4. Materials and Methods
All non-aqueous reactions were carried out under an atmosphere of nitrogen or argon using oven-dried glassware that was cooled in a desiccator prior to use. Unless otherwise noted, starting materials and reagents were obtained from commercial suppliers and were used without further purification. Anhydrous solvents were either obtained from commercial suppliers or were obtained from a GlassContour solvent purification system (SPS), using activated alumina. Aqueous solutions of inorganic salts are represented as (volume; % aq. or w/w aq.). Saturated aqueous solutions of inorganic salts are represented as (volume; sat. aq.).
Nuclear magnetic resonance (NMR) spectra were recorded at ambient temperature (298 K, unless otherwise stated) on a Bruker AVA400, AVA500 or AVA600 spectrometer running at 400, 500, or 600 MHz (1H spectra) or 101, 126, 151 Hz (13C spectra, respectively. Chemical shifts (δ values) are reported in parts-per-million (ppm) relative to tetramethylsilane (1H and 13C spectra; δTMS = 0) and are corrected to the residual solvent peak. 1H NMR data are reported as follows: chemical shift, relative intensity, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, p = pentet, m = multiplet, br = broad), coupling constants (J value, Hz), and interpretation. 13C NMR data are reported as follows: chemical shift, relative intensity and assignment (C = quaternary, CH = methane, CH2 = methylene, CH3 = methyl).
Infra-red spectra (IR) were recorded neat on Shimadzu IRAffinity-1. The value of peaks at maximum absorbance (νmax) are quoted in wavenumbers (cm−1), relative intensity (str = strong, med = medium, w = weak, br = broad).
Optical rotations were obtained at ambient temperature with a cell of 1 dm length using an Optical Activity Ltd. AA series polAAr 20 spectrometer operating at a wavelength of 589 nm (sodium D-line). Concentrations (c) for the derived specific rotations ([α]D) are expressed in g/100 mL.
Melting points (mp) were determined on a Gallenkamp Electrothermal Melting Point apparatus and are uncorrected, the temperature range and whether the substance undergoes decomposition (dec.) over this range is reported.
Rf values (Rf) were recorded using Merck Silicagel 60 F254 aluminium backed plates. Flash chromatography was carried out using Merck Kieselgel 60 (Merck 9385) under positive pressure. Eluent compositions are quoted as v/v ratios (+ additive if used).
Mass spectra were obtained by: electrospray (ESI) on a Bruker 12 T SolariX, Waters Synapt G2 or Bruker microTOF II; electron ionisation (EI) on a MAT 900 XP mass spectrometer. Mass-to-charge ratios (m/z) of all parent (molecular) ions ([M]+/−) and their intensities are reported, followed by (major) fragment or adduct ions and their intensities. MS/MS experiments were recorded on a Waters Synapt G2, using CID fragmentation.
Preparative RP-HPLC was performed using a Waters® 600E pump connected to a Waters® 486 tunable absorbance detector equipped with a Phenomenex Luna C18(2), 5 µm, 250 × 21.2 mm column. Solvents used for preparative chromatography were A: H2O (+0.1% v/v TFA) and B: MeCN (+0.1% v/v TFA) and the flow rate was 18 mL min−1. Samples were introduced by a Rheodyne, and were either dissolved in the starting purification solvent or the minimum amount of DMSO. Purifications used isocratic elutions where the % B (MeCN + 0.1% TFA) and retention time (Rt) are stated.
Histone H4K12C was prepared as described previously [
4]. SCP-2L was expressed and purified as described previously [
32].
Benzyl 2-azidoethanoate (
2a), benzyl bromoacetate
1a (3.97 mL, 25.0 mmol) was added dropwise to NaN
3 (30 mL; 1 M in DMSO) at 5 °C and was stirred at r.t. for 18 h. The reaction was cooled to around 10 °C, diluted with H
2O (80 mL) and extracted with Et
2O (3 × 30 mL), the combined organics were washed with brine (50 mL) dried over MgSO
4 and concentrated in vacuo. The product was used without further purification yielding
2a as a colourless oil (4.81 g, quantitative); R
f (1:1 Hex/EtOAc) = 0.35; IR (neat, cm
−1) 2104 (str, N
3), 1741 (str, C=O);
1H NMR δ (600 MHz, CDCl
3) 7.47–7.30 (5H, m, 5 × Ar
H), 5.23 (2H, s, ArC
H2), 3.91 (2H, s, C
H2N
3);
13C NMR δ (126 MHz, CDCl
3) 168.3 (C), 135.0 (C), 128.8 (3 × Ar CH), 128.7 (2 × Ar CH), 67.7 (CH
2), 50.5 (CH
2). Spectroscopic data are in good agreement with the literature [
39].
![Molecules 26 05461 i002]()
Benzyl 2-[4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl]ethanoate (
3a), benzyl 2-azidoacetate
2a (4.81 g, 25.0 mmol) was dissolved in anhydrous, degassed DCM/MeOH (50 mL). Cu(MeCN)
4·BF
4 (370 mg, 5 mol%) and propargyl alcohol (1.75 mL, 30.0 mmol,) were added and the solution stirred at r.t. for 5 h under argon. Celite was added to the reaction mixture which was then concentrated in vacuo. Dry loading the celite layer allowed the product to be purified by FCC (EtOAc) to yield
3b as a colourless solid (4.52 g, 73% yield); R
f (EtOAc) = 0.3; mp 133–135 °C [lit., 130–131 °C]; IR (neat, cm
−1) 3242 (br, OH), 1747 (str, C=O);
1H NMR δ (500 MHz, CDCl
3) 7.70 (1H, br s, C=C
H), 7.43–7.32 (5H, m, 5 × Ar
H), 5.24 (2H, s, C
H2CO
2Bn), 5.20 (2H, s, C
H2Ar), 4.84 (2H, br s, C
H2OH), 2.12 (1H, br s, O
H).
