
The Structural and Dynamical Properties of the Hydration of SNase Based on a Molecular Dynamics Simulation

Abstract
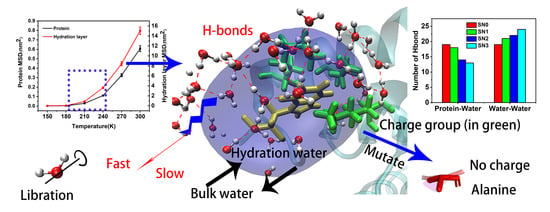
:
1. Introduction
2. Results and Discussion
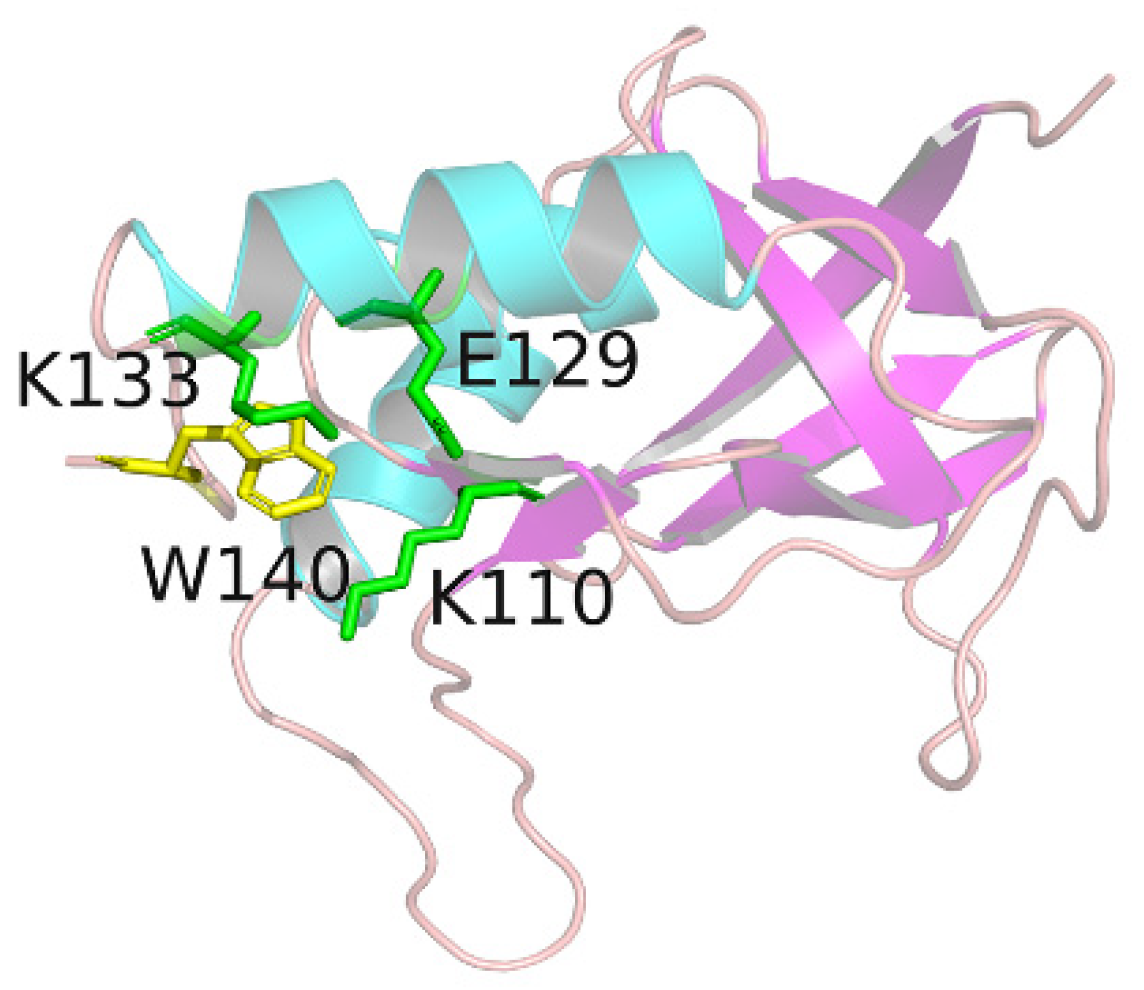
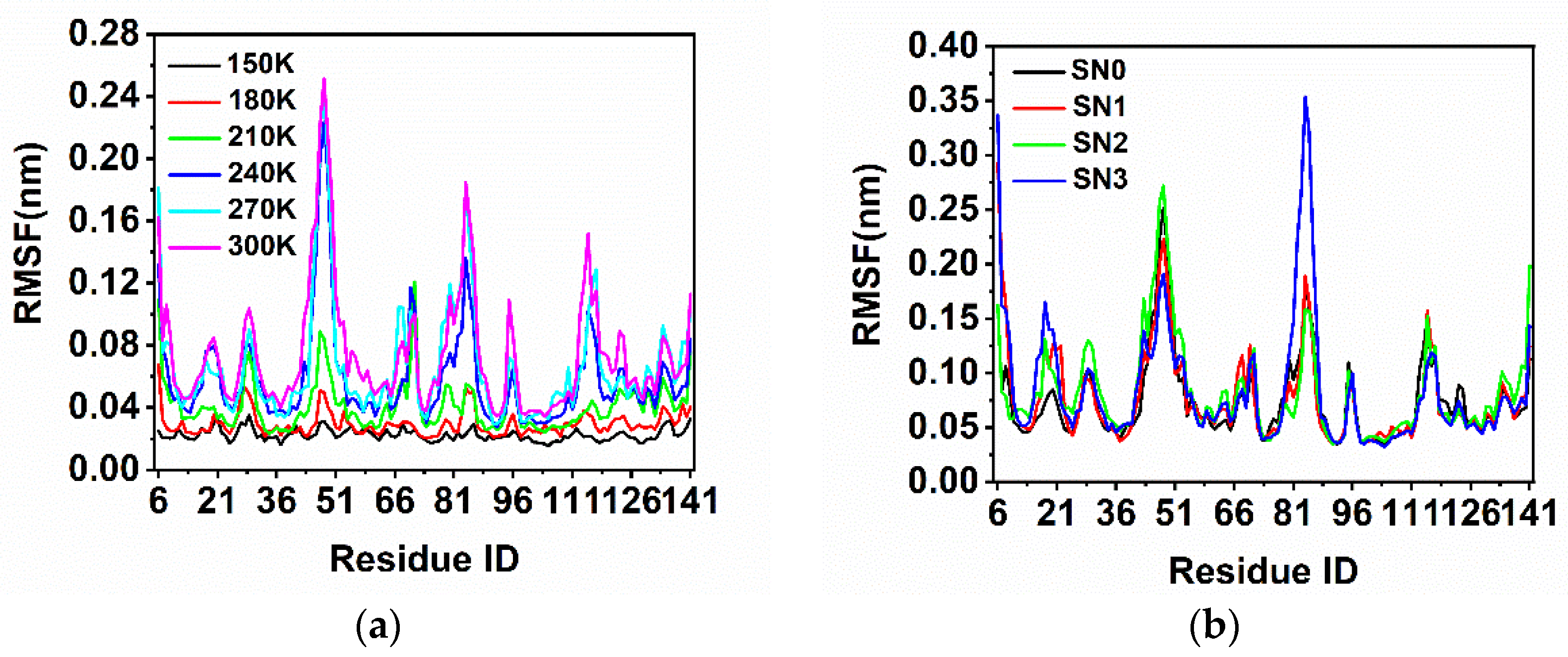
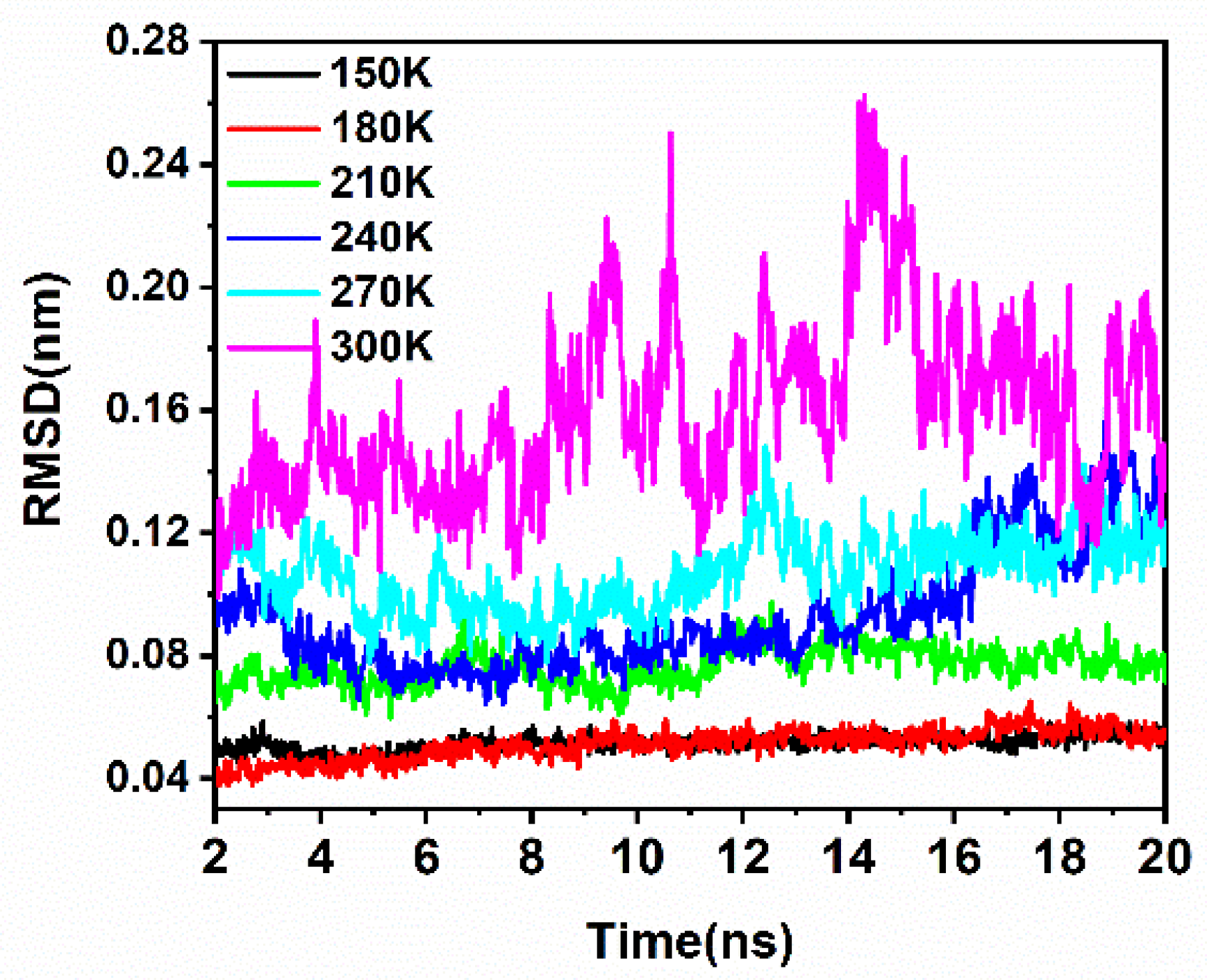
2.1. Structural Fluctuations
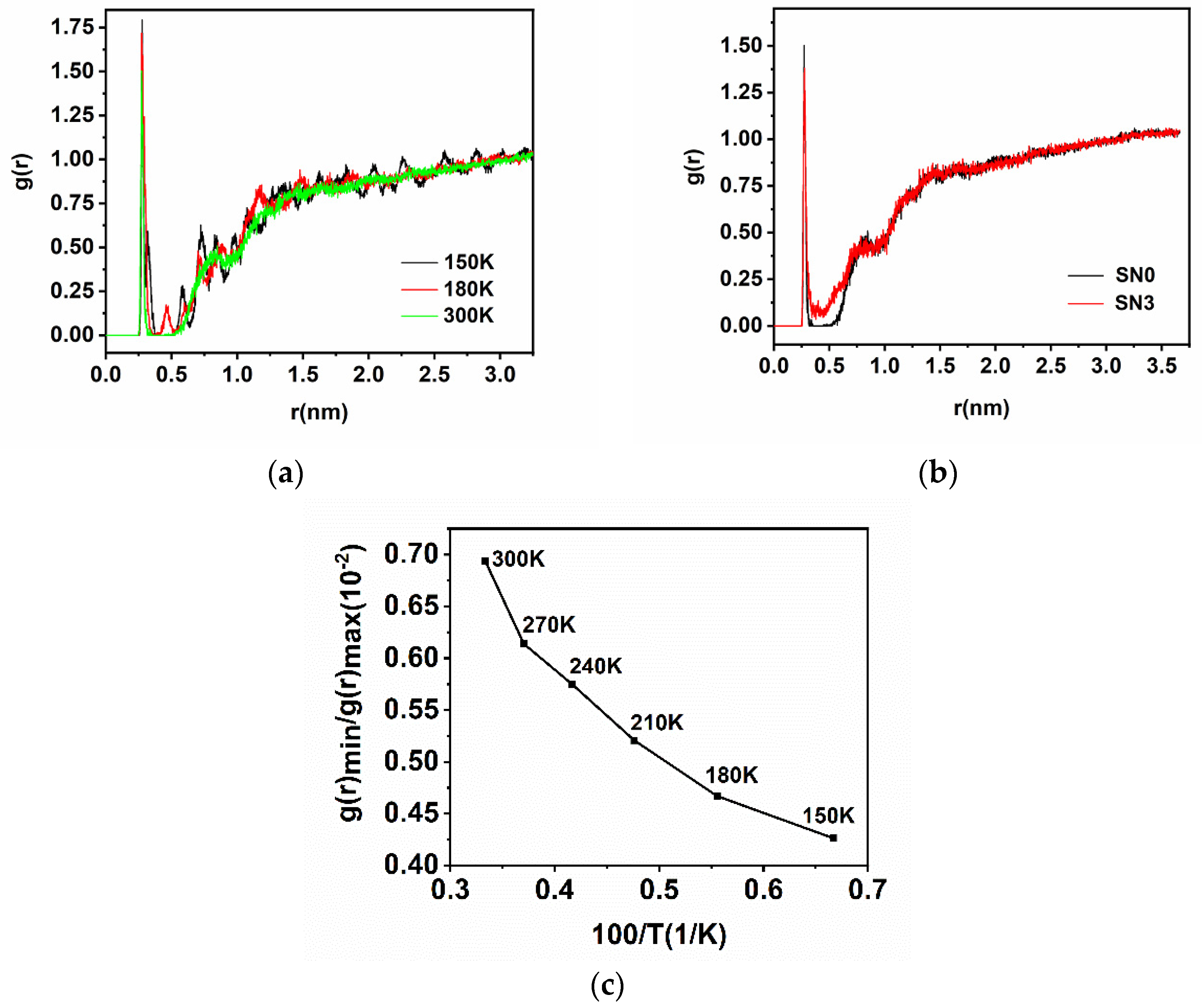
2.2. Radial Distributions Function
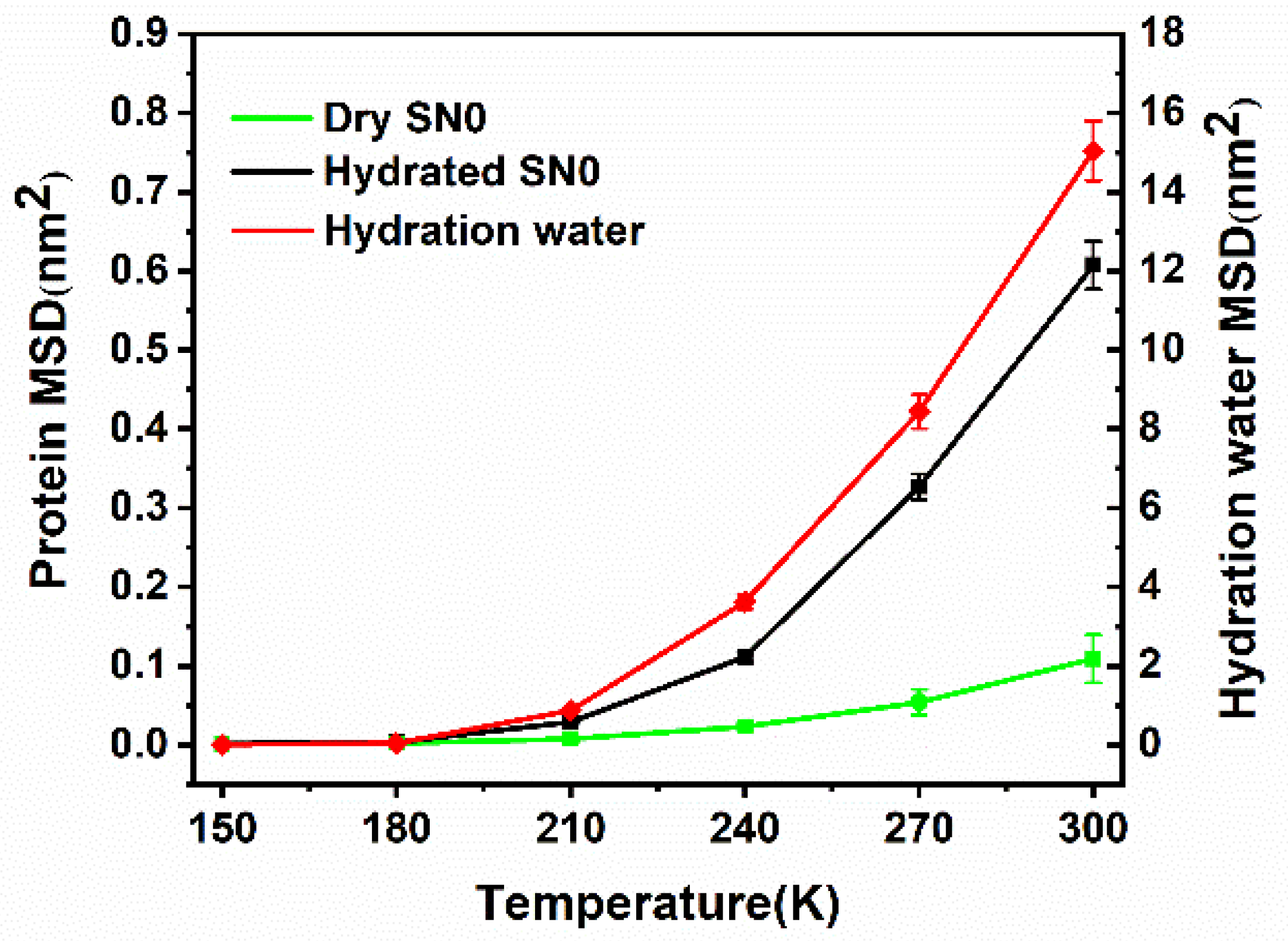
2.3. Mean Square Displacement of SNase and Hydration Water
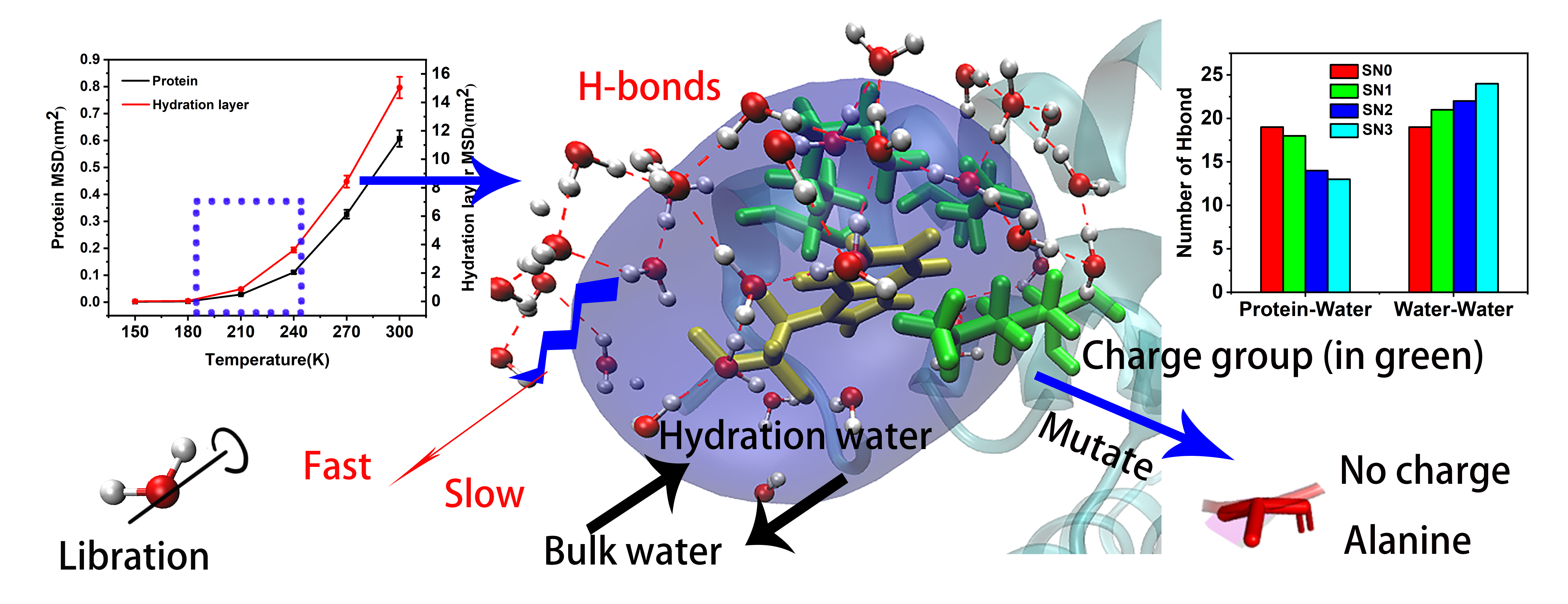
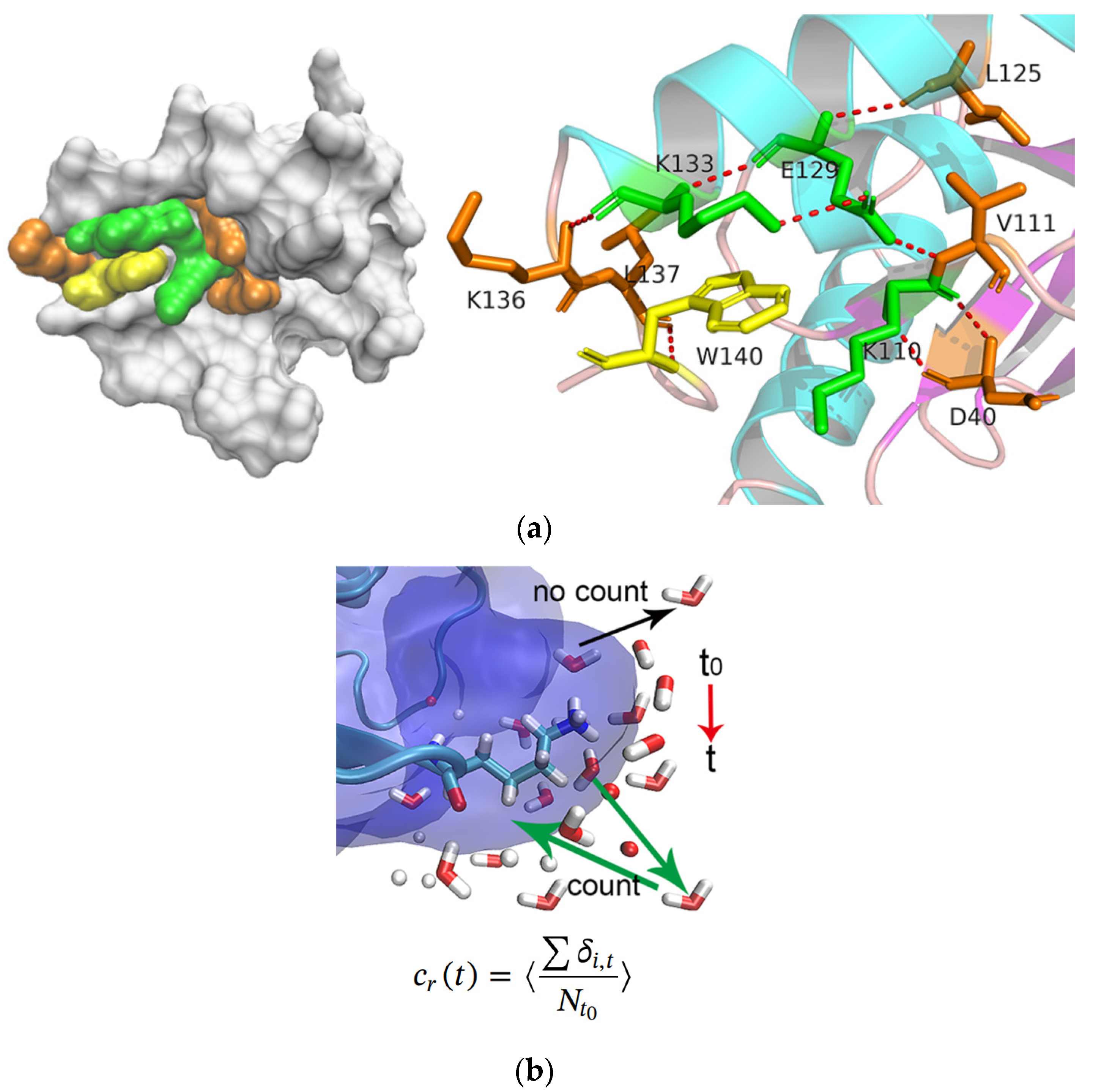
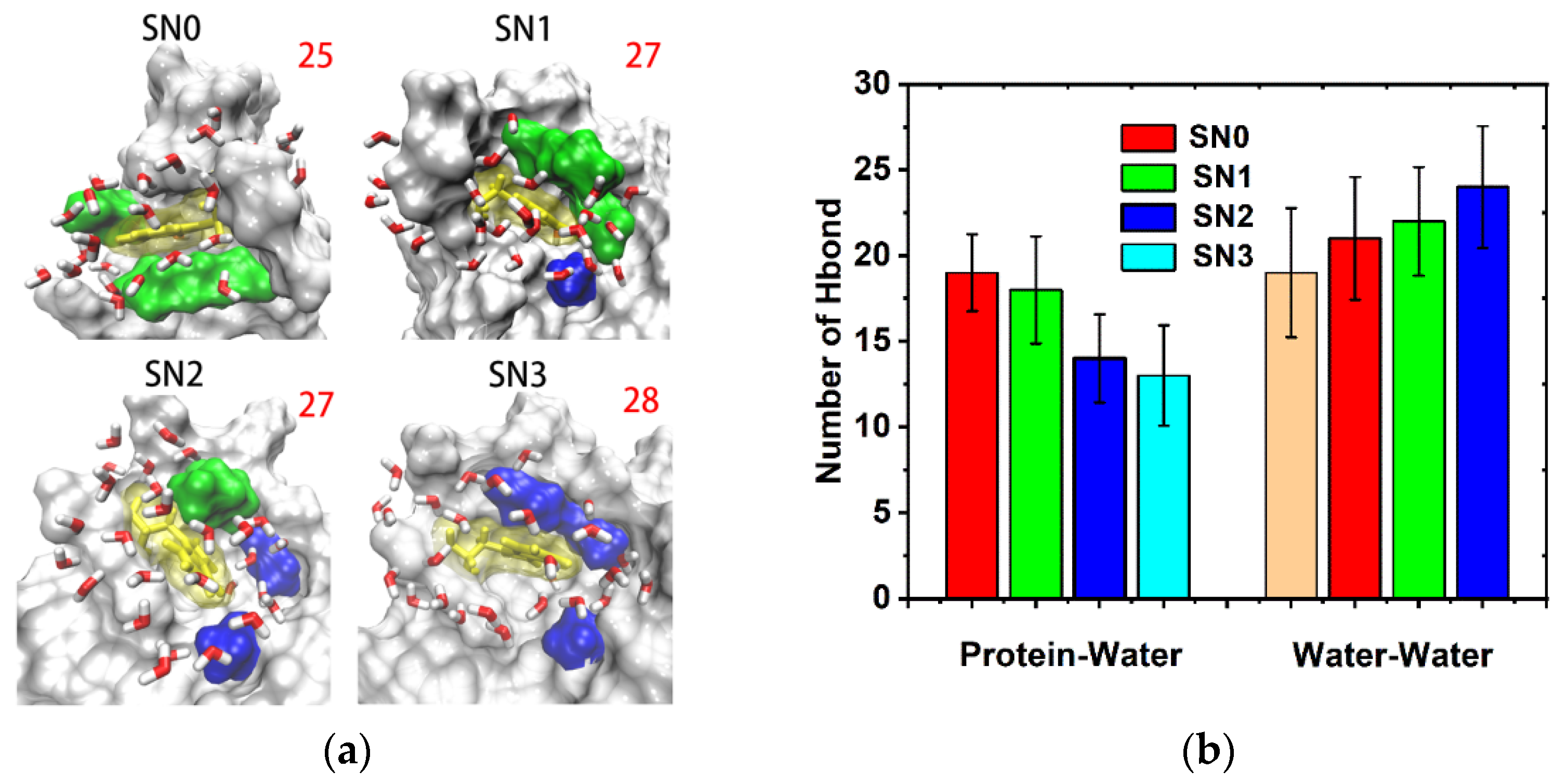
2.4. Hydration Dynamics Based on Mutation
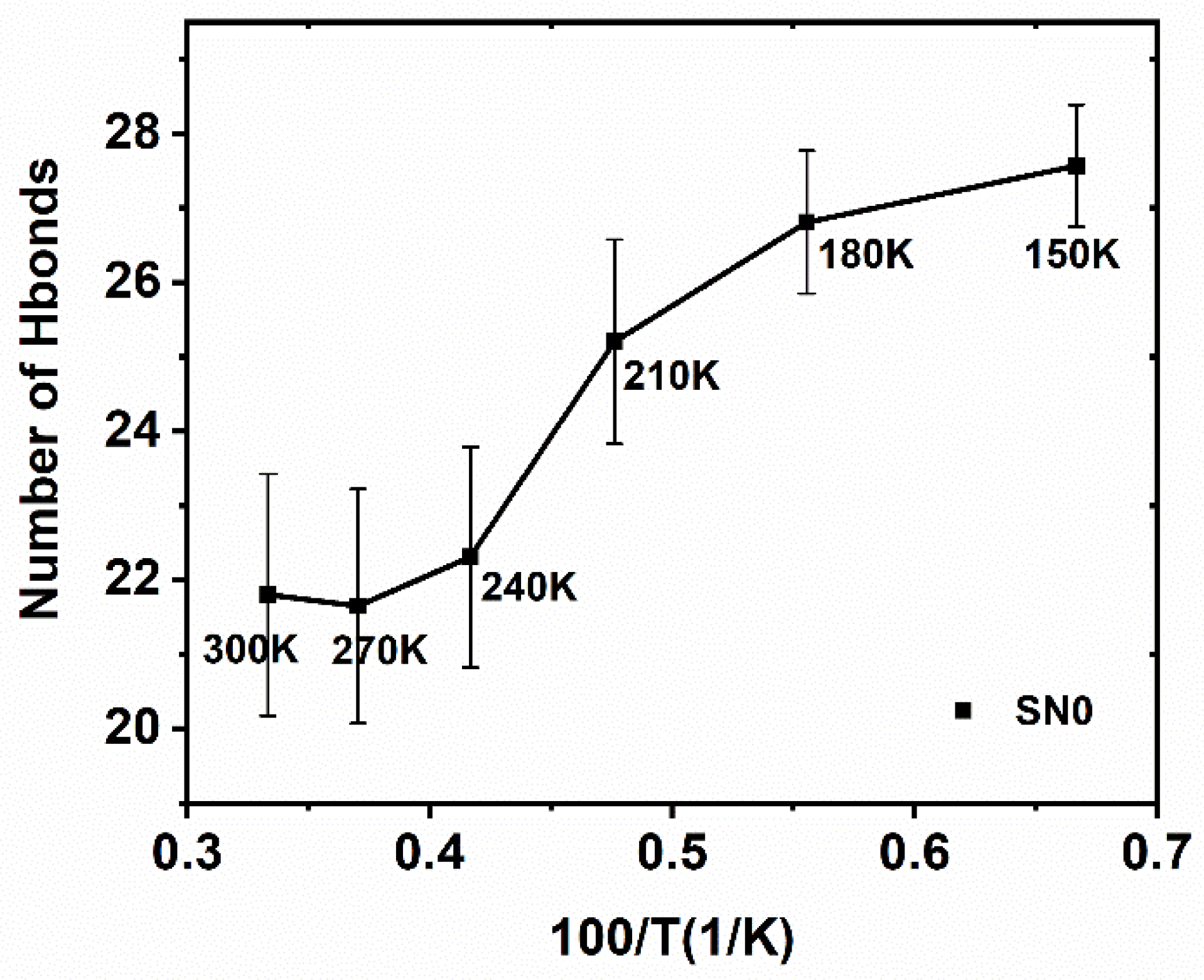
