saha, B. Critical design of heterogeneous catalysts for biomass valorization: Current thrust and emerging prospects. Catal. Sci. Technol. 2016, 6, 7364–7385. [Google Scholar] [CrossRef] [Green Version]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Delidovich, I.; Leonhard, K.; Palkovits, R. Cellulose and hemicellulose valorisation: An integrated challenge of catalysis and reaction engineering. Energy Environ. Sci. 2014, 7, 2803–2830. [Google Scholar] [CrossRef]
- Fogassy, G.; Ke, P.; Figueras, F.; Cassagnau, P.; Rouzeau, S.; Courault, V.; Gelbard, G.; Pinel, C. Catalyzed ring opening of epoxides: Application to bioplasticizers synthesis. Appl. Catal. A 2011, 393, 1–8. [Google Scholar] [CrossRef]
- Fabiano, D.P.; Hamad, B.; Cardoso, D.; Essayem, N. On the understanding of the remarkable activity of template-containing mesoporous molecular sieves in the transesterification of rapeseed oil with ethanol. J. Catal. 2010, 276, 190–196. [Google Scholar] [CrossRef]
- Hamad, B.; Lopes de Souza, R.O.; Sapaly, G.; Carneiro Rocha, M.G.; Pries de Oliveira, P.G.; Gonzalez, W.A.; Andrade Sales, E.; Essayem, N. Transesterification of rapeseed oil with ethanol over heterogeneous heteropolyacids. Catal. Commun. 2008, 10, 92–97. [Google Scholar] [CrossRef]
- Morin, P.; Hamad, B.; Sapaly, G.; Carneiro Rocha, M.G.; Pries de Oliveira, P.G.; Gonzalez, W.A.; Andrade Sales, E.; Essayem, N. Transesterification of rapeseed oil with ethanol. I. Catalysis with homogeneous Keggin heteropolyacids. Appl. Catal. A 2007, 330, 69–76. [Google Scholar] [CrossRef]
- Mesnager, J.; Quettier, C.; Lambin, A.; Rataboul, F.; Perrard, A.; Pinel, C. Telomerization of butadiene with starch in water: Role of the surfactants. Green Chem. 2010, 12, 475–482. [Google Scholar] [CrossRef]
- Mesnager, J.; Quettier, C.; Lambin, A.; Rataboul, F.; Pinel, C. Telomerization of butadiene with starch under mild conditions. ChemSusChem 2009, 2, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.B.; Kachkarova-Sorokina, S.L.; Donze, C.; Pinel, C.; Gallezot, P. From native starch to hydrophilic and hydrophobic products: A catalytic approach. Top. Catal. 2004, 27, 67–76. [Google Scholar] [CrossRef]
- Donze, C.; Pinel, C.; Gallezot, P.; Taylor, P.L. Palladium-catalyzed telomerization of butadiene with starch. Adv. Synth. Catal. 2002, 344, 906–910. [Google Scholar] [CrossRef]
- Braga, T.P.; Essayem, N.; Valentini, A. Synthesis of Cu-MxOy/Al2O3 (M = Fe, Zn, W or Sb) catalysts for the conversion of glycerol to acetol: Effect of texture and acidity of the supports. RSC Adv. 2015, 5, 93394–93402. [Google Scholar] [CrossRef]
- Ftouni, J.; Villandier, N.; Auneau, F.; Besson, M.; Djakovitch, L.; Pinel, C. From glycerol to lactic acid under inert conditions in the presence of platinum-based catalysts: The influence of support. Catal. Today 2015, 257, 267–273. [Google Scholar] [CrossRef]
- Garcia, R.; Besson, M.; Gallezot, P. Chemoselective catalytic oxidation of glycerol with air on platinum metals. Appl. Catal. A 1995, 127, 165–176. [Google Scholar] [CrossRef]