1H NMR δ (500 MHz,
d6-DMSO) 7.98 (1H, s, C=C
H), 7.44–7.32 (5H, m, 5 × Ar
H), 5.44 (2H, s, NC
H2), 5.23 (1H, t,
J = 5.7 Hz, O
H), 5.21 (2H, s, ArC
H2), 4.54 (2H, d,
J = 5.6 Hz, C
H2OH);
13C NMR δ (126 MHz,
d6-DMSO) 167.3 (C), 148.1 (C), 135.4 (C), 128.5 (2 × ArCH), 128.3 (ArCH), 128.1 (2 × ArCH), 124.1 (CH), 66.7 (CH
2), 55.0 (CH
2), 50.2 (CH
2);
m/
z (EI) 247 ([M]
+•, 10%), 91 ([M − C
5H
6N
3O
3]
+•, 100); HRMS (EI) [M]
+• found 247.0936, C
12H
13O
3N
3 requires 247.0951. Spectroscopic data are in good agreement with the literature [
40].
![Molecules 26 05461 i003]()
Benzyl 2-[4-chloromethyl-1H-1,2,3-triazol-1-yl]ethanoate (4a), to alcohol 3a (988 mg, 4.00 mmol), suspended CHCl3 (30 mL), was added SOCl2 (1 mL) which caused the solid to dissolve. The solution was stirred for 1 h at r.t. and the solvent was removed under a stream of N2 yielding a brown oil. CHCl3 (20 mL) was added followed by NaHCO3 (20 mL; sat. aq.). The organic layer was separated and the aqueous phase was extracted with CHCl3 (20 mL). The combined organics were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo giving compound 4a as a colourless solid (902 mg, 85% yield); Rf (1:1 Hexane/EtOAc) = 0.5; mp 130–131 °C; IR (neat, cm−1) 1747 (str, C=O); 1H NMR δ (600 MHz, d6-DMSO) 8.22 (1H, s, C=CH), 7.42–7.33 (5H, m, 5 × ArH), 5.49 (2H, s, CH2CO2Bn), 5.21 (2H, s, ArCH2), 4.87 (2H, s, CH2Cl); 13C NMR δ (126 MHz, d6-DMSO) 167.1 (C), 143.4 (C), 135.3 (C), 128.5 (2 × ArCH), 128.3 (CH), 128.0 (2 × ArCH), 125.7 (CH), 66.8 (CH2), 50.4 (CH2), 36.3 (CH2); m/z (EI) 267 ([37ClM]+•, 7%) 265 ([35ClM]+•, 21), 148 (48), 131 (100); HRMS (EI) [35ClM]+• found 265.0608, C12H1235ClO2N3 requires 265.0613.
![Molecules 26 05461 i004]()
2-[4-Methyl-1H-1,2,3-triazol-1-yl]ethanoic acid (6), chloride 4a (53.0 mg, 0.20 mmol) was dissolved in methanol (2 mL) with palladium on carbon (5.3 mg, 10% w/w) under an argon atmosphere with stirring. Triethylsilane (322 µL, 2.00 mmol) was added dropwise to the mixture over a period of 20 min via syringe at r.t. TLC indicated no starting material remained. The mixture was filtered through celite, washed with MeOH (50 mL) and concentrated in vacuo to yield a brown oil that was triturated with Et2O (10 mL) and CHCl3 (10 mL) yielding 6 as a brown oil (20 mg, 71% yield); 1H NMR δ (500 MHz, CD3OD) 8.43 (1H, s, C=CH), 5.54 (2H, s, CH2), 2.54 (3H, s, CH3); 13C NMR δ (126 MHz, CD3OD) 168.0 (C), 141.7 (C), 129.5 (CH), 54.0 (CH2), 8.9 (CH3); m/z (ESI-, MeOH) 140 ([M − H]−, 100); HRMS (EI) [M − H]+• found 140.0471, C5H6N3O2 requires 140.0466.
![Molecules 26 05461 i005]()
Benzyl 2-[4-methanesulfonylmethyl-1H-1,2,3-triazol-1-yl]ethanoate (S1), alcohol 3a (494 mg, 2.00 mmol) was dissolved in THF (50 mL) with Ms2O (1.04 g, 6.00 mmol) and cooled to 0 °C with stirring. Triethylamine (1.67 mL, 6.00 mmol) was added dropwise and the reaction stirred at 0 °C for 30 min. The reaction was concentrated in vacuo and DCM (20 mL) and NH4Cl (30 mL; sat. aq.) were added. The organic layer was separated and the aqueous phase extracted with DCM (2 × 20 mL). The organics were combined, dried over MgSO4 and concentrated in vacuo yielding S1 as a brown oil that was used without further purification (650 mg, quantitative); Rf (1:1 Hexane/EtOAc) = 0.2; IR (neat, cm−1) 1751 (str, C=O), 1352 (str, S=O); 1H NMR δ (500 MHz, CDCl3) 7.86 (1H, s, C=CH), 7.40–7.33 (5H, m, 5 × ArH), 5.40 (2H, s, CH2OMs), 5.24 (2H, s, CH2), 5.23 (2H, s, CH2), 2.98 (3H, s, OMs); 13C NMR δ (126 MHz, CDCl3) 166.0 (C), 141.7 (C), 134.5 (C), 129.1 (CH), 129.0 (2 × ArCH), 128.8 (2 × ArCH), 126.0 (CH), 68.4 (CH2), 62.4 (CH), 51.1 (CH2), 38.7 (CH3); m/z (EI) 325.1 ([M]+•, 1%) 91 ([M − C6H8N3O5S]+•, 100).
![Molecules 26 05461 i006]()
Benzyl 2-[4-bromomethyl-1H-1,2,3-triazol-1-yl]ethanoate (7a), mesylate S1 (650 mg, 2.00 mmol) was dissolved in acetone (30 mL) at 0 °C and lithium bromide (430 mg, 5.00 mmol) was added. The solution was stirred for 30 min at r.t. and H2O (20 mL) was added. The solution was extracted with DCM (3 × 50 mL) and the combined organics were washed with Na2S2O3 (20 mL, 5% aq. w/w), dried over MgSO4 and concentrated in vacuo yielding 7a as colourless crystals (570 mg, 92% yield over 2 steps); Rf (1:1 Hexane/EtOAc) = 0.5; mp 122–124 °C; IR (neat, cm−1) 1737 (C=O); 1H NMR δ (500 MHz, CDCl3) 7.73 (1H, s, C=CH), 7.43–7.31 (5H, m, 5 × ArH), 5.23 (2H, s, ArCH2), 5.19 (2H, s, CH2CO2Bn), 4.59 (2H, s, CH2Br); 13C NMR δ (126 MHz, CDCl3) 166.0 (C), 145.3 (C), 134.5 (C), 129.1 (CH), 128.9 (2 ×ArCH), 128.7 (2 × ArCH), 124.3 (CH), 68.3 (CH2), 51.1 (CH2), 21.5 (CH2); m/z (EI) 311 ([81BrM]•, 3%), 309 ([79BrM]+•, 3), (230 ([M − 79Br]+•, 100), 145 (80); HRMS (EI) [79BrM]+• found 309.0111, C12H12O2N379Br requires 309.0107.