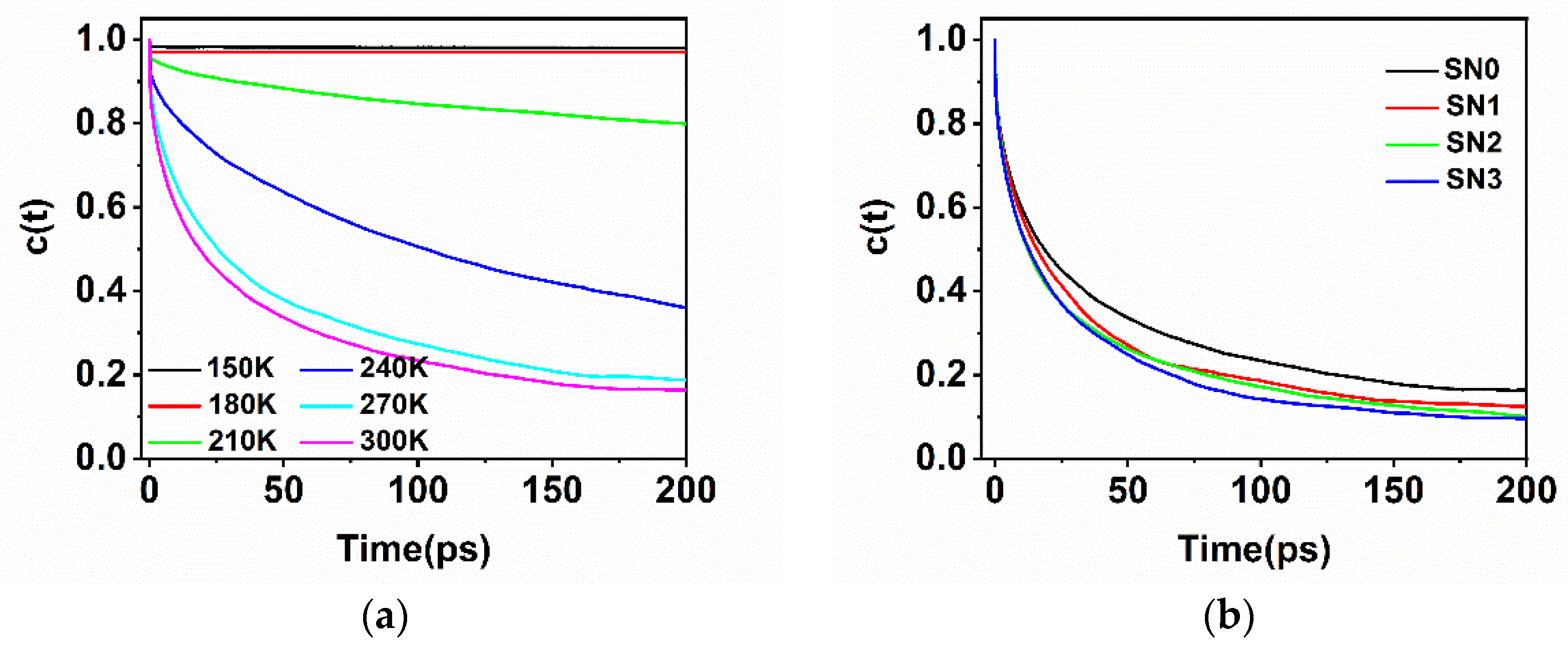
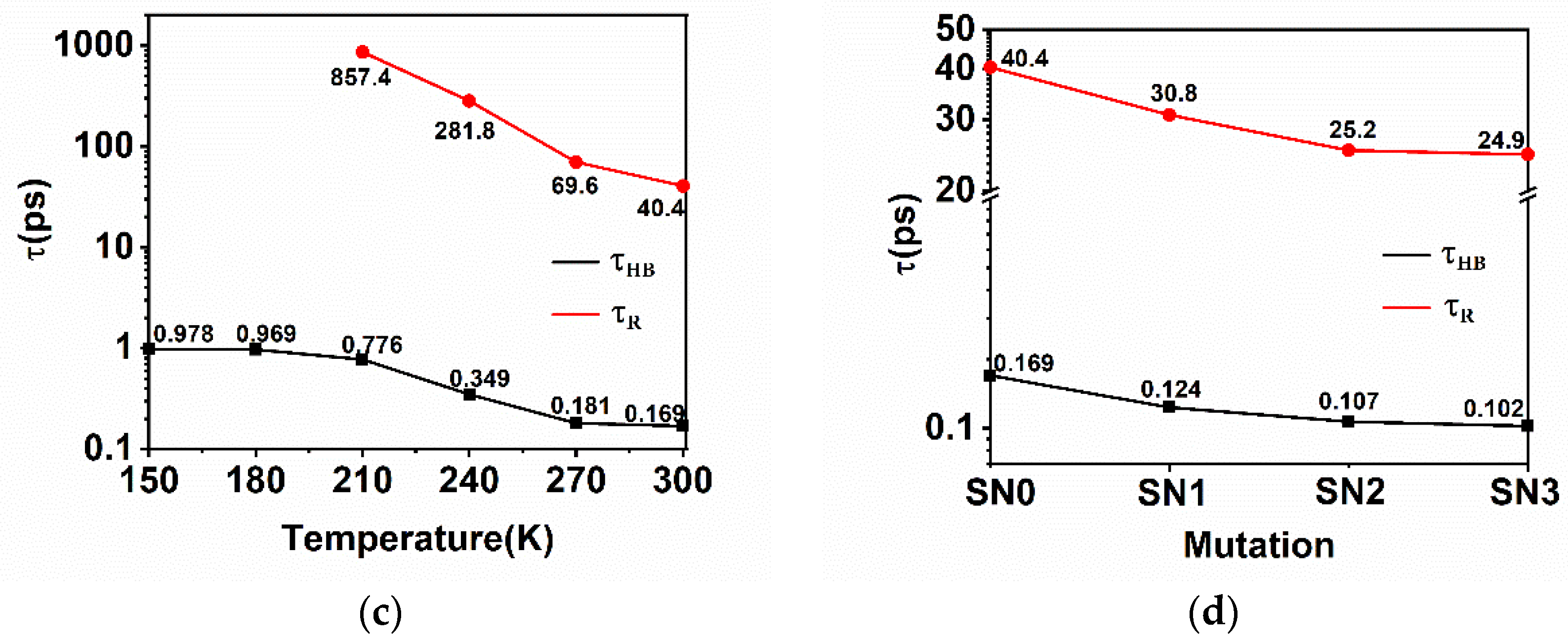
2.5. Hydrogen Bonding Dynamics
3. Materials and Methods
3.1. Molecule Dynamics Simulation
3.2. Analysis Methods
3.2.1. Root Mean Square Fluctuation
3.2.2. Root Mean Square Deviation
3.2.3. Mean Square Displacement
3.2.4. Radial Distributions Function
3.2.5. Hydration Dynamics Analysis Based on Mutation
3.2.6. Hydration Bonding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ball, P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N. Water mediation in protein folding and molecular recognition. Annu Rev. Biophys Biomol Struct 2006, 35, 389–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, A.C.; Duboué-Dijon, E.; Sterpone, F.; Hynes, J.T.; Laage, D. Biomolecular