- Auneau, F.; Noel, S.; Aubert, G.; Besson, M.; Djakovitch, L.; Pinel, C. On the role of the atmosphere in the catalytic glycerol transformation over iridium-based catalysts. Catal. Commun. 2011, 16, 144–149. [Google Scholar] [CrossRef]
- Auneau, F.; Michel, C.; Delbecq, F.; Pinel, C.; Sautet, P. Unravelling the mechanism of glycerol hydrogenolysis over rhodium catalyst through combined experimental-theoretical investigations. Chem. Eur. J. 2011, 17, 14288–14299. [Google Scholar] [CrossRef] [PubMed]
- Essayem, N.; Lopes de Souza, R.O.; Hamad, B.; Sapaly, G.; Pries de Oliveira, P.G.; de Aurojo Gonzales, W. Simultaneous Glyceride Transesterification-Glycerol Etherification for Manufacture of Biodiesel and Fuel Additives; PCT FR2930779; CNRS: Tsukuba, Japan, 2009. [Google Scholar]
- Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C.; Rosier, C. Glycerol hydrogenolysis on heterogeneous catalysts. Green Chem. 2004, 6, 359–361. [Google Scholar] [CrossRef]
- Abdouli, I.; Eternot, M.; Dappozze, F.; Guillard, C.; Essayem, N. Comparison of hydrothermal and photocatalytic conversion of glucose with commercial TiO2: Superficial properties-activities relationships. Catal. Today 2021, 367, 268–277. [Google Scholar] [CrossRef]
- Riviere, M.; Perret, N.; Delcroix, D.; Cabiac, A.; Pinel, C.; Besson, M. Solvent effect in hydrogenolysis of xylitol over bifunctional Ru/MnO/C catalysts under alkaline-free conditions. ACS Sustain. Chem. Eng. 2018, 6, 4076–4085. [Google Scholar] [CrossRef]
- Doiseau, A.C.; Rataboul, F.; Burel, L.; Essayem, N. Synergy effect between solid acid catalysts and concentrated carboxylic acids solutions for efficient furfural production from xylose. Catal. Today 2014, 226, 176–184. [Google Scholar] [CrossRef]
- Auneau, F.; Berchu, M.; Aubert, G.; Pinel, C.; Besson, M.; Todaro, D.; Bernardi, M.; Ponsetti, T.; Di Felice, R. Exploring the reaction conditions for Ru/C catalyzed selective hydrogenolysis of xylitol alkaline aqueous solutions to glycols in a trickle-bed reactor. Catal. Today 2014, 234, 100–106. [Google Scholar] [CrossRef]
- Karaki, M.; Karout, A.; Toufaily, J.; Rataboul, F.; Essayem, N.; Lebeau, B. Synthesis and characterization of acidic ordered mesoporous organosilica SBA-15: Application to the hydrolysis of cellobiose and insight into the stability of the acidic functions. J. Catal. 2013, 305, 204–216. [Google Scholar] [CrossRef]
- Souza, R.O.L.; Fabiano, D.P.; Feche, C.; Rataboul, F.; Cardoso, D.; Essayem, N. Glucose-fructose isomerisation promoted by basic hybrid catalysts. Catal. Today 2012, 195, 114–119. [Google Scholar] [CrossRef]
- Perrard, A.; Gallezot, P.; Joly, J.-P.; Durand, R.; Baljou, C.; Coq, B.; Trens, P. Highly efficient metal catalysts supported on activated carbon cloths: A catalytic application for the hydrogenation of D-glucose to D-sorbitol. Appl. Catal. A 2007, 331, 100–104. [Google Scholar] [CrossRef]
- Gallezot, P.; Nicolaus, N.; Fleche, G.; Fuertes, P.; Perrard, A. Glucose hydrogenation on ruthenium catalysts in a trickle-bed reactor. J. Catal. 1998, 180, 51–55. [Google Scholar] [CrossRef]
- Dechamp, N.; Gamez, A.; Perrard, A.; Gallezot, P. Kinetics of glucose hydrogenation in a trickle-bed reactor. Catal. Today 1995, 24, 29–34. [Google Scholar] [CrossRef]