![Molecules 26 05461 i007]()
Benzyl 2-[4-iodomethyl-1H-1,2,3-triazol-1-yl]ethanoate (8a), mesylate S1 (650 mg, 2.00 mmol) was dissolved in acetone (30 mL) at 0 °C and sodium iodide (745 mg, 5.00 mmol) was added. The solution was stirred for 30 min at r.t. and H2O (20 mL) was added. The solution was extracted with DCM (3 × 50 mL) and the combined organics were washed with Na2S2O3 (20 mL; 5% aq. w/w), dried over MgSO4 and concentrated in vacuo yielding 8a as a pale yellow solid (490 mg, 71% yield over 2 steps); Rf (decomposes on silica); IR (neat, cm−1) 1747 (str, C=O); 1H NMR δ (500 MHz, CDCl3) 7.70 (1H, s, C=CH), 7.40–7.32 (5H, m, 5 × ArH), 5.23 (2H, s, ArCH2), 5.16 (2H, s, CH2CO2Bn), 4.48 (2H, s, CH2I); 13C NMR δ (126 MHz, CDCl3) 166.0 (C), 146.2 (C), 134.5 (C), 129.0 (ArCH), 128.9 (2 × ArCH), 128.7 (2 × ArCH), 123.6 (CH), 68.2 (CH2), 51.0 (CH2), -9.1 (CH2); m/z (EI) 334 (3%), 332 (3), 230 ([M − I]+•, 10), 205 (10), 145 (23); HRMS (EI) [M]+• found 356.9963, C12H12O2N3127I requires 356.9969.
![Molecules 26 05461 i008]()
2-[4-Chloromethyl-1H-1,2,3-triazol-1-yl]ethanoic acid (5a), chloride 4a (331 mg, 1.25 mmol) was dissolved in a mix of HCl (15 mL; 3 M, aq.) and THF (5 mL) and was stirred for 16 h at 50 °C. The solvent was removed in vacuo and HCl (10 mL; 1 M aq.) was added and extracted with EtOAc (3 × 10 mL). The combined organics were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo to give a slightly yellow oil that precipitated on addition of CHCl3 (1 mL). The product was purified by FCC (1:1 Hex/EtOAc + 5% AcOH) to yield 5a as a colourless oil, which was then lyophilised to remove traces of AcOH, giving a colourless solid (159 mg, 85% yield); Rf (1:1 Hex/EtOAc + 5% AcOH) = 0.21; mp 132–134 °C; IR (neat, cm−1) 2935 (br, OH), 1685 (str, C=O); 1H NMR δ (500 MHz, d6-DMSO) 8.16 (1H, s, C=CH), 5.22 (2H, s, CH2CO2H), 4.85 (2H, s, CH2Cl); 13C NMR δ (126 MHz, d6-DMSO) 168.4 (C), 143.2 (C), 125.6 (CH), 50.8 (CH2), 36.5 (CH2); m/z (ESI-, MeOH) 375 ([237ClM + Na − 2H]−, 4%), 373 ([37Cl35Cl2M + Na − 2H]−, 22), 371 ([235ClM + Na − 2H]−, 30), 176 ([37ClM − H]−, 31), 174 ([35ClM − H]−, 100); HRMS (ESI-, MeOH) [35ClM − H]− found 174.0080, C5H535ClN3O2 requires 174.0076.
![Molecules 26 05461 i009]()
2-[4-Bromomethyl-1H-1,2,3-triazol-1-yl]ethanoic acid (9a), bromide 7a (500 mg, 1.62 mmol) was dissolved in a mix of LiOH (100 mL; 0.1 M aq.) and THF (50 mL) and was stirred for 5 h at r.t. The solution was acidified with AcOH, concentrated in vacuo and purified by RP-HPLC (15% B, Rt = 7.0 min) then lyophilised giving 9a as colourless solid (30 mg, 8% yield); mp 120–122 °C; IR (neat, cm−1) 2470 (br, COOH), 1714 (str, C=O); 1H NMR δ (601 MHz, CD3OD) 8.07 (1H, s, C=CH), 5.27 (2H, s, CH2CO2H), 4.64 (2H, s, CH2Br); 13C NMR δ (126 MHz, CD3OD) 169.6 (C), 145.9 (C), 126.5 (CH), 51.7 (CH2), 21.6 (CH2); m/z (ESI-, MeOH) 440 ([281BrM − H]−, 4%), 438 ([81Br79Br2M − H]−, 9), 436 ([279BrM − H]−, 4), 220 ([81BrM − H]−, 97), 218 ([79BrM − H]−, 100); HRMS (ESI-, MeOH) [79BrM − H]− 217.9575, C5H579BrN3O2 requires 217.9571.
![Molecules 26 05461 i010]()
2-[4-Iodomethyl-1H-1,2,3-triazol-1-yl]ethanoic acid (10a), iodide 8a (450 mg, 1.26 mmol) was dissolved in a mix of LiOH (100 mL; 0.1 M aq.) and THF (50 mL) and was stirred for 5 h at r.t. The solution was acidified with AcOH, concentrated in vacuo and purified by RP-HPLC (20% B, Rt = 7.7 min) then lyophilised giving 10a as colourless solid (305 mg, 91% yield); mp 124–126 °C; IR (neat, cm−1) 2470 (br, COOH), 1712 (str, C=O); 1H NMR δ (600 MHz, CD3OD) 8.02 (1H, s, C=CH), 5.24 (2H, s, CH2CO2H), 4.54 (2H, s, CH2I). 13C NMR δ (126 MHz, CD3OD) 169.6 (C), 147.0 (C), 125.5 (CH), 51.7 (CH2), −9.7 (CH2); m/z (ESI-, MeOH) 554 ([2M + Na − 2H]−, 4%), 532 ([2M − H]−, 32), 265 ([M − H]−, 100), 126 ([M − C6H8N3O2]−, 11); HRMS (ESI-, MeOH) [M − H]− found 265.9428, C5H5IN3O2 requires 265.9432.