hydration dynamics: A jump model perspective. Chem Soc. Rev. 2013, 42, 5672–5683. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef] [Green Version]
- Amann-Winkel, K.; Bellissent-Funel, M.; Bove, L.E.; Loerting, T.; Nilsson, A.; Paciaroni, A.; Schlesinger, D.; Skinner, L. X-ray and Neutron Scattering of Water. Chem. Rev. 2016, 116, 7570–7589. [Google Scholar] [CrossRef] [Green Version]
- Brotzakis, Z.F.; Groot, C.C.M.; Brandeburgo, W.H.; Bakker, H.J.; Bolhuis, P.G. Dynamics of Hydration Water around Native and Misfolded α-Lactalbumin. J. Phys. Chem. B 2016, 120, 4756–4766. [Google Scholar] [CrossRef]
- Pronk, S.; Lindahl, E.; Kasson, P.M. Dynamic heterogeneity controls diffusion and viscosity near biological interfaces. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, E.W.; Muegge, I. Heterogeneous diffusion of water at protein surfaces: Application to BPTI. J. Phys. Chem. 1993, 97, 11339–11343. [Google Scholar] [CrossRef]
- Sedlmeier, F.; von Hansen, Y.; Mengyu, L.; Horinek, D.; Netz, R.R. Water Dynamics at Interfaces and Solutes: Disentangling Free Energy and Diffusivity Contributions. J. Stat. Phys. 2011, 145, 240–252. [Google Scholar] [CrossRef]
- Tan, P.; Huang, J.; Mamontov, E.; Garcia, S.V.; Merzel, F.; Liu, Z.; Ye, Y.; Hong, L. Decoupling between the translation and rotation of water in the proximity of a protein molecule. Phys. Chem. Chem. Phys. 2020, 22, 18132–18140. [Google Scholar] [CrossRef]