- Besson, M.; Lahmer, F.; Gallezot, P.; Fuertes, P.; Fleche, G. Catalytic oxidation of glucose on bismuth-promoted palladium catalysts. J. Catal. 1995, 152, 116–121. [Google Scholar] [CrossRef]
- Ait Rass, H.; Essayem, N.; Besson, M. Selective aerobic oxidation of 5-HMF into 2,5-furandicarboxylic acid with Pt catalysts supported on TiO2- and ZrO2-based supports. ChemSusChem 2015, 8, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Ait Rass, H.; Essayem, N.; Besson, M. Selective aqueous phase oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Pt/C catalysts: Influence of the base and effect of bismuth promotion. Green Chem. 2013, 15, 2240–2251. [Google Scholar] [CrossRef]
- Lopes de Souza, R.O.; Rataboul, F.; Essayem, N. Hydroxymethylfurfural (5-HMF) production from hexoses: Limits of heterogeneous catalysis in hydrothermal conditions and potential of concentrated aqueous organic acids solutions as reactive solvent system. Challenges 2012, 3, 212–232. [Google Scholar] [CrossRef]
- Olivier-Bourbigou, H.; Chizallet, C.; Dumeignil, F.; Fongarland, P.; Geantet, C.; Granger, P.; Launay, F.; Löfberg, A.; Massiani, P.; Maugé, F.; et al. The pivotal role of catalysis in France: Selected examples of recent advances and future prospects. ChemCatChem 2017, 9, 2029–2064. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Cabiac, A.; Guillon, E.; Chambon, F.; Pinel, C.; Rataboul, F.; Essayem, N. Cellulose reactivity and glycosidic bond cleavage in aqueous phase by catalytic and non catalytic transformations. Appl. Catal. A 2011, 402, 1–10. [Google Scholar] [CrossRef]
- Lanzafame, P.; Temi, D.M.; Perathoner, S.; Spadaro, A.N.; Centi, G. Direct conversion of cellulose to glucose and valuable intermediates in mild reaction conditions over solid acid catalysts. Catal. Today 2012, 179, 178–184. [Google Scholar] [CrossRef]
- Pang, J.; Wang, A.; Zheng, M.; Zhang, T. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperatures. Chem. Commun. 2010, 46, 6935–6937. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, S.; Nakajima, K.; Kiyano, M.; Yamaguchi, D.; Kato, H.; Hayashi, S.; Hara, M. Hydrolysis of cellulose by amorphous carbon bearing SO3H, COOH, and OH groups. J. Am. Chem. Soc. 2008, 130, 12787–12793. [Google Scholar] [CrossRef] [PubMed]
- Onda, A.; Ochi, T.; Yanagisawa, K. Selective hydrolysis of cellulose into glucose over solid acid catalysts. Green Chem. 2008, 10, 1033–1037. [Google Scholar] [CrossRef]
- Van de Vyver, S.; Peng, L.; Geboers, J.; Schepers, H.; de Clippel, F.; Gommes, C.J.; Goderis, B.; Jacobs, P.A.; Sels, B.F. Sulfonated silica/carbon nanocomposites as novel catalysts for hydrolysis of cellulose to glucose. Green Chem. 2010, 12, 1560–1563. [Google Scholar] [CrossRef]
- Vilcocq, L.; Castilho, P.C.; Carvalheiro, F.; Duarte, L.C. Hydrolysis of oligosaccharides over solid acid catalysts: A review. ChemSusChem 2014, 7, 1010–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, R.; Schüth, F. Acid hydrolysis of cellulose as the entry point into biorefinery schemes. ChemSusChem 2009, 2, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A.M. New frontiers in the catalytic synthesis of levulinic acid: From sugars to raw and waste biomass as starting feedstock. Catalysts 2016, 6, 196. [Google Scholar] [CrossRef]