![Molecules 26 05461 i011]()
Ethyl 3-azidopropanoate (
2b), ethyl 3-bromopropionate
1b (1.27 mL, 10.0 mmol) was mixed with NaN
3 (12 mL; 1 M in DMSO) and heated to 45 °C for 18 h. TLC confirmed complete consumption of starting material (R
f (1:2 Hex/EtOAc) = 0.8). The reaction was cooled to around 10 °C, diluted with H
2O (40 mL) and extracted with Et
2O (3 × 20 mL). The combined organics were dried over MgSO
4 and concentrated
in vacuo yielding
2b as a colourless oil that was used without further purification (950 mg, 67%); R
f (1:2 Hex/EtOAc) = 0.5; IR (neat, cm
−1) 2100 (str, N
3), 1732 (str, C=O);
1H NMR δ (500 MHz, CDCl
3) 4.21 (1H, q,
J = 7.1 Hz, OC
H2CH
3), 3.59 (1H, t,
J = 6.5 Hz, N
3C
H2), 2.60 (1H, t,
J = 6.5 Hz, N
3CH
2C
H2), 1.30 (1H, t,
J = 7.1 Hz, C
H3);
13C NMR δ (126 MHz, CDCl
3) 171.0 (C), 61.1 (CH
2), 47.0 (CH
2), 34.2 (CH
2), 14.3 (CH
3). Spectroscopic data are in good agreement with the literature [
41].
![Molecules 26 05461 i012]()
Ethyl 3-[4-hydroxymethyl-1H-1,2,3-triazol-1-yl]propanoate (3b), ethyl 3-azidopropanoate 2b (715 mg, 5.00 mmol) was dissolved in anhydrous, degassed DCM (10 mL). Cu(MeCN)4·BF4 (157 mg, 10 mol%) and propargyl alcohol (320 µL, 5.50 mmol) were added. The solution turned brown and opaque but after 3 h had turned green and clear. TLC indicated starting material remained so propargyl alcohol (160 µL, 2.25 mmol) was added and the reaction left for 16 h. Silica was added to the flask and the solvent removed in vacuo. Dry loading the silica allowed the product to be purified by FCC (9:1 EtOAc/MeOH) to yield 3b as a colourless oil (700 mg, 71% yield); Rf (9:1 EtOAc/MeOH) = 0.2); IR (neat, cm−1) 3334 (br, OH), 1728 (str, C=O); 1H NMR δ (500 MHz, CDCl3) 7.63 (1H, s, C=CH), 4.77 (2H, br s, HOCH2), 4.64 (2H, t, J = 6.4 Hz, NCH2), 4.15 (2H, q, J = 7.1 Hz, OCH2CH3), 2.95 (2H, t, J = 6.4 Hz, NCH2CH2), 2.63 (1H, br s, OH), 1.24 (3H, t, J = 7.1 Hz, CH3); 13C NMR δ (126 MHz, CDCl3) 170.6 (C), 147.2 (C), 122.7 (CH), 61.4 (CH2), 56.6 (CH2), 45.7 (CH2), 34.8 (CH2), 14.2 (CH3); m/z (EI) 199 ([M]+•, 4%), 154 ([M − C2H5O]+•, 22), 86 (100), 78 (71), 49 (100); HRMS (EI) [M]+• found 199.0580, C8H13O3N3 requires 199.0951.
![Molecules 26 05461 i013]()
Ethyl 3-[4-chloromethyl-1H-1,2,3-triazol-1-yl]propanoate (4b), to alcohol 3b (497 mg, 2.50 mmol) in CHCl3 (10 mL) was added SOCl2 (0.5 mL). The solution was stirred for 1 h and the solvent was removed under a stream of N2 yielding a brown oil. CHCl3 (20 mL) was added followed by NaHCO3 (20 mL, sat. aq.). The organic layer was separated and the aqueous phase was extracted with CHCl3 (2 × 20 mL). The combined organics were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo giving a yellow oil. The product was purified by FCC (1:1 Hex/EtOAc) to yield 4b as a colourless oil (503 mg, 92% yield); Rf (1:1 Hex/EtOAc) = 0.3); IR (neat, cm−1) 1728 (s, C=O); 1H NMR δ (500 MHz, CDCl3) 7.69 (1H, s, C=CH), 4.69 (2H, ClCH2), 4.65 (2H, t, J = 6.4 Hz, NCH2), 4.16 (2H, q, J = 7.1 Hz, OCH2CH3), 2.96 (2H, t, J = 6.4 Hz, NCH2CH2), 1.25 (3H, t, J = 7.1 Hz, CH3); 13C NMR δ (126 MHz, CDCl3) 170.6 (C), 144.7 (C), 123.8 (CH), 61.4 (CH2), 45.8 (CH2), 36.3 (CH2), 34.8 (CH2), 14.2 (CH3); m/z (EI) 219 ([37ClM]+•, 3%), 217 ([35ClM]+•, 7), 174 ([37ClM − C2H5O]+•, 3), 172 ([35ClM − C2H5O]+•, 10), 154 (57), 101 (85), 84 (100); HRMS (EI) [35ClM]+• found 217.0622, C8H1235ClO2N3 requires 217.0613.