- Ji, M.; Odelius, M.; Gaffney, K.J. Large Angular Jump Mechanism Observed for Hydrogen Bond Exchange in Aqueous Perchlorate Solution. Science 2010, 328, 1003–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Biswas, R.; Mukherjee, B. Reorientational Jump Dynamics and Its Connections to Hydrogen Bond Relaxation in Molten Acetamide: An All-Atom Molecular Dynamics Simulation Study. J. Phys. Chem. B 2014, 119, 274–283. [Google Scholar] [CrossRef]
- Srivastava, A.; Malik, S.; Karmakar, S.; Debnath, A. Dynamic coupling of a hydration layer to a fluid phospholipid membrane: Intermittency and multiple time-scale relaxations. Phys. Chem. Chem. Phys. 2020, 22, 21158–21168. [Google Scholar] [CrossRef] [PubMed]
- Pluhařová, E.; Marsalek, O.; Schmidt, B.; Jungwirth, P. Ab Initio Molecular Dynamics Approach to a Quantitative Description of Ion Pairing in Water. J. Phys. Chem. Lett. 2013, 4, 4177–4181. [Google Scholar] [CrossRef]
- Mattea, C.; Qvist, J.; Halle, B. Dynamics at the Protein-Water Interface from 17O Spin Relaxation in Deeply Supercooled Solutions. Biophys. J. 2008, 95, 2951–2963. [Google Scholar] [CrossRef] [Green Version]
- Conti Nibali, V.; Havenith, M. New Insights into the Role of Water in Biological Function: Studying Solvated Biomolecules Using Terahertz Absorption Spectroscopy in Conjunction with Molecular Dynamics Simulations. J. Am. Chem. Soc. 2014, 136, 12800–12807. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water Dynamics in the Hydration Shells of Biomolecules. Chem. Rev. 2017, 117, 10694–10725. [Google Scholar] [CrossRef]
