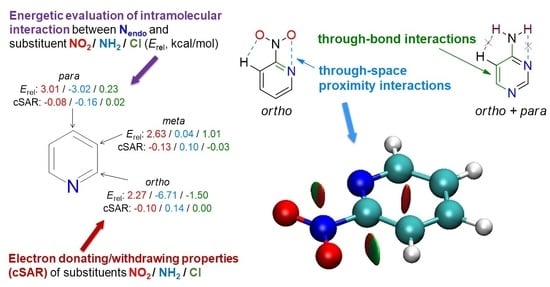
3.1. Energetic Characteristics of Substituents
The energy of the substituent,
Erel, can be calculated concerning various reference systems. However, by using benzene as a reference system (
Scheme 1), it is possible to differentiate signs between the electron-withdrawing group, NO
2 (+), and electron-donating group, NH
2 (−), as shown in
Table 1. In the case of X = NH
2, the
Erel values range from 1.01 to −15.8 kcal/mol. 5-X-quinoline is the only case where
Erel is substantially positive. On the other hand, the nitro substitution of the studied heterocycles results in the positive energy effect only,
Erel, ranging from 0.59 to 8.55 kcal/mol. Chlorine substitution leads to
Erel values varying from −2.25 to 2.60 kcal/mol. So, contrary to the strongly electron-donating or withdrawing amino and nitro groups, the values are positive or negative. It can be concluded that swapping one or more carbon atoms for nitrogen atoms in the aromatic ring generally results in a decrease in the stability of the nitro-substituted systems and an increase in the stability of the amino-substituted compounds. Therefore, regarding X = NO
2 and NH
2, electronic interactions between the substituent and the substituted system are a major factor affecting the stability of the substituted molecule. Other effects, such as the substitution position, especially the
ortho effect, can change the extent of the stabilization but cannot change the character (stabilizing/destabilizing) of the interaction between the X group and the substituted N-heterocyclic moiety.
Interestingly, the
Erel values derived from additivity of interactions (
Erel-add) in some cases differ only slightly from the values obtained from Eq. (1). In order to calculate
Erel-add, three cases of intramolecular interactions with endocyclic nitrogen atom were considered: (i)
ortho (o), as in 2-X-pyridine, (ii)
meta (m), as in 3-X-pyridine, and (iii)
para (p), as in 4-X-pyridine. For example, the
Erel-add for 4-NO
2-pyrimidine (
o,
p) was calculated as the sum of the
Erel in the
ortho (2-X-pyridine, 2.27 kcal/mol) and
para (4-X-pyridine, 3.01 kcal/mol) systems:
Erel-add (o,p) =
Erel(o) +
Erel(p). Hence, more complicated intramolecular interactions can be approximately separated into a sum of simpler ones. For 5-, 6-, and 7-X- quinolines, the
Erel values differ only slightly from the
Erel values for 1- and 2-X- naphthalenes (
Erel-add given in
Table 1) [
14]. This indicates that the N atom in the adjacent ring does not strongly affect the substituent energy.
The systems in which
Erel-add highly differs from
Erel are characterized by significant proximity interactions. For example, in 2-NO
2-pyrimidine, the existence of two O∙∙∙N interactions causes a substantial rotation of the nitro group around the C−N bond (by 56°), which further increases the destabilization of this system. Consequently, the additivity of interactions predicts a lower than the actual
Erel value. Other systems where such rotation occurs are the 4-NO
2-quinoline (35°), 5-NO
2-quinoline (28°), 8-NO
2-quinoline (58°), and NO
2-triazine (66°). Substantial difference between
Erel and
Erel-add also occurs in 8-NH
2-quinoline, where N∙∙∙HN H-bond is present (see
Section 3.3). In the case of X = Cl, such differences can be observed for (
o,
o) systems (X-triazine, 2-pyrimidine).