![Molecules 26 05461 i014]()
Ethyl 3-[4-methanesulfonylmethyl-1H-1,2,3-triazol-1yl]propanoate (S2), alcohol 3b (1.79 g, 9.00 mmol) was dissolved in THF (50 mL) with Ms2O (2.35 g, 13.5 mmol) and cooled to 0 °C with stirring. Triethylamine (3.76 mL, 27.00 mmol) was added dropwise and the reaction stirred at 0 °C for 30 min. The reaction was concentrated in vacuo and EtOAc (20 mL) and NH4Cl (30 mL; sat. aq.) were added. The organic layer was separated and the aqueous phase extracted with EtOAc (2 × 20 mL). The organics were combined, dried over Na2SO4 and concentrated in vacuo yielding S1 as a brown oil that was used without further purification. (2.50 g, quantitative); Rf (1:1 Hex/EtOAc) = 0.1; IR (neat, cm−1) 1728 (str, C=O), 1348 (str, S=O); 1H NMR δ (500 MHz, CDCl3) 7.82 (1H, s, C=CH), 5.36 (s, CH2OMs), 4.67 (2H, t, J = 6.3 Hz, NCH2), 4.16 (2H, q, J = 7.1 Hz, OCH2CH3), 3.03 (3H, s, OMs), 2.97 (2H, t, J = 6.3 Hz, CH2CO), 1.25 (3H, t, J = 7.1 Hz, CH3); 13C NMR δ (126 MHz, CDCl3) 170.5 (C), 141.0 (CH), 125.4 (CH2), 62.5 (CH2), 61.5 (CH2), 45.9 (CH2), 38.5 (CH3), 34.7 (CH2), 14.2 (CH3); m/z (EI) 277 ([M]+•, 4%), 232 ([M − C2H5O]+•, 36), 101 ([M − C4H6N3O3S]+•, 100); HRMS (EI) [M]+• found 277.0716, C9H15O5N3S requires 277.0727.
![Molecules 26 05461 i015]()
Ethyl 3-[4-bromomethyl-1H-1,2,3-triazol-1-yl]propanoate (7b), mesylate S2 (499 mg, 1.8 mmol) was dissolved in acetone (30 mL) at 0 °C and lithium bromide (430 mg, 5.00 mmol) was added. The solution was stirred for 30 min at r.t. and was concentrated in vacuo. The residue was triturated between EtOAc (30 mL) and H2O (20 mL). The organic layer was separated and the aqueous phase extracted with EtOAc (2 × 10 mL). The combined organics were washed with Na2S2O3 (20 mL; 5% aq. w/w), dried over Na2SO4 and concentrated in vacuo yielding 7b as a brown oil (385 mg, 82% yield); Rf (1:1 Hex/EtOAc) = 0.3; IR (neat, cm−1) 1730 (str, C=O); 1H NMR δ (500 MHz, CDCl3) 7.69 (1H, s, C=CH), 4.64 (2H, t, J = 6.3 Hz, NCH2), 4.56 (2H, s, BrCH2), 4.16 (2H, q, J = 7.1 Hz, OCH2CH3), 2.96 (2H, t, J = 6.3 Hz, CH2CO), 1.25 (3H, t, J = 7.1 Hz, CH3); 13C NMR δ (126 MHz, CDCl3) δ 170.6 (C), 144.7 (C), 123.9 (CH), 61.5 (CH2), 45.9 (CH2), 34.8 (CH2), 21.7 (CH2), 14.3 (CH3); m/z (EI) 218 ([81BrM]+•, 12%), 216 ([79BrM]+•, 12), 178 (100), 154 (54), 145 (64), 101 ([M-C3H3BrN3]+•, 52), 73 ([M − C5H7BrN3]+•, 52); HRMS (EI) [M]+• found 261.0117, C8H1279BrO2N3 requires 261.0107.
![Molecules 26 05461 i016]()
Ethyl 3-[4-iodomethyl-1H-1,2,3-triazol-1-yl]propanoate (8b), mesylate S2 (1.99 g, 7.2 mmol) was dissolved in acetone (100 mL) and sodium iodide (3.22 g, 21.6 mmol) was added. The solution was stirred for 30 min and was concentrated in vacuo. The residue was triturated between EtOAc (50 mL) and H2O (50 mL). The organic layer was separated and the aqueous phase extracted with EtOAc (2 × 30 mL). The combined organics were washed with Na2S2O3 (50 mL; 5% aq. w/w), dried over Na2SO4 and concentrated in vacuo yielding 8b as a brown oil (1.96 g, 70% yield); Rf (1:1 Hex/EtOAc) = 0.3; IR (neat, cm−1) 1733 (str, C=O); 1H NMR δ (500 MHz, CDCl3) 7.65 (1H, s, C=CH), 4.61 (2H, t, J = 6.4 Hz, NCH2), 4.45 (2H, s, ICH2), 4.16 (3H, q, J = 7.1 Hz, OCH2CH3), 2.95 (2H, t, J = 6.4 Hz, CH2CO), 1.25 (3H, t, J = 7.1 Hz, CH3); 13C NMR δ (126 MHz, CDCl3) δ 170.6 (C), 145.6 (C), 123.2 (CH), 61.4 (CH2), 45.8 (CH2), 34.8 (CH2), 14.3 (CH3), −8.7 (CH2); m/z (EI) 264 ([M − C2H5O]+•, 58%), 182 ([M − I]+•, 92), 154 (100), 108 (90), 101 ([M − C3H3IN3]+•, 95); HRMS (EI) [M]+• found 308.9938, C8H12121IO2N3 requires 308.9969.
![Molecules 26 05461 i017]()
3-[4-(Chloromethyl)-1H-1,2,3-triazol-1-yl]propanoic acid (5b), compound 4b (217 mg, 1.00 mmol) was dissolved in HCl (15 mL; 3 M aq.) and THF (5 mL) and was stirred for 16 h at r.t. The solvent was removed in vacuo and HCl (10 mL; 1 M aq.) was added. The aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organics were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo to give a slightly yellow oil that precipitated on addition of CHCl3 (1 mL). The product was purified by FCC (1:1 Hex/EtOAc + 5% AcOH) to yield 5b as a colourless oil, which was then lyophilised to remove traces of AcOH, giving a colourless solid (159 mg, 85% yield); Rf (1:1 Hex/EtOAc + 5% AcOH) = 0.2; mp = 114–116 °C; IR (neat, cm−1) 2935 (br, OH), 1686 (s, C=O); 1H NMR δ (500 MHz, d6-DMSO) 12.50 (1H, s, COOH), 8.17 (1H, s, C=CH), 4.82 (2H, s, ClCH2), 4.55 (2H, t, J = 6.7 Hz, NCH2), 2.89 (2H, t, J = 6.7 Hz, CH2CO2H); 13C NMR δ (126 MHz, d6-DMSO) 171.7 (C), 143.2 (C), 124.4 (CH), 45.5 (CH2), 36.5 (CH2), 34.0 (CH2); m/z (ESI-, MeOH) 403 ([237ClM + Na − 2H]¯, 3%), 401 ([37Cl35Cl2M + Na − 2H]¯, 21%), 399 ([235ClM + Na − 2H]¯, 33), 190 ([37ClM − H]¯, 32), 188 ([35ClM − H]¯, 100); HRMS (ESI-, MeOH) [35ClM − H]¯ found 188.0219, C6H735ClN3O2 requires 188.0221.