- Rinaldi, R.; Meine, N.; vom Stein, J.; Palkovits, R.; Schüth, F. Which controls the depolymerization of cellulose in ionic liquids: The solid acid catalyst or cellulose? ChemSusChem 2010, 3, 266–276. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Vigier, K.; Chatel, G.; Jérôme, F. Contribution of deep eutectic solvents for biomass processing: Opportunities, challenges, and limitations. ChemCatChem 2015, 7, 1250–1260. [Google Scholar] [CrossRef]
- Jérôme, F.; Chatel, G.; De Oliveira Vigier, K. Depolymerization of cellulose to processable glucans by non-thermal technologies. Green Chem. 2016, 18, 3903–3913. [Google Scholar] [CrossRef]
- Fukuoka, A.; Dhepe, P.L. Catalytic conversion of cellulose into sugar alcohols. Angew. Chem. Int. Ed. 2006, 45, 5161–5163. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, A.M.; Weinberg, K.; Palkovits, R. Hydrogenolysis goes bio: From carbohydrates and sugar alcohols to platform chemicals. Angew. Chem. Int. Ed. 2012, 51, 2564–2601. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Komanoya, T.; Guha, S.K.; Hara, K.; Fukuoka, A. Conversion of cellulose into renewable chemicals by supported metal catalysis. Appl. Catal. A 2011, 409–410, 13–20. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, T.; Zheng, M.; Wang, A.; Wang, H.; Wang, X.; Chen, J.G. Direct catalytic conversion of cellulose into ethylene glycol using nickel-promoted tungsten carbide catalysts. Angew. Chem. Int. Ed. 2008, 47, 8510–8513. [Google Scholar] [CrossRef] [PubMed]
- Jollet, V.; Chambon, F.; Rataboul, F.; Cabiac, A.; Pinel, C.; Guillon, E.; Essayem, N. Non-catalyzed and Pt/γ-Al2O3-catalyzed hydrothermal cellulose dissolution-conversion: Influence of the reaction parameters and analysis of the unreacted cellulose. Green Chem. 2009, 11, 2052–2060. [Google Scholar] [CrossRef]
- Möller , M.; Nilges , P.; Harnisch, F.; Schröder, U. Subcritical water as reaction environment: Fundamentals of hydrothermal biomass transformation. ChemSusChem 2011, 4, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Shrotri, A.; Fukuoka, A. Synthesis of cello-oligosaccharides by depolymerization of cellulose: A review. Appl. Catal. A 2021, 621, 118177. [Google Scholar] [CrossRef]
- Fongarland, P.; Essayem, N.; Rataboul, F. Noncatalyzed liquefaction of celluloses in hydrothermal conditions: Influence of reactant physicochemical characteristics and modeling studies. Ind. Eng. Chem. Res. 2017, 56, 126–134. [Google Scholar] [CrossRef]
- Chambon, F.; Essayem, N.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E. Process for Converting Cellulose or Lignocellulosic Biomass Using Stable Non-Zeolite Solid Lewis Acids Based on Tin or Antimony Alone or as a Mixture; PCT WO2012085361; IFP Energies Nouvelles and CNRS: Tsukuba, Japan, 2012. [Google Scholar]