- Charkhesht, A.; Regmi, C.K.; Mitchell-Koch, K.R.; Cheng, S.; Vinh, N.Q. High-Precision Megahertz-to-Terahertz Dielectric Spectroscopy of Protein Collective Motions and Hydration Dynamics. J. Phys. Chem. B 2018, 122, 6341–6350. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef]
- Perticaroli, S.; Comez, L.; Paolantoni, M.; Sassi, P.; Morresi, A.; Fioretto, D. Extended Frequency Range Depolarized Light Scattering Study of N-Acetyl-leucine-methylamide–Water Solutions. J. Am. Chem. Soc. 2011, 133, 12063–12068. [Google Scholar] [CrossRef]
- Perticaroli, S.; Ehlers, G.; Stanley, C.B.; Mamontov, E.; O’Neill, H.; Zhang, Q.; Cheng, X.; Myles, D.A.A.; Katsaras, J.; Nickels, J.D. Description of Hydration Water in Protein (Green Fluorescent Protein) Solution. J. Am. Chem. Soc. 2017, 139, 1098–1105. [Google Scholar] [CrossRef]
- Schiro, G.; Fichou, Y.; Gallat, F.X.; Wood, K.; Gabel, F.; Moulin, M.; Hartlein, M.; Heyden, M.; Colletier, J.P.; Orecchini, A.; et al. Translational diffusion of hydration water correlates with functional motions in folded and intrinsically disordered proteins. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- King, J.T.; Kubarych, K.J. Site-Specific Coupling of Hydration Water and Protein Flexibility Studied in Solution with Ultrafast 2D-IR Spectroscopy. J. Am. Chem. Soc. 2012, 134, 18705–18712. [Google Scholar