![Molecules 26 05461 i018]()
3-[4-(Iodomethyl)-1H-1,2,3-triazol-1-yl]propanoic acid (10b), iodide 8b (500 mg, 1.62 mmol) was dissolved in a mix of LiOH (70 mL; 0.1 M aq.) and THF (30 mL) and was stirred for 5 h at r.t. The solution was acidified with AcOH, concentrated in vacuo, purified by RP-HPLC (15% B, Rt = 14.0 min) and lyophilised to give 10b as colourless solid (103 mg, 22% yield); mp = 113–115 °C; IR (neat, cm−1) 2472 (br, COOH), 1714 (str, C=O); 1H NMR δ (500 MHz, CD3OD) 8.00 (1H, s, C=CH), 4.62 (2H, t, J = 6.6 Hz, NCH2), 4.51 (2H, s, ICH2), 2.95 (2H, t, J = 6.6 Hz, CH2CO2H); 13C NMR δ (126 MHz, CD3OD) 173.7 (C), 146.8 (C), 124.5 (CH), 47.2 (CH2), 35.0 (CH2), −9.7 (CH2); m/z (ESI-, MeOH) 561 ([2M − H]¯, 44%), 280 ([M − H]¯, 100), 127 ([M − C6H8N3O2]¯, 52); HRMS (ESI-, MeOH) [M − H]¯ found 279.9600, C6H7IN3O2 requires 279.9588.
Compound
12 and the azide derivative were synthesized following the literature procedure [
28].
![Molecules 26 05461 i019]()
1-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-β-d-glucopyranosyl)-4-hydroxymethyl-1H-1,2,3-triazole (
13), peracetylated GlcNAc azide (1.00 g, 2.70 mmol) was dissolved in degassed DCM/MeOH (10 mL) under an argon atmosphere. Propargyl alcohol (236 μL, 4.05 mmol) was added, followed by Cu(MeCN)
4·BF
4 (84.8 mg, 10 mol%) and the solution was stirred for 16 h. Celite was added to the green solution and the solvent was removed in vacuo. Dry loading the celite allowed the product to be purified by FCC (19:1–9:1 DCM/MeOH) yielding
13 as a colourless solid (1.09 g, 95% yield); R
f (9:1, DCM/MeOH) = 0.4; mp 220 °C [lit. [
42] 230–232 °C]; [α]
D = −40.0 (c, 0.4 in CHCl3), [lit. [
42] [α]
D = −26.0 (c, 1.00 in CHCl
3)]; IR (neat, cm
−1) 1741 (C=O). 1728 (C=O), 1668 (C=O);
1H NMR δ (500 MHz, CD
3OD) 8.12 (1H, s, C=C
H), 6.08 (1H, d,
J = 10.0 Hz,
H-1), 5.47 (1H, br t,
J = 10.0 Hz,
H-3), 5.23 (1H, br t,
J = 9.7 Hz,
H-4), 4.67 (2H, br s, C
H2OH), 4.58 (1H, br t,
J = 10.2 Hz,
H-2), 4.32 (1H, dd,
J = 12.4, 4.8 Hz,
H-6′), 4.20–4.12 (2H, m,
H-5, H-6), 2.05 (3H, s, AcO-4), 2.04 (3H, s, AcO-6), 2.01 (3H, s, AcO-3), 1.72 (3H, s, NAc);
13C NMR δ (126 MHz, CD
3OD) 173.4 (C), 172.2 (C), 171.7 (C), 171.2 (C), 149.6 (C), 122.9 (CH), 87.1 (CH), 76.0 (CH), 73.8 (CH), 69.6 (CH), 63.1 (CH
2), 56.4 (br, CH
2), 54.6 (CH), 22.4 (CH
3), 20.6 (2 × CH
3), 20.5 (CH
3);
m/
z (ESI+, MeCN) 879 ([2M + Na]
+, 40%), 451 ([M + Na]
+, 100), 330 ([M − C
3H
4N
3O]
+, 80); HRMS (ESI+, MeCN) [M + Na]
+ found 451.1435, C
17H
24N
9O
9Na requires 451.1435. Spectroscopic data are in good agreement with the literature [
42].
![Molecules 26 05461 i020]()
1-(2-Acetamido-2-deoxy-3, 4, 6-tri-O-acetyl-β-D-glucopyranosyl)-4-methanesulfonylmethyl-1H-1,2,3-triazole (S3), alcohol 13 (898 mg, 2.1 mmol) was dissolved in THF (100 mL) with Ms2O (730 mg, 4.2 mmol) and cooled to 0 °C. Triethylamine (880 µL, 6.3 mmol) was added dropwise to the reaction, which was left to stir for 30 min. HPLC analysis confirmed complete consumption of starting material (Rt = 8.2 min) and a single new peak (Rt = 9.6 min) The solution was concentrated in vacuo and DCM (60 mL) and NH4Cl (40 mL; sat aq.) were added. The organic layer was extracted and the aqueous phase extracted with DCM (2 × 60 mL). The organics were combined, dried over Na2SO4 and concentrated in vacuo yielding mesylate S3 as a colourless solid (1.06 g, quantitative) that was used without further purification; Rf (19:1, DCM/MeOH) = 0.3; mp 150–152 °C; [α]D = −3.00 (c, 0.2 in CHCl3); IR (neat, cm−1) 1741 (str, C=O), 1666 (str, C=O); 1H NMR δ (500 MHz, CDCl3) 8.05 (1H, s, C=CH), 6.84 (1H, d, J = 9.3 Hz, NH), 6.01 (1H, d, J = 9.9 Hz, H-1), 5.41 (1H, dd, J = 10.2, 9.6 Hz, H-3), 5.22 (1H, dd, J = 9.7 Hz, H-4), 4.53 (1H, dd, J = 10.2 Hz, H-2), 4.28 (1H, dd, J = 12.6, 4.8 Hz, H-6′), 4.13 (1H, dd, J = 12.6, 2.2 Hz, H-6), 4.01 (1H, ddd, J = 10.2, 4.8, 2.2 Hz, H-5), 3.03 (3H, s, MsO), 2.07 (3H, s, AcO), 2.05 (3H, s, AcO), 2.04 (3H, s, AcO), 1.73 (3H, s, NAc); 13C NMR δ (126 MHz, CDCl3) 170.9 (C), 170.8 (C), 170.8 (C), 169.5 (C), 141.4 (C), 123.7 (CH), 86.3 (CH), 75.1 (CH), 72.4 (CH), 68.0 (CH), 61.8 (CH2), 53.4 (CH), 46.3 (CH2), 38.5 (CH3), 22.7 (CH3), 20.8 (CH3), 20.7 (CH3), 20.7 (CH3); m/z (ESI+, MeCN) 529 ([M + Na]+, 13%), 507 ([M + H]+, 8), 405 (7), 330 ([M − C4H6N3O3S]+, 100); HRMS (ESI+, MeCN) [M + Na]+ found 529.1211, C18H26N4O11SNa requires 529.1211.