- Chambon, F.; Essayen, N.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E. Process for Conversion of Lignocellulosic Biomass or Cellulose by Using Supported Tungsten-Based Solid Lewis Acids as Catalysts; PCT WO2011098683; IFP Energies Nouvelles, CNRS and University Claude-Bernard Lyon 1: Villeurbanne, France, 2011. [Google Scholar]
- Nguyen, V.C.; Dandach, A.; Vu, T.T.H.; Fongarland, P.; Essayem, N. ZrW catalyzed cellulose conversion in hydrothermal conditions: Influence of the calcination temperature and insights on the nature of the active phase. Molecular Catal. 2019, 476, 110518. [Google Scholar] [CrossRef]
- Chambon, F.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E.; Essayem, N. Cellulose hydrothermal conversion promoted by heterogeneous Brønsted and Lewis acids: Remarkable efficiency of solid Lewis acids to produce lactic acid. Appl. Catal. B 2011, 105, 171–181. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Simakova, I.L.; Salmi, T.; Murzin, D.Y. Production of lactic acid/lactates from biomass and their catalytic transformations to commodities. Chem. Rev. 2014, 114, 1909–1971. [Google Scholar] [CrossRef] [PubMed]
- West, R.M.; Holm, M.S.; Saravanamurugan, S.; Xiong, J.; Beversdorf, Z.; Taarning, E.; Christensen, C.H. Zeolite H-USY for the production of lactic acid and methyl lactate from C3-sugars. J. Catal. 2010, 269, 122–130. [Google Scholar] [CrossRef]
- Swesi, Y.; Nguyen, C.; Vu, T.T.H.; Rataboul, F.; Eternot, M.; Fongarland, P.; Essayem, N. Direct solid Lewis acid catalyzed wood liquefaction into lactic acid: Kinetic evidences that wood pretreatment might not be a prerequisite. ChemCatChem 2017, 9, 2377–2382. [Google Scholar] [CrossRef] [Green Version]
- Chambon, F.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E.; Essayem, N. Cellulose conversion with tungstated-alumina-based catalysts: Influence of the presence of platinum and mechanistic studies. ChemSusChem 2013, 6, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Chambon, F.; Essayem, N.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E. Process for Conversion of Lignocellulosic Biomass or Cellulose Using Solid Lewis Acid Catalysts Containing an Oxide of Tungsten and a Metal from Groups 8 to 11; PCT WO2012022853; IFP Energies Nouvelles, CNRS and University Claude-Bernard Lyon 1: Villeurbanne, France, 2012. [Google Scholar]
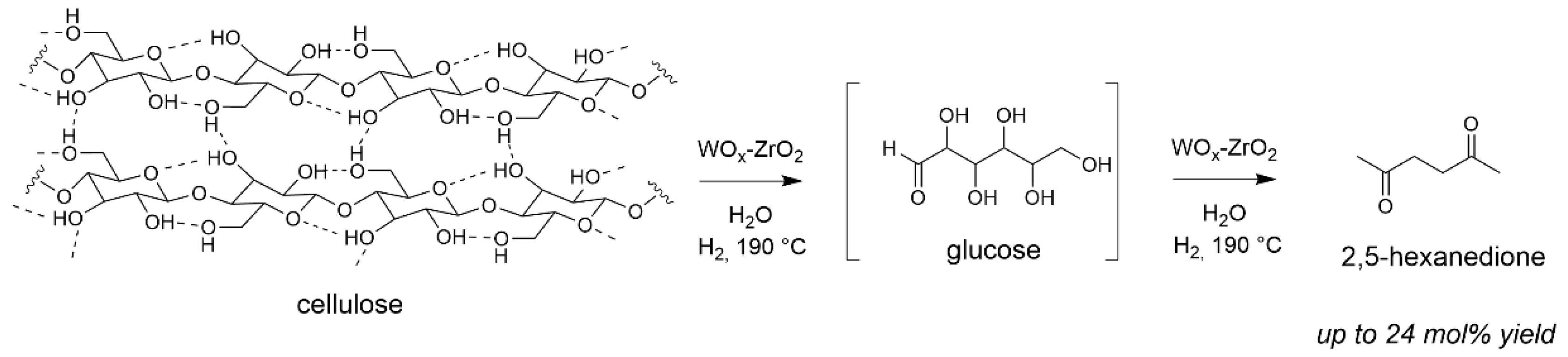
- Chambon, F.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E.; Essayem, N. Conversion of cellulose to 2,5-hexanedione using tungstated zirconia in hydrogen atmosphere. Appl. Catal. A 2015, 504, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, V.C.; Bui, N.Q.; Eternot, M.; Vu, T.T.H.; Fongarland, P.; Essayem, N. Kinetic of ZrW catalyzed cellulose hydrothermal conversion: Deeper understanding of reaction pathway via analytic tools improvement. Mol. Catal. 2018, 458, 171–179. [Google Scholar] [CrossRef]
- Liu, Y.; Li, G.; Hu, Y.; Wang, A.; Lu, F.; Zou, J.-J.; Cong, Y.; Li, N.; Zhang, T. Integrated conversion of cellulose to high-density aviation fuel. Joule 2019, 3, 1028–1036. [Google Scholar] [CrossRef]
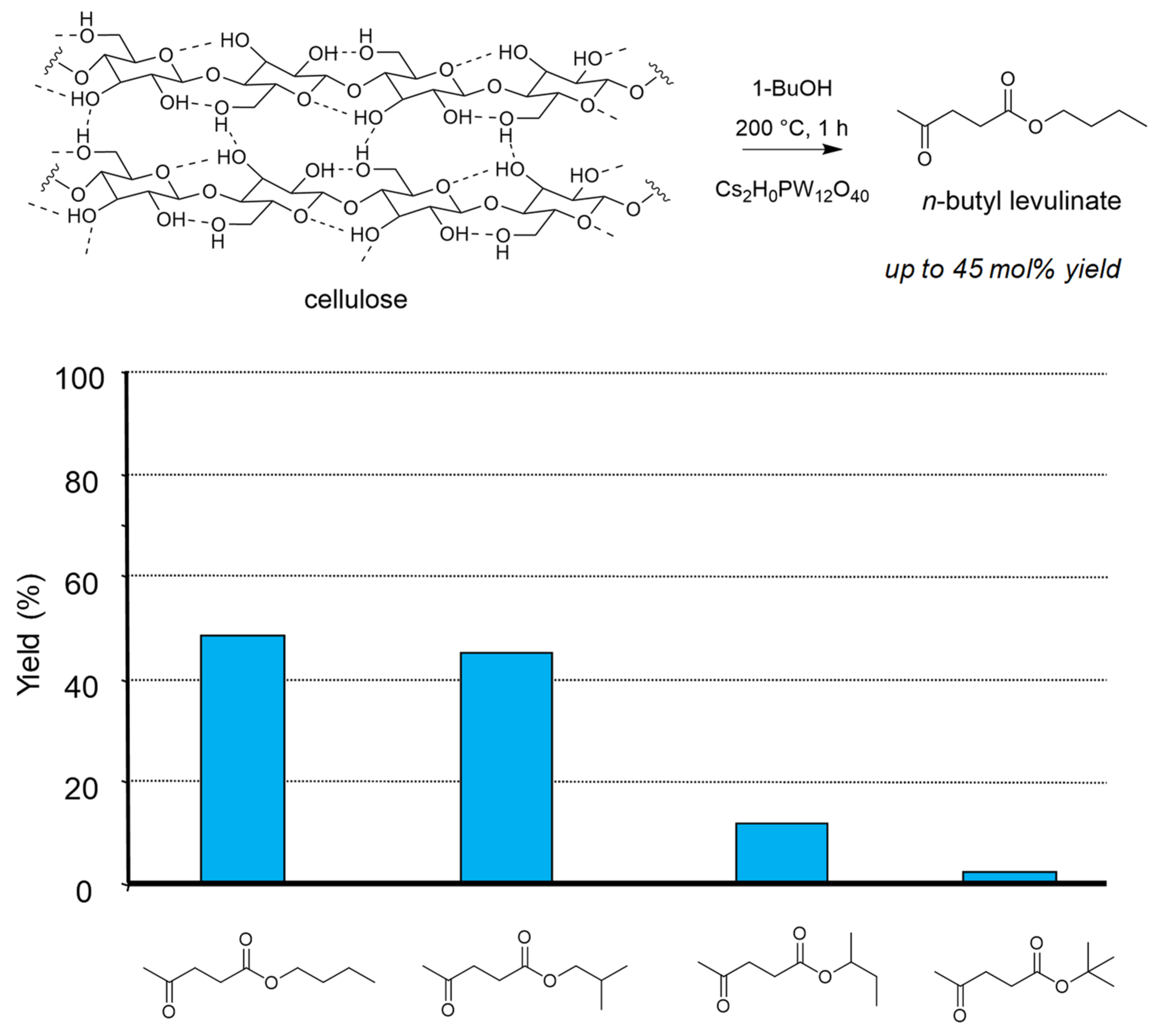