Despite the repulsive O∙∙∙N contacts occurring in ortho nitro substituted heterocycles, the energetic effect of such interactions is smaller than in meta and para systems. This is clearly seen by comparing 2-, 3-, 4-NO2-pyridines and 2-, 3-, 4-NO2-quinolines. Another example of this effect can be seen in 4- and 5-NO2-pyrimidines. The (m, m) 5-derivative has the Erel value higher by 0.32 kcal/mol than the (o, p) 4-system. The difference is similar to that between 2- and 3-NO2-pyridine (0.36 kcal/mol).
As the Erel values show, the para interaction between the nitro group and the endocyclic N atom leads to the highest destabilizing effect in the considered heterocycles.
Substitution by the chlorine results in values of Erel that are both positive and negative. The latter are observed only in the ortho systems. It should also be noticed that Cl derivatives are characterized by a smaller range of Erel variability than NH2 and NO2.
Regarding the substitution by the electron-donating amino group, the most energetically favored is the
ortho position with respect to the endocyclic nitrogen. The exchanging of more carbons for nitrogens in the ring results in better stabilization, as shown for pyrimidines and triazine in
Table 1. Interestingly, unlike in the nitro derivatives, the
meta interaction does not lead to any energetic effects (
Erel ~ 0 kcal/mol). It means that the
meta interaction with endo nitrogen (or double
meta, as in 5-NH
2-pyrimidine) is energetically the same as if the ring consisted of only carbon atoms (benzene). The
Erel values for 4-NO
2- and 4-NH
2- pyridines indicate that
para interactions of nitro and amino groups have roughly the same absolute energetic effect (~3 kcal/mol). However, the direction of effects is opposite—stabilization in amino and destabilization for nitro derivatives. A simple explanation of this phenomenon comes from the analysis of resonance structures of 4-NO
2 and 4-NH
2-pyridine, shown in
Scheme 2. In a very unlikely structure (2), with a positive charge at the highly electronegative nitrogen atom, the resonance effect of the NO
2 group has to be significantly disturbed. On the other hand, in the NH
2 derivative, the structure (6) has a negative charge on the nitrogen atom, which is more likely than a negative charge on the less electronegative carbon atom (as in aniline). Hence, the resonance effect of the NH
2 group is likely to be strengthened compared to that in aniline. Both the disruption of resonance for the NO
2 and the strengthening for the NH
2 are reflected in the electron-donating/withdrawing properties of substituents (see below).
3.2. Electronic and Geometric Properties of Substituents
Electronic properties of substituents, described by the cSAR index, provide further information on the reverse substituent effect. Data gathered in
Table 2 show that electronic properties are highly dependent on the position of substitution. The electron-donating/accepting strength of considered substituents in heterocycles can be compared with the appropriate strength in nitro-(cSAR(X) = −0.138), amino-(cSAR(X) = +0.094), and chlorobenzene (cSAR(X) = −0.051). All but one substitution of heterocycles with the NO
2 group result in lower EA strength of the NO
2 than in nitrobenzene. In 3-NO
2-quinoline, its slightly higher EA ability may come from the existence of a second aromatic ring, which can lend its π-electrons to the NO
2 group. In this case (and other quinolines), it is more reasonable to compare cSAR(X) to the appropriate value in 2-nitronaphthalene (−0.148) [
14]. Thus, replacing carbon with nitrogen highly influences the electronic interactions of NO
2 with the aromatic system, lowering the EA strength of this group. This is also reflected in the ∆pEDA values, which indicate a lower (relative to all-carbon substituted system—X-benzene or 2-X-naphthalene) occupancy of 2p
z natural atomic orbitals of atoms in the substituent (i.e., less electrons are transferred from the substituted moiety to the NO
2 group). On the other hand, all NH
2 substituted heterocycles have a higher ED strength as compared to aminobenzene, so the opposite effect to the nitro substitution is observed. This is clearly seen from all-negative ∆pEDA(X) values (
Table 2) (i.e., more electrons are transferred from the NH
2 group to the substituted moiety). This further supports the interpretation of heteroatom effect given in
Scheme 2.