![Molecules 26 05461 i021]()
1-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-β-D-glucopyranosyl)-4-iodomethyl-1H-1,2,3-triazole (14), mesylate S3 (1.06 g, 2.1 mmol) was suspended in acetone (100 mL) and cooled to 0 °C. Sodium iodide (939 mg, 6.3 mmol) was added and the solution was stirred for 30 min at 0 °C and then concentrated in vacuo. Water (50 mL) and DCM (50 mL) were added and the organic layer extracted. The aqueous layer was extracted with DCM (2 × 50 mL) and the combined organics washed with Na2S2O3 (50 mL; 5% aq. w/w), dried over Na2SO4 and concentrated in vacuo to give iodide 14 as a colourless solid (1.10 g, 97% yield); Rf (19:1, DCM/MeOH) = 0.3; mp 184–188 °C; [α]D = −61.0 (c, 1.00 in CHCl3); IR (neat, cm−1) 1743 (C=O), 1666 (C=O); 1H NMR δ (500 MHz, CDCl3) 7.85 (1H, s, C=CH), 5.95 (1H, d, J = 9.9 Hz, NH), 5.76 (1H, d, J = 9.3 Hz, H-1), 5.41 (1H, br t, J = 10.0, 9.7 Hz, H-3), 5.24 (1H, br t, J = 9.7 Hz, H-4), 4.54 (1H, br q, J = 9.9 Hz, H-2), 4.47 (1H, d, J = 11.2 Hz, CHaHbI), 4.44 (1H, d, J = 11.2 Hz, CHaHbI), 4.30 (1H, dd, J = 12.6, 5.0 Hz, H-6′), 4.15 (1H, dd, J = 12.6, 2.2 Hz, H-6), 3.99 (1H, ddd, J = 10.2, 5.0, 2.2 Hz, H-5), 2.09 (3H, s, AcO-6), 2.07 (3H, s, AcO-4), 2.07 (3H, s, AcO-3), 1.79 (3H, s, NAc); 13C NMR δ (126 MHz, CDCl3) 171.0 (C), 170.7 (C), 170.5 (C), 169.4 (C), 146.1 (C), 121.3 (CH), 86.2 (CH), 75.4 (CH), 72.3 (CH), 67.9 (CH), 61.8 (CH2), 53.7 (CH), 23.1 (CH3), 20.9 (CH3), 20.8 (CH3), 20.7 (CH3), −9.40 (CH2); m/z (ESI+, MeCN) 561 ([M + Na]+, 100%), 330 ([M − C3H3IN3]+, 24); HRMS (ESI+, MeCN) [M + Na]+ found 561.0455 C17H23IN4O8Na requires 561.0453.
![Molecules 26 05461 i022]()
1-(2-Acetamido-2-deoxy-β-D-glucopyranosyl)-4-iodomethyl-1H-1,2,3-triazole (15), iodide 14 (1.00 g, 1.86 mmol) was dissolved in anhydrous MeOH (100 mL) and MeONa (400 µL, 10 mol%; 0.5 M in MeOH) was added. The solution was stirred in the dark for 2 h. The reaction was neutralised with NH4Cl (400 µL; 0.5 M aq.), concentrated in vacuo, re-dissolved in H2O (40 mL) and washed with EtOAc (20 mL). The solution was concentrated in vacuo and purified by RP-HPLC (10% B, Rt = 9.2 min). The compound was lyophilised to yield 15 as a colourless solid (405 mg, 53% yield); mp 150–154 °C; [α]D = −40.0 (c, 0.40 in MeOH); IR (neat, cm−1) 3269 (str, br, OH), 1651 (str, C=O); 1H NMR δ (600 MHz, CD3OD) 8.17 (1H, s, C=CH), 5.76 (1H, d, J = 10.0 Hz, H-1), 4.52 (1H, d, J = 11.1 Hz, CHaHbI), 4.50 (1H, d, J = 11.1 Hz, CHaHbI), 4.19 (1H, br t, J = 10.0 Hz, H-2), 3.90 (1H, dd, J = 12.3, 2.1 Hz, H-6′), 3.75 (1H, dd, J = 12.3, 5.3 Hz, H-6), 3.67 (1H, dd, J = 10.0, 8.6 Hz, H-4), 3.58 (1H, ddd, J = 10.0, 5.3, 2.1 Hz, H-5), 3.53 (1H, dd, J = 10.0, 8.6 Hz, H-3), 1.78 (3H, s, NAc); 13C NMR δ (126 MHz, CD3OD) 173.4 (C), 147.0 (C), 122.7 (CH), 88.2 (CH), 81.3 (CH), 75.7 (CH), 71.4 (CH), 62.3 (CH2), 56.7 (CH), 22.5 (CH3), −9.7 (CH2); m/z (ESI+, MeOH) 451 ([M + K]+, 28%), 435 ([M + Na]+, 72), 413 ([M + H]+, 9), 204 ([M − C3H3IN3]+, 100); (ESI-, MeOH) 525 ([M – H + C2O2F3]¯, 100%), 457 ([M – H + CH1O2]¯, 43), 411 ([M − H]−, 37); HRMS (ESI-, MeCN) [M − H]¯ found 411.0175, C11H16IN4O5 requires 411.0171.