- Démolis, A.; Essayem, N.; Rataboul, F. Synthesis and applications of alkyl levulinates. ACS Sustain. Chem. Eng. 2014, 2, 1338–1352. [Google Scholar] [CrossRef]
- Garves, K. Acid catalyzed degradation of cellulose in alcohols. J. Wood Chem. Technol. 1988, 8, 121–134. [Google Scholar] [CrossRef]
- Rataboul, F.; Essayem, N. Cellulose reactivity in supercritical methanol in the presence of solid acid catalysts: Direct synthesis of methyl-levulinate. Ind. Eng. Chem. Res. 2011, 50, 799–805. [Google Scholar] [CrossRef]
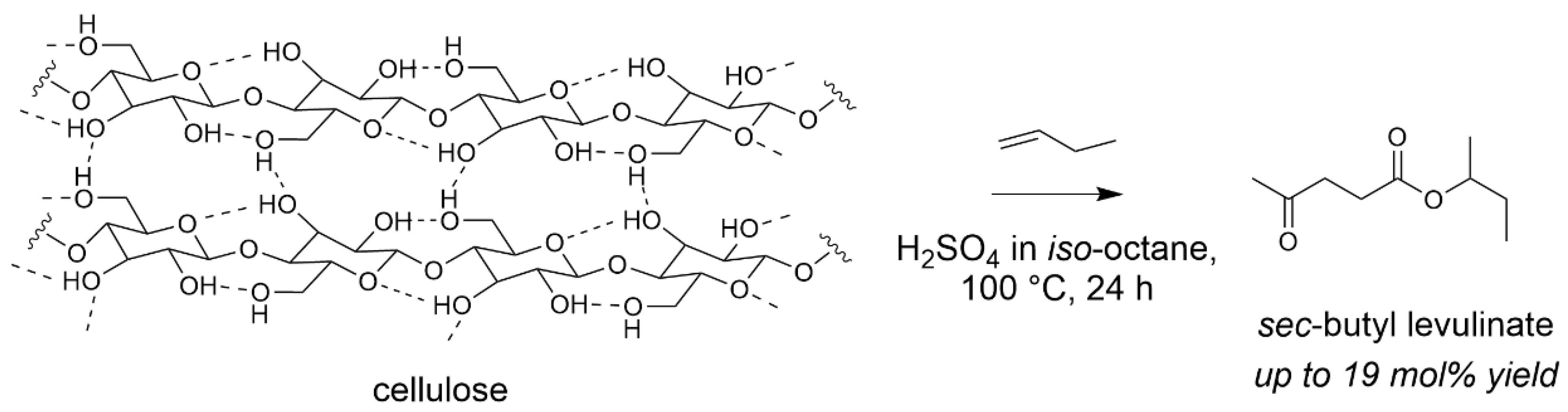
- Démolis, A.; Eternot, M.; Essayem, N.; Rataboul, F. Influence of butanol isomers on the reactivity of cellulose towards the synthesis of butyl levulinates catalyzed by liquid and solid acid catalysts. New J. Chem. 2016, 40, 3747–3754. [Google Scholar] [CrossRef]
- Guerbuez, E.I.; Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Reactive extraction of levulinate esters and conversion to γ-valerolactone for production of liquid fuels. ChemSusChem 2011, 4, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Démolis, A.; Eternot, M.; Essayem, N.; Rataboul, F. New insights into the reactivity of biomass with butenes for the synthesis of butyl levulinates. ChemSusChem 2017, 10, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Strappaveccia, G.; Luciani, L.; Bartollini, E.; Marrocchi, A.; Pizzo, F.; Vaccaro, L. γ-Valerolactone as an alternative biomass-derived medium for the Sonogashira reaction. Green Chem. 2015, 17, 1071–1076. [Google Scholar] [CrossRef]
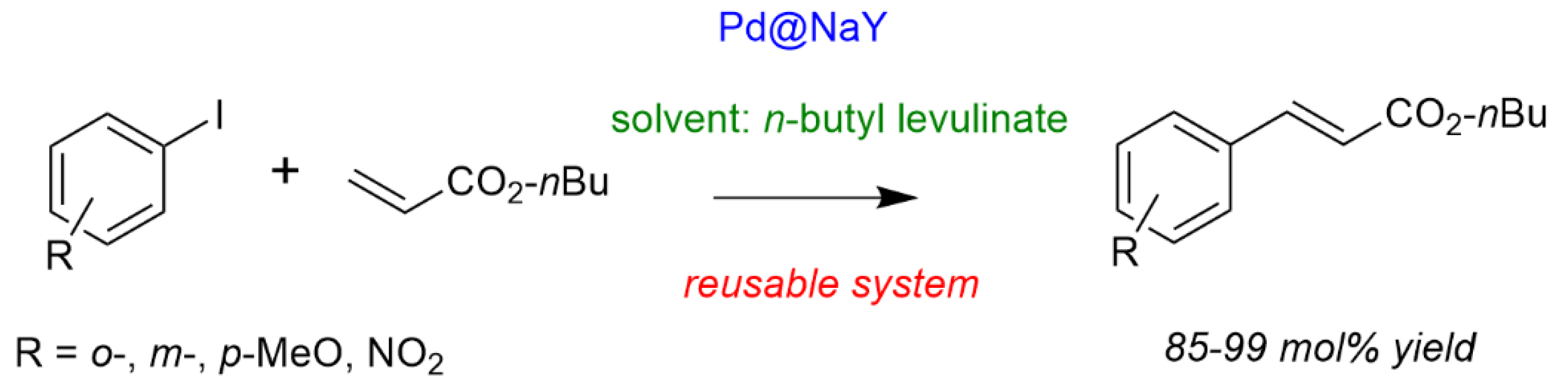
- Marcel, R.; Durillon, T.; Djakovitch, L.; Fache, F.; Rataboul, F. First example of the use of biosourced alkyl levulinates as solvents for synthetic chemistry: Application to the heterogeneously catalyzed Heck coupling. ChemistrySelect 2019, 4, 3329–3333. [Google Scholar] [CrossRef]
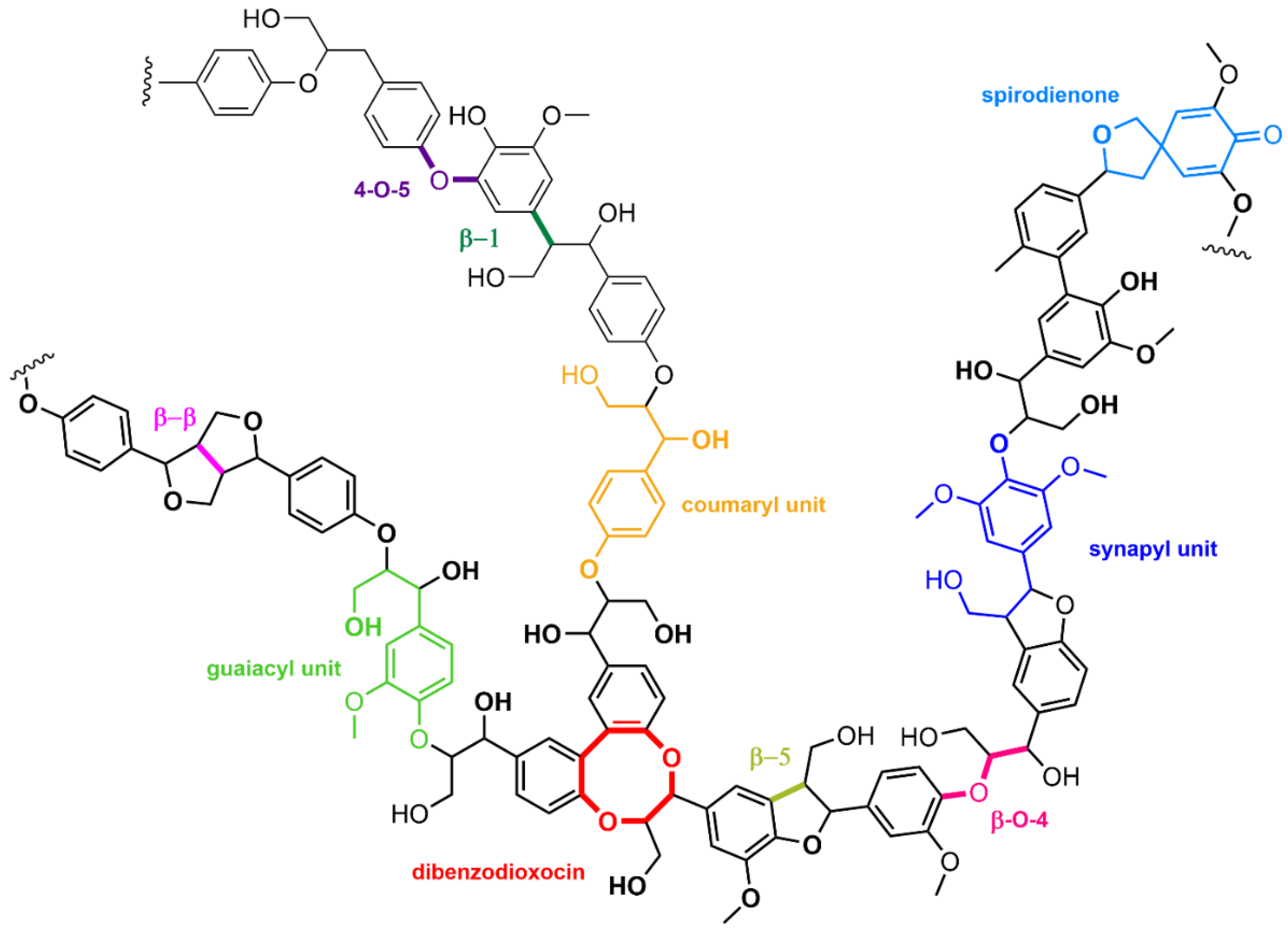
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
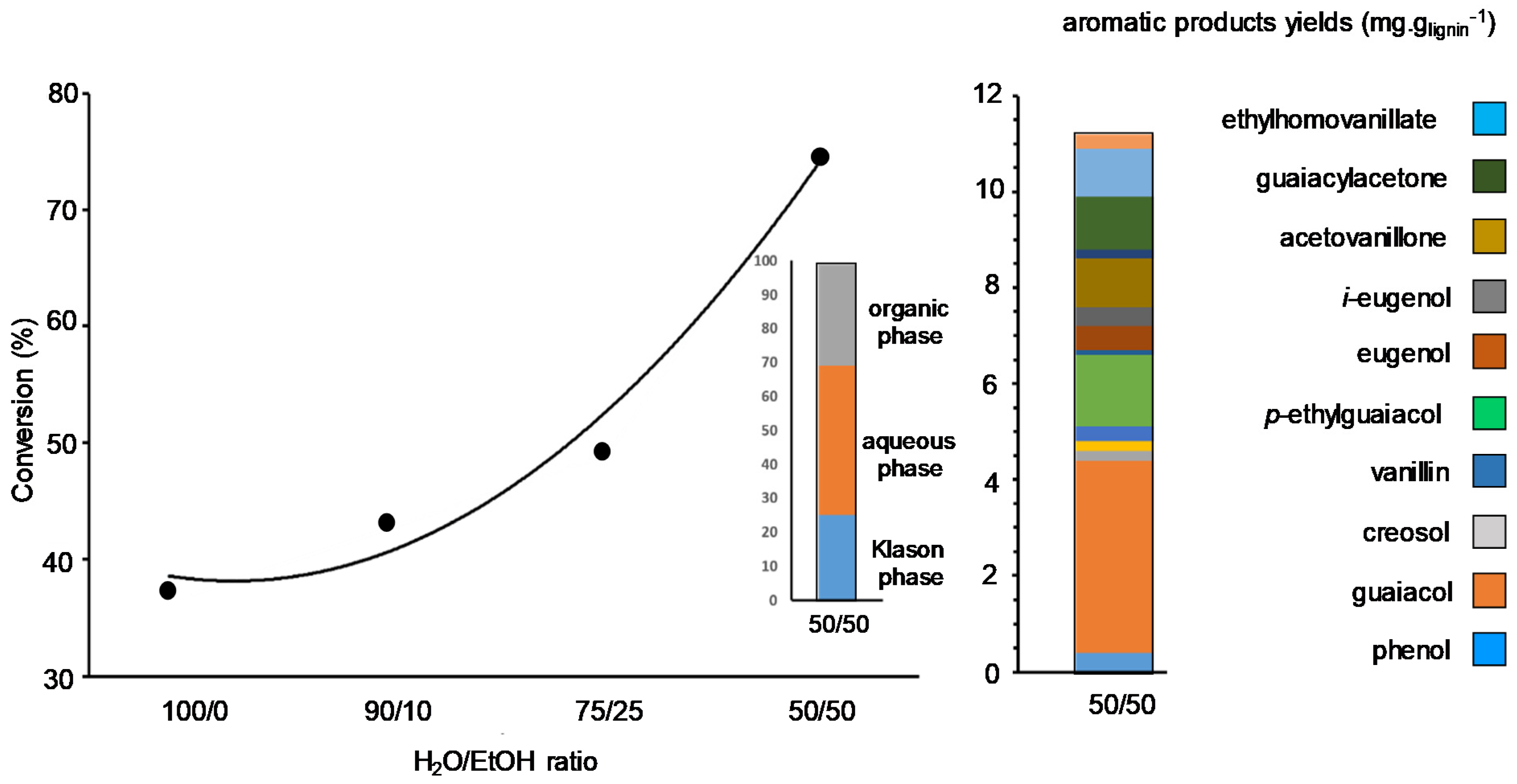
- Sebhat, W.; El-Roz, A.; Crepet, A.; Ladaviere, C.; Perez, D.D.S.; Mangematin, S.; Cabral Almada, C.; Vilcocq, L.; Djakovitch, L.; Fongarland, P. Comparative study of solvolysis of technical lignins in flow reactor. Biomass Conv. Biorefin. 2020, 10, 351–366. [Google Scholar] [CrossRef]
- Sebhat, W.; El Roz, A.; Fongarland, P.; Vilcocq, L.; Djakovitch, L. Catalytic liquefaction of Kraft lignin with solvothermal approach. Catalysts 2021, 11, 875. [Google Scholar] [CrossRef]
- Bourbiaux, D.; Pu, J.; Rataboul, F.; Djakovitch, L.; Geantet, C.; Laurenti, D. Reductive or oxidative catalytic lignin depolymerization: An overview of recent advances. Catal. Today 2021, 373, 24–37. [Google Scholar] [CrossRef]
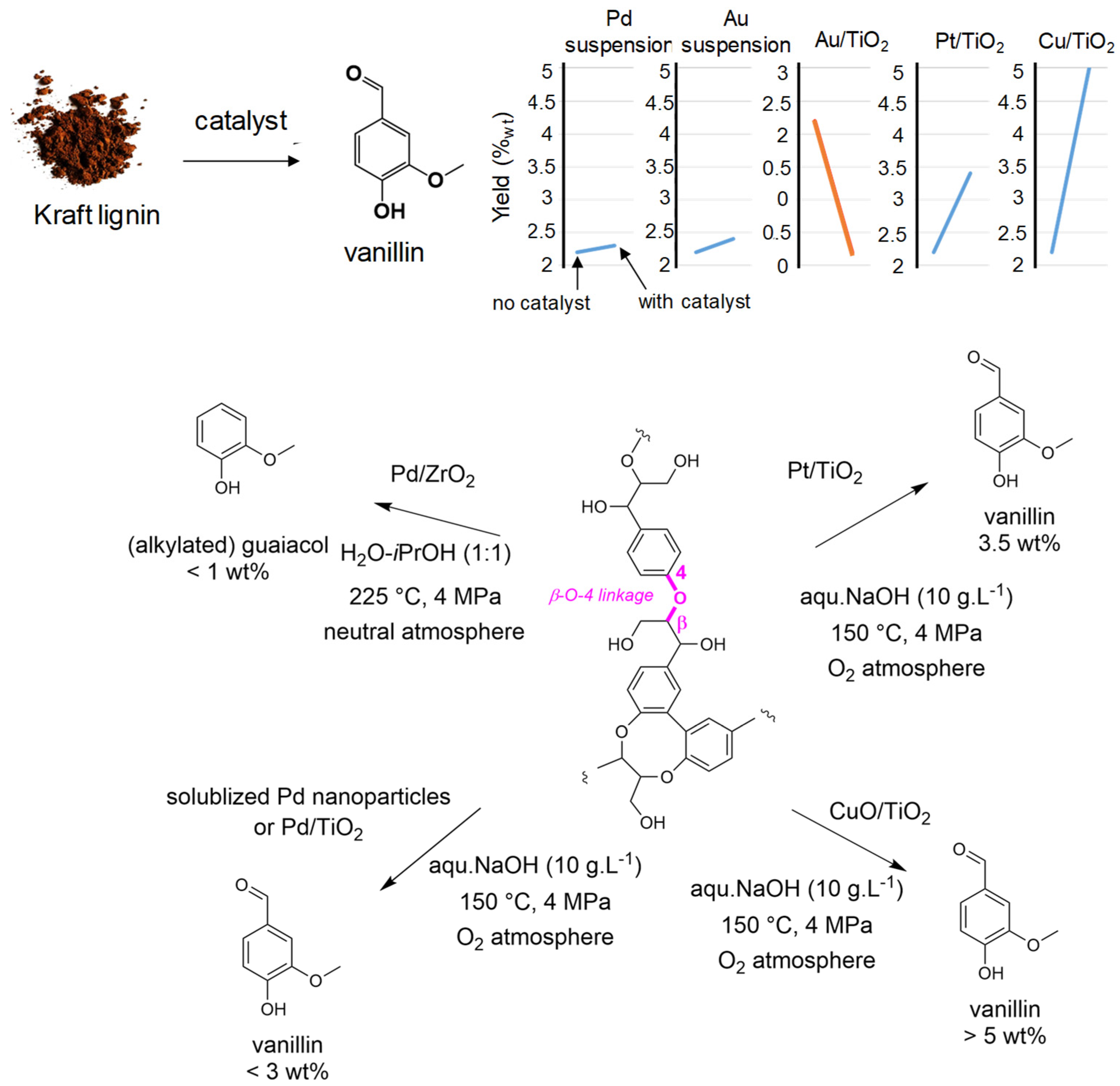
- Zhang, J.; Deng, H.; Lin, L. Wet aerobic oxidation of lignin into aromatic aldehydes catalyzed by a perovskite-type-oxide: LaFe1-xCuxO3 (x = 0, 0.1, 0.2). Molecules 2009, 14, 2747–2757. [Google Scholar] [CrossRef]
- Hdidou, L.; Kouisni, B.; Manoun, H.; Hannache, H.; Solhy, A.; Barakat, A. Oxidative conversion of lignin over cobalt-iron mixed oxides prepared via the alginate gelation. Catal. Commun. 2018, 117, 99–104. [Google Scholar] [CrossRef]