Substitution by Cl leads to cSAR(X) values higher than that in chlorobenzene, even positive for
ortho derivatives. Thus, the electronic properties of Cl are slightly shifted towards electron donation. Additionally, relations in
Figure 2 show that cSAR(X) and ∆pEDA(X) electronic parameters are well correlated, with a clear separation of six-membered heterocycles and quinolines; however, for the nitro derivatives, only planar systems are considered. The cSAR(X) parameter does not correlate well with the
Erel energetic parameter for X = Cl and NO
2; however, it correlates well (
R2 = 0.948) for the X = NH
2 series (
Figure 3). It shows that changes in the electronic structure of the substituent are only partly responsible for
Erel variability. In other words, cSAR is the local substituent effect parameter, while
Erel characterizes the global substituent effect, including proximity interactions. In the case of X = NH
2,
ortho NH∙∙∙N interactions are responsible for a high stability increase, which is accompanied by an increase in the ED power of the substituent due to favorable electronic interactions (
Scheme 2). Thus, for the NH
2 series, these two parameters are well-correlated.
Susceptibility of ED/EA properties of X groups to the reverse substituent effect can be illustrated by ranges of variation of the cSAR(X) index. Thus, in terms of electronic properties, the NH2 group is the most sensitive to changes in the substituted moiety, the NO2 group is slightly less, and the Cl substituent is the least susceptible (the ranges of their cSAR variation are 0.190, 0.186, and 0.159, respectively).
Considering the particular substitution positions for the NO
2 group, the substitution in the
ortho position to the endocyclic N atom gives the lowest absolute values of the cSAR index. In the case of such “double
ortho” substitution, in 2-NO
2-pyrimidine and NO
2-triazine, the cSAR values are even slightly positive. Apart from
ortho interactions, the sterically induced rotation of the nitro group is another factor that weakens its electron-withdrawing power in these systems. Comparing the
meta and
para positions is particularly insightful, as any differences result from various electronic interactions. In the
meta substitution, the nitro group possesses the highest EA strength. However, looking at ∆pEDA values, this should be rather attributed to a weaker disruption of the resonance effect by N-heteroatom in the
meta position than to the enhancement of other electronic effects. As mentioned earlier, the
para substitution is energetically unfavorable, resulting in the weakest EA properties of the NO
2. It indicates a weakened π conjugation with the aromatic ring, also supported by lower occupancies of 2pz orbitals on the substituent, compared to the
meta derivatives, as shown by ∆pEDA (
Table 2). Furthermore, the weaker conjugation leads to an increase in CN bond lengths (
Table 3), lower
ρBCP(CN) values (
Table S1), and an increased
Erel. Hence, the conclusion drawn from the simple analysis of the resonance structures (
Scheme 2) can be confirmed by electronic descriptors obtained from the ground state wavefunction. An interesting observation can be made by looking at ∆pEDA(X) and ∆sEDA(X) values of systems where rotation of NO
2 occurs. Rotation significantly weakens electron-withdrawing by resonance (highly negative values of ∆pEDA), but this seems to be somewhat compensated by an enhanced inductive effect (positive values of ∆sEDA).
The amino group, as already mentioned, is stabilized in N-heterocycles with respect to benzene. Exceptional stabilization occurs in the
ortho position, where favorable proximity interactions are present. The NH
2 group is highly electron-donating in the
ortho position, as shown by the cSAR values in
Table 2. Low values of pEDA also point out to high ED resonance effect of this group. Moreover, CN bonds are relatively short (
Table 3), especially in double
ortho derivatives where two attractive NH∙∙∙N interactions are present. Shortening of the CN bond and a significant increase in ED properties also occur in
para derivatives. In this case, purely electronic interaction between the NH
2 group and the
endo N atom can be assumed. Its stabilizing and ED enhancing nature can be explained by resonance structures, shown in
Scheme 2. Moreover, the most robust ED character of the amino group is observed for amino-triazine, where double
ortho and
para effects are present. It is documented by both the largest cSAR(X) (0.292,
Table 2) and the shortest CN bond length (1.347 Å,
Table 3).