Peptide synthesis was performed as previously described [
4].
(16): Ac-MVLCEGEW-NH2; m/z (ESI+, MeCN) 1029 ([M + Na]+, 100%), 1007 ([M + H]+, 33); HRMS (ESI+, MeCN) [M + Na]+ found 1029.4176, C44H66N10O13S2Na requires 1029.4144.
Alkylation of Histone H4K12C: A 1 mM stock solution of H4K12C was prepared by dissolving H4K12C (2.2 mg) in Alkylation Buffer (196 µL; guanidine·HCl (4 M), HEPES (pH 7.8, 1 M), d/l-methionine (10 mM)). TCEP or DTT (4 µL; 1 M aq.) and the solution incubated at 37 °C for 1 h.
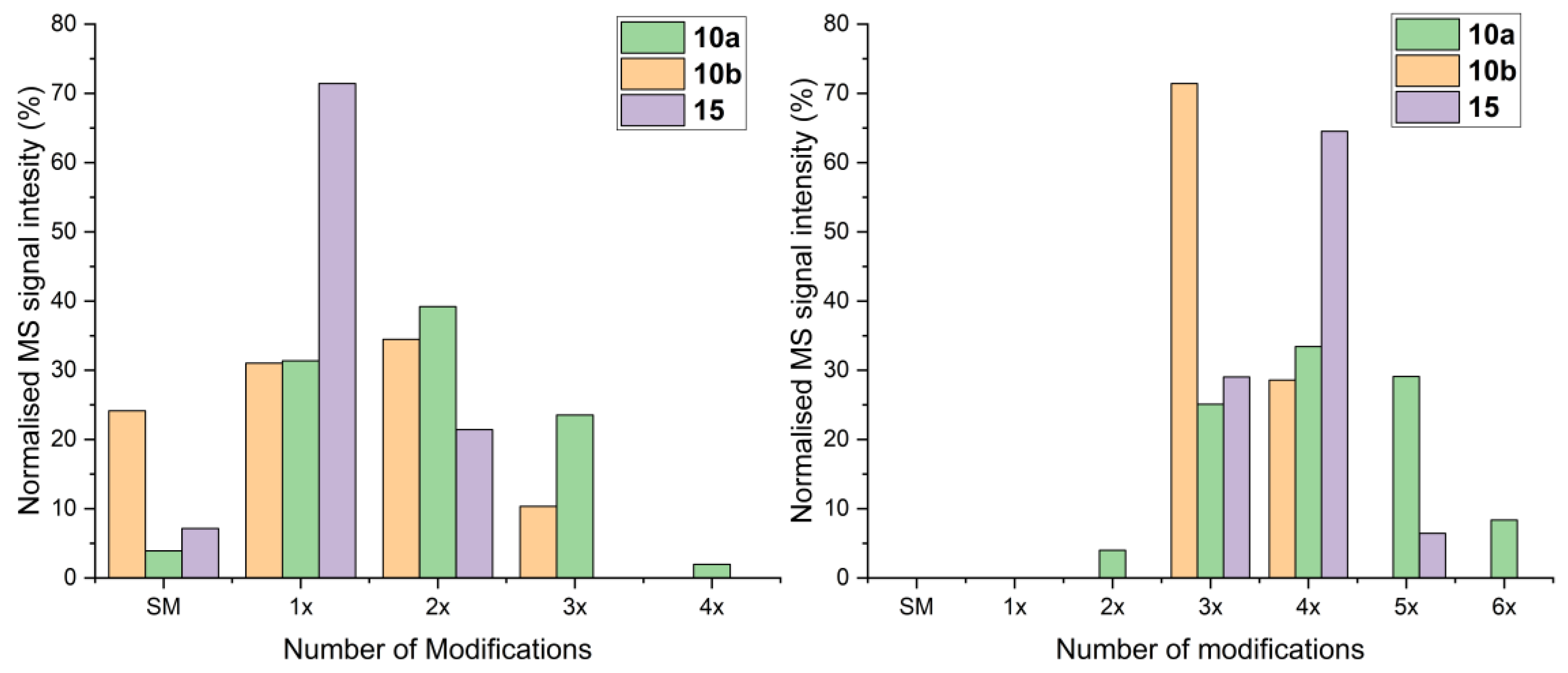
Alkylation reagents 5a, 5b, 9a, 10a, 10b and 15 were prepared as stock solutions in DMSO (1 M) and were used within 10 min of preparation.
Reactions were performed on a 20 µL scale in PCR tubes. Alkylation reagent (1 M, DMSO) was added to the tube, followed by the appropriate quantity of water to take the volume to 10 µL. H4K12C (90 µL, 1 mM) was added to the alkylation reagent and aspirated using a pipette. The reaction was left in darkness at r.t. for the stated time. An aliquot (1 µL) was removed from the reaction mixture and diluted with H2O (99 µL) to quench the reaction.
Alkylation of SCP-2L Q111C: To 90 µL a 110 µM stock of SCP-2L in HEPES (50 mM, 50 mM NaCl; pH 8) or MES (20 mM, 30 mM NaCl) buffer was added alkylating reagent (10–100 eq. from a 100 mM stock in DMSO; reducing agent was not used as the protein does not form disulfide bonds). The reaction was left at room temperature in the dark for 1 h (HEPES) or 16 h (MES) before being quenched by addition of 1 µL beta mercaptoethanol. Samples were analyzed by LC-MS.
LC-MS analysis of protein samples was performed using a Waters Acquity I Class UPLC, A = H2O (+0.1% FA), B = MeCN (+0.1%FA) using a Waters BEH C4 column 2.1 × 100 mm, using 5% B for 3 mins, followed by a linear gradient to 95% B over 5 min at 0.2 mL/min. The eluent was infused into a Waters Synapt G2 mass spectrometer.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




