- Sales, F.G.; Maranhao, L.C.A.; Lima filho, N.M.; Abreu, C.A.M. Kinetic evaluation and modeling of lignin catalytic wet oxidation to selective production of aromatic aldehydes. Ind. Eng. Chem. Res. 2006, 45, 6627–6631. [Google Scholar] [CrossRef]
- Bourbiaux, D.; Mangematin, S.; Djakovitch, L.; Rataboul, F. Selective aerobic oxidation of benzyl alcohols with palladium(0) nanoparticles suspension in water. Catal. Lett. 2021, 151, 3239–3249. [Google Scholar] [CrossRef]
- Bourbiaux, D.; Xu, Y.; Goc, F.; Fongarland, P.; Philippe, R.; Aubert, G.; Aymonier, C.; Rataboul, F.; Djakovitch, L. Investigating (pseudo)-heterogeneous Pd-catalysts for Kraft lignin depolymerisation under mild aqueous basic conditions. Catalysts 2021, 11, 1311. [Google Scholar] [CrossRef]
- Cabral Almada, C.; Kazachenko, A.; Fongarland, P.; Da Silva Perez, D.; Kuznetsov, B.N.; Djakovitch, L. Oxidative depolymerization of lignins for producing aromatics: Variation of botanical origin and extraction methods. Biomass Conv. Biorefin. 2020, in press. [Google Scholar] [CrossRef]
- Cabral Almada, C.; Kazachenko, A.; Fongarland, P.; Da Silva Perez, D.; Kuznetsov, B.N.; Djakovitch, L. Supported-metal catalysts in upgrading lignin to aromatics by oxidative depolymerization. Catalysts 2021, 11, 467. [Google Scholar] [CrossRef]
- Hernandez Manas, A.; Mangematin, S.; Vilcocq, L.; Fongarland, P.; Djakovitch, L. From technical lignins to aromatics: A study on oxidative depolymerization in batch and continuous reactor. In Proceedings of the 5th International Congress on Catalysis for Biorefineries (CatBior-5), Turku, Finland, 23–27 September 2019. [Google Scholar]
- Hernandez Manas, A.; Zhou, S.; Djakovitch, L.; Mangematin, S.; Liu, S.; Vilcocq, L.; Fongarland, P. Lignin depolymerization in oxidative conditions: From batch to continuous reactor. In Proceedings of the 4th Iberoamerican Congress on Biorefineries, Jaén, Spain, 24–26 October 2018. [Google Scholar]
- Yamaguchi, A.; Sato, O.; Mimura, N.; Hirosaki, Y.; Kobayashi, H.; Fukuoka, A.; Shirai, M. Direct production of sugar alcohols from wood chips using supported platinum catalysts in water. Catal. Commun. 2014, 54, 22–26. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, T.; Liao, Y.; Cai, C.; Tan, J.; Wang, T.; Qiu, S.; He, M.; Ma, L. Production of C5/C6 sugar alcohols by hydrolytic hydrogenation of raw lignocellulosic biomass over Zr based solid acids combined with Ru/C. ACS Sustain. Chem. Eng. 2017, 5, 5940–5950. [Google Scholar] [CrossRef]
- Li, C.; Zheng, M.; Wang, A.; Zhang, T. One-pot catalytic hydrocracking of raw woody biomass into chemicals over supported carbide catalysts: Simultaneous conversion of cellulose, hemicellulose and lignin. Energy Environ. Sci. 2012, 5, 6383–6390. [Google Scholar] [CrossRef]