Meta interaction, which is energetically neutral, only slightly affects the electronic properties of the substituent. In 3-NH
2-pyridine, the cSAR value is higher by about 0.01 than that of aniline, which is much less than in
ortho (0.07) and
para (0.05) pyridine derivatives. Similar conclusions can be drawn from analyzing amino-substituted quinolines and pyrimidines. Additionally, when comparing
meta systems to
para and
ortho, the CN bond for
meta is longer by about 0.01 Å in pyridine and quinoline, and 0.02 Å in pyrimidine. However, it is still shorter by ~0.005 Å than that in aminobenzene. The shortened CN bonds are another implication of enhanced π-resonance with respect to aminobenzene.
Chlorine differs from the NH
2 and NO
2 groups due to the much weaker resonance effect. It is well known, however, that it can act in two directions, i.e., withdraw electrons by inductive effect and donate by resonance. As mentioned earlier,
ortho substitution leads to the positive values of cSAR(X), indicating ED properties, and its highest values are present in double
ortho systems (2-Cl-pyrimidine, Cl-triazine). Interestingly, these are the only cases for X = Cl when
Erel is negative. In contrast,
meta derivatives are characterized by the lowest values (negative), i.e., the strongest EA properties.
Para interaction weakens the EA properties of the chlorine, as can be seen, for example, by comparing the cSAR(X) of 3-and 4-Cl-pyridine (
Table 2). In
meta and
para cases, cSAR(X) values are positive, indicating electron donation. It follows that, in Cl derivatives, its ED properties are associated with negative
Erel (stabilization), whereas EA with positive
Erel (destabilization). It is well illustrated by the two groups of points for X = Cl laying in two quarters of the coordinate system in
Figure 3.
For all groups, the substitution of various positions in the full-carbon ring of quinoline (5-, 6-, 7-, 8-) leads to cSAR(X) values that do not differ substantially.
The substituent X can also be described by its geometry (
Figure 4). The geometry of substituent carries information about electronic interactions, as well as the proximity interactions, such as steric effects and hydrogen bonding. The obtained structural parameters are collected in
Table 3,
Table 4 and
Table S2; the ∆α is the difference between the angle α for substituted and unsubstituted molecules.
The data summarized in
Table 3 and
Table 4 show that the geometry of the substituent depends not only on the substituted moiety but also influences the angle
α (at
ipso ring carbon atom). The ED substituent (NH
2) reduces the
α angle with respect to the unsubstituted system (negative ∆
α values). The nitro group has an opposite effect, causing its increase (positive ∆
α values). Substitution by chlorine atom leads to an increase of
α, but a lower extent than the NO
2 group. It is consistent with the weaker EA strength of Cl (Hammett substituent constant,
σp = 0.23), than NO
2 (
σp = 0.78) [
34]. Additionally, the ∆
α ranges of variation are higher for NO
2 (1.35°) and NH
2 (1.12°) than for Cl (0.68°). It can also be attributed to the fact that the NO
2 and NH
2 groups exhibit a much higher variation in electronic properties than Cl and participate in specific
ortho interactions. Bond length
dCX (X = N or Cl) has the highest variability for X = NH
2 (0.0492 Å, 3.6% of the average bond length); for X = NO
2, it is slightly lower (0.0415 Å, 2.8%), and it is the lowest for X = Cl (0.0260 Å, 1.5%). It is understandable since the Cl group does not participate in through-space proximity interactions, as NH
2 and NO
2, and its electronic properties are less susceptible to change due to lack of the resonance effect.
As shown in
Table 3, NH
2 substitution in the
ortho position results in the smallest changes in α angle.
Meta substitution results in a 23% larger change, whereas
para substitution causes the highest decrease of α (43%). In the case of 8-NH
2-quinoline, the most significant change in α occurs, which is probably a geometric distortion related to the formation of a favorable NH∙∙∙N hydrogen bond. This hydrogen bond fulfills the Koch-Popelier criteria [
35] (
ρBCP = 0.0170, ∇²
ρBCP = 0.0766 a.u.). In previously investigated polycyclic aromatic hydrocarbons [
14], the
Erel value obtained for the structurally similar 1-aminonaphthalene (differing only in the endo-N atom) equals 0.96 kcal/mol. Hence, the difference between
Erel for 8-NH
2-quinoline and 1-aminonaphthalene (−4.34 kcal/mol) can be considered an approximation of the stability gain due to the existence of a hydrogen bond. The value of the ∠HNH angle is highly related to the electronic properties of the NH
2 group (
Table 5). The most important factors in this case, however, are proximity interactions.
Ortho systems show the highest values of ∠HNH, which indicates how the NH
2 group geometry is changed upon the formation of attractive NH∙∙∙N interactions. Generally, when strongly ED enhancing and stabilizing
ortho and
para electronic interactions occur, the ∠HNH angle is greater. For the substituted pyridine derivatives, it is 115.3° and 114.1°, respectively, while, for 3-NH
2-pyridine, it is 112.5°. The highest value corresponds to a system with double
ortho and single
para interaction—NH
2-triazine (120.9°). Additionally, the differences in its values between 4-NH
2-pyrimidine (117.6°) and NH
2-pyrazine (115.7°), or 3- and 4-NH
2-pyridine, can be solely associated with stronger
para interactions in 4-NH
2 derivatives than
meta in 3-NH
2-pyridine and NH
2-pyrazine.
Regarding the NO2 substitution, the most significant changes in the α angle occur for double-ortho systems. In these molecules, the NO2 group undergoes rotation along the CN bond to reduce steric strain. It significantly disturbs the resonance effect of the NO2 group, due to which the highest ∠ONO angles are observed.
As already mentioned above, the range of variability of ∆α is the smallest for the Cl substitution (
Table 4). Moreover, in all cases, the ∆α values are lower than for chlorobenzene (∆α = 1.42°,
Table S1). The same goes for its electron-accepting ability (in Cl-Ph cSAR(Cl) = −0.0505,
Table S2). Thus, the ring nitrogen atom(s) decreases the EA strength of the Cl group.
Changes in geometry resulting from the electronic interactions with the endo-N atom can be noticed by looking at the α angles. According to the Bent-Walsh rule [
36,
37], α angle should be well-correlated with the electronic properties of the substituent. As shown in
Figure S2, the determination coefficients (
R2) of the obtained relations are less than 0.7.
However, considering only six-membered derivatives, the
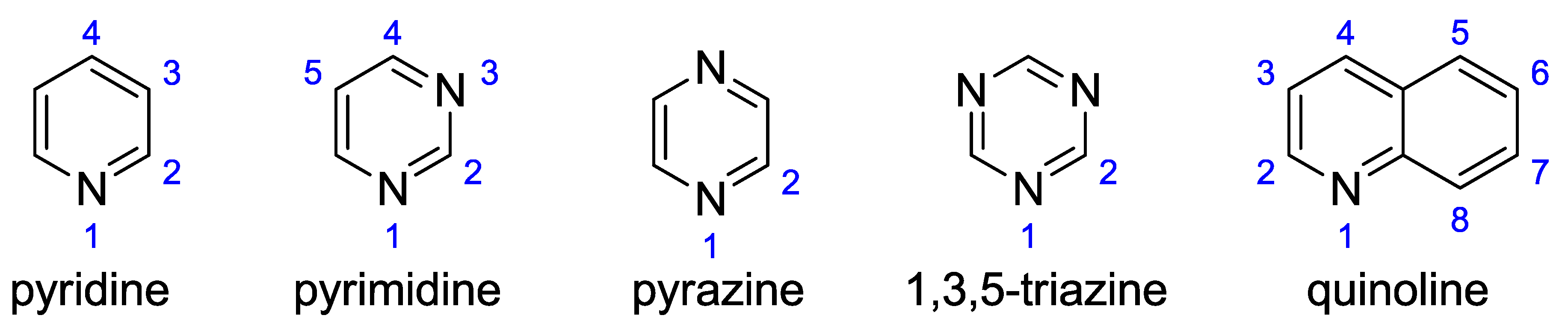
R2 increases and is greater than 0.75. So, the Bent-Walsh rule works. In the case of quinolines, both the position of the substitution and the additional ring influence the geometry and properties of the substituents. For example, for 4, 5, and 8 positions in quinoline (as marked on
Scheme 1), a significant steric strain is imposed upon substitution, which perturbs the geometric parameters and lowers determination coefficients.