- Li, X.; Guo, T.; Xia, Q.; Liu, X.; Wang, Y. One-pot catalytic transformation of lignocellulosic biomass into alkylcyclohexanes and polyols. ACS Sustain. Chem. Eng. 2018, 6, 4390–4399. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.-K.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Bui, N.Q.; Fongarland, P.; Rataboul, F.; Dartiguelongue, C.; Charon, N.; Vallée, C.; Essayem, N. FTIR as a simple tool to quantify unconverted lignin from chars in biomass liquefaction process: Application to SC ethanol liquefaction of pine wood. Fuel Process. Technol. 2015, 134, 378–386. [Google Scholar] [CrossRef]
- Putro, J.N.; Soetaredjo, F.E.; Lin, S.-Y.; Ju, Y.-H.; Ismadji, S. Pretreatment and conversion of lignocellulose biomass into valuable chemicals. RSC Adv. 2016, 6, 46834–46852. [Google Scholar] [CrossRef]
- Silveira, M.H.L.; Morais, A.R.C.; da Costa Lopes, A.M.; Olekszyszen, D.N.; Bogel-Łukasik, R.; Andreaus, J.; Pereira Ramos, L. Current pretreatment technologies for the development of cellulosic ethanol and biorefineries. ChemSusChem 2015, 8, 3366–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eternot, M.; Rataboul, F.; Essayem, N. SC organic solvents coupled to heterogeneous catalysis: A unique tool for selective wood components liquefaction into chemicals. In Proceedings of the 4th International Congress on Catalysis for Biorefineries (CatBior-4), Lyon, France, 11–15 December 2017. [Google Scholar]
- Bui, N.Q.; Fongarland, P.; Rataboul, F.; Dartiguelongue, C.; Charon, N.; Vallée, C.; Essayem, N. Controlled pinewood fractionation with supercritical ethanol: A prerequisite toward pinewood conversion into chemicals and biofuels. Comptes Rendus Chim. 2018, 21, 555–562. [Google Scholar] [CrossRef]
- Eternot, M.; Rataboul, F.; Essayem, N. Sequential wood components liquefaction using supercritical organic solvents mixtures implemented in a flowthrough reactor. In Proceedings of the 18th European Meeting on Supercritical Fluids (Webconference), online, 4–6 May 2021. [Google Scholar]
- Essayem, N.; Sapaly, G.; Eternot, M.; Rataboul, F. Method for Preparing Levulinic Acid Esters; PCT WO2014001486; CNRS and University Claude-Bernard Lyon 1: Villeurbanne, France, 2014. [Google Scholar]
- Rataboul, F.; Sapaly, G.; Eternot, M.; Essayem, N. Acid-catalyzed direct formation of sec-butyl-levulinate from the reaction of cellulose with 2-butene. In Proceedings of the 15th International Congress on Catalysis (ICC-15), Munich, Germany, 1–6 July 2012. [Google Scholar]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djakovitch, L.; Essayem, N.; Eternot, M.; Rataboul, F. A Landscape of Lignocellulosic Biopolymer Transformations into Valuable Molecules by Heterogeneous Catalysis in C’Durable Team at IRCELYON. Molecules 2021, 26, 6796. https://doi.org/10.3390/molecules26226796
Djakovitch L, Essayem N, Eternot M, Rataboul F. A Landscape of Lignocellulosic Biopolymer Transformations into Valuable Molecules by Heterogeneous Catalysis in C’Durable Team at IRCELYON. Molecules. 2021; 26(22):6796. https://doi.org/10.3390/molecules26226796
Chicago/Turabian StyleDjakovitch, Laurent, Nadine Essayem, Marion Eternot, and Franck Rataboul. 2021. "A Landscape of Lignocellulosic Biopolymer Transformations into Valuable Molecules by Heterogeneous Catalysis in C’Durable Team at IRCELYON" Molecules 26, no. 22: 6796. https://doi.org/10.3390/molecules26226796