Pearson correlation coefficients (
cc) between the pairs of the considered geometric, electronic, and energetic parameters of the six-membered heterocyclic derivatives are gathered in
Table 5 and
Table 6. It can be immediately noticed that, for the NH
2 group,
cc values are higher, and more parameters are mutually correlated. For the NO
2 substitution, less parameters are well correlated. In this case, worse correlations may result from significant proximity interactions of the bulky NO
2 group, that are present in
ortho derivatives. The least significant correlations exist for Cl, where only relations between ∆α, cSAR, and pEDA have an absolute value of
cc above 0.80. This is probably due to smaller variability of the parameters in Cl derivatives than in NH
2 and NO
2.
Indicative are not only the absolute values of
cc but also their signs. The sign of
cc is consistent with the sign of the slope in the linear equation between the parameters under consideration. For example, in the
Erel vs cSAR relation for nitro derivatives, the slope is positive (
cc = 0.82), while negative for amine systems (
cc = −0.99). It is illustrated, but for all tested derivatives, in
Figure 2.
In the case of nitro derivatives, changes in the structural parameters are generally consistent with the Bent-Walsh rule. Increasing the α angle causes the CN bond to be lengthened (cc = 0.73), which results in a dNY(N) bond shortening (cc = −0.95). Increasing the angle α also increases the angle ∠YNA (cc = 0.86). The latter is also observed for amine systems, but changes in other geometric parameters of the amine group do not comply with the Bent-Walsh rule. For example, increasing the α angle shortens the CN bond (cc = −0.81). However, much better correlations between pairs of its structural, electronic, and energy parameters were found.
3.3. Proximity Interactions
Proximity interactions are clearly noticeable when analyzing NO and NH bond lengths, as well as ∠YNY and ∠CNY angles. In nitro-substituted heterocycles with one N atom in the
ortho position (2-X-pyridine, X-pyrazine, 2-X-quinoline, 4-X-pyrimidine) the NO bonds directed towards the
endo N atom are shorter by about 0.014 Å than those directed towards the H atom (
Table 3). Additionally, the ∠ONO angles are slightly larger, and there is a marked discrepancy between the two ∠CNO angles. In
ortho amino-substituted heterocycles, differences in length between the two NH bonds are very slight (few thousands of Å); however, the NH(N) bonds are always longer, as a consequence of NH∙∙∙N interaction. The ∠CNH(N) angles are lower than ∠CNH(H), which is another proof of the attractive character of the NH∙∙∙N interaction.
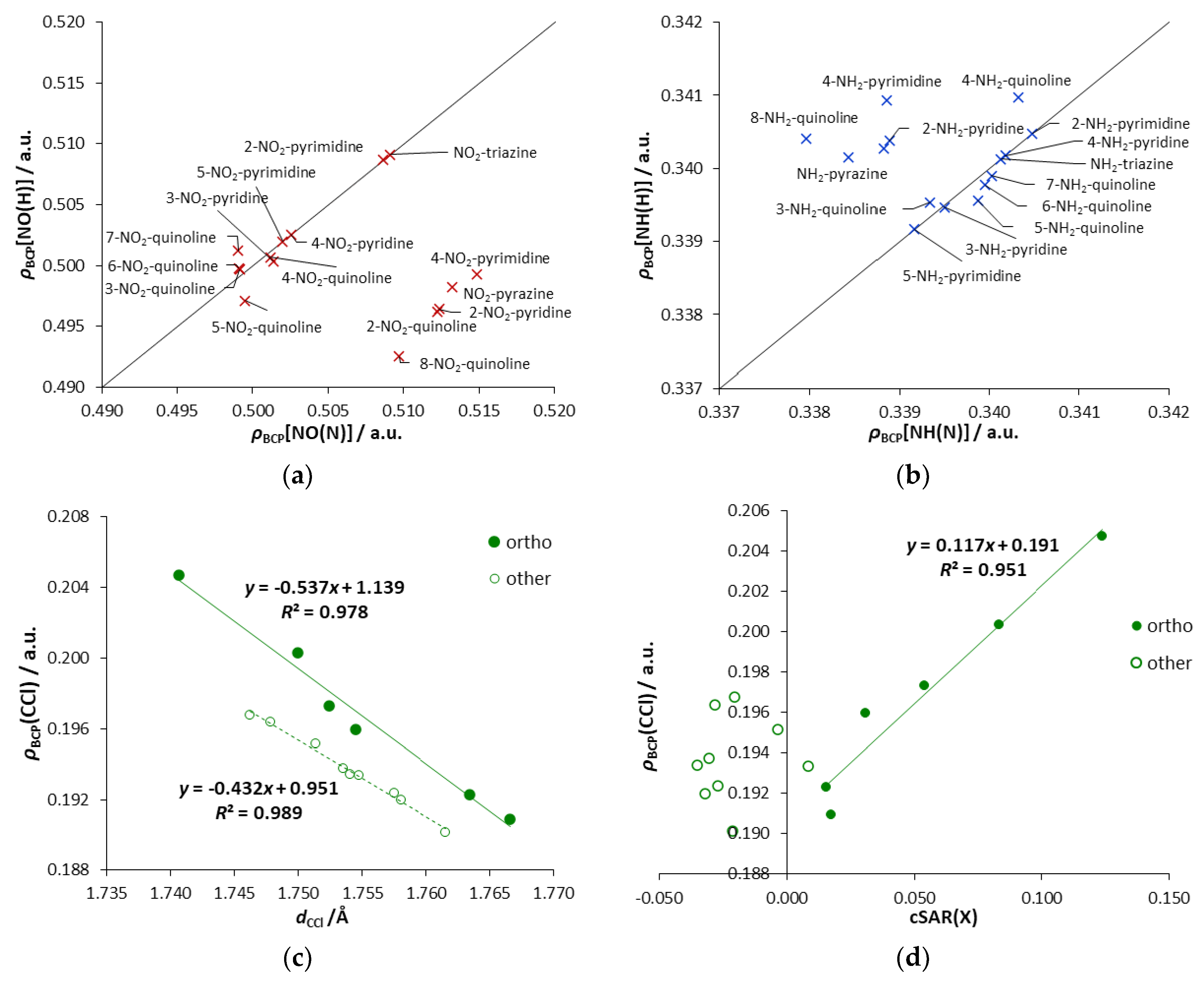
Asymmetricity of interactions can also be expressed with the values of
ρBCP(NY), as shown in
Figure 5a,b. Moreover, the character of these interactions can also be seen in these plots. Points below the y = x line have an increased electron density at BCP[NY(N)] compared to BCP[NY(H)]. It is connected with the NO(N) bond’s shortening due to the repulsive interaction, for derivatives when only one
endo N atom is in the
ortho position. Points above the y = x line are slightly depleted of electron density, indicating the presence of an attractive interaction. The reduction of the electron density is related to the elongation of the NH(N) bond (
Table 3), which may be due to its participation in hydrogen bonding. Such depletion is one of the criteria for the existence of hydrogen bond, as pointed out by Koch and Popelier [
35].
However, in this case, other criteria, such as the existence of a bond critical point between the two hydrogen-bonded atoms, are not fulfilled. Moreover, the reduced density gradient (RDG) function analysis does not reveal a bonding character of NH∙∙∙N interactions (
Figure 6; data for all systems are shown in
Figure S3). The low NH∙∙∙N angle and the H∙∙∙N distance (in 2-NH
2-pyridine, respectively: 71° and 2.430 Å) are possible reasons for this. The only NH∙∙∙N interaction detected by RDG analysis (at 0.50 isosurface) appears in 8-NH
2-quinoline (H∙∙∙N distance: 2.286 Å, NH∙∙∙N angle: 104°). As mentioned earlier, for this interaction, a bond critical point was detected. Detection by both approaches indicates its superior strength compared to other proximity interactions. Close NH∙∙∙HC contacts in 4- and 5-NH
2-quinolines (respectively, 2.056 and 2.120 Å) may indicate dihydrogen interactions. These interactions are detected by RDG as van der Waals in character (
Figure 6). Moreover, in 4-NH
2-quinoline, where closer contact exists, QTAIM analysis indicates the existence of a bond critical point between the two hydrogens (
ρBCP = 0.0111; ∇
2ρBCP = 0.0469 a.u.). However, the short distance between the BCP and RCP (0.397 Bohr) puts in doubt the stability of this critical point.
Regarding X = Cl, despite the stabilization of the
ortho derivatives mentioned above (
Table 1), no
ortho interactions, such as the halogen bonding [
38], are observed. Precisely, no spatial requirements for its creation are fulfilled because the σ-hole of Cl is located in a different region than the lone pair of the N atom (Lewis base) is directed, as seen on the electrostatic potential (ESP) map (
Figure S4c, where σ-hole appears as a reddish spot above the Cl atom). Therefore, the observed stabilization comes from the electronic interactions between Cl and the endo N atom. An interesting observation can be made from the plot
ρBCP (CCl) versus
dCCl, shown in
Figure 5c. Two distinctive series of points are noticeable. One of them corresponds to the
ortho systems, while the second to other systems (
meta,
para, quinolines substituted in all-carbon ring). It indicates the existence of some specific stabilizing electronic interactions occurring between the endo-N and the Cl atom in the
ortho position. Their stabilizing character is confirmed by
Erel (negative values,
Table 1); for the remaining derivatives, the obtained
Erel values are positive. Taking into account
ρBCP(CCl), its range of variability for
ortho systems is more than twice as extensive as for other derivatives. Almost the same applies to changes in the length of the CCl bonds. The most substantial stabilizing electronic interactions are observed for Cl-triazine (highest electron density at BCP of CCl bond), followed by 2-Cl- and 4-Cl-pyrimidine. In the first case, the two
ortho interactions are enhanced by the
para endo-N effect. In 2-Cl-pyrimidine, there are two
ortho interactions, while, in 4-Cl-pyrimidine, there are
ortho and
para interactions. Moreover, in all
ortho systems, the obtained cSAR values reveal the ED ability of the Cl group (
Figure 5d).
The only proximity interactions of Cl detected by RDG analysis are close Cl∙∙∙H contacts in 4- and 5-Cl-quinolines, classified as van der Waals interactions (
Figure 6). Additionally, in 8-Cl-quinoline, Cl∙∙∙N contact is classified as partly steric and partly van der Waals in character.
The results of the RDG function analysis reveal interesting proximity effects of the NO
2 group. As shown in
Figure 6, the NO∙∙∙HC interaction is detectable in the RDG plots as an attractive interaction with strength in between the hydrogen bonding and the van der Waals interaction (sign[λ
2(r)]
∙ρ(r) < 0). For NH∙∙∙N interaction, no hydrogen bond critical point is observed, but, instead, the O∙∙∙HC angle and O∙∙∙H distance are more favorable (in 2-NO
2-pyridine, respectively, 95° and 2.396 Å) compared to the NH∙∙∙N ones. Although the two O∙∙∙H interactions are present in
meta and
para derivatives, the O∙∙∙H distance is 0.03 to 0.05 Å longer (
Table S1). RDG plots, presented in
Figure 6, show that they can be classified as van der Waals interactions. The geometry of the
ortho NO
2 group is deformed by the interaction with the endo-N atom, which strengthens the NO∙∙∙HC interaction. Interestingly, RDG isosurfaces suggest that NO∙∙∙N contacts are not purely repulsive (sign[λ
2(r)]
∙ρ(r) > 0). A contribution of the van der Waals interaction can be seen as a green-colored fragment (corresponding to the sign[λ
2(r)]
∙ρ(r) ~ 0) on the RDG isosurface between the O and N atoms (
Figure 6). On the other hand, there is no indication of attractive interaction between these atoms on ESP plots (
Figure S4a). Namely, the ESP value on the surface in the vicinity of the O and N atoms is negative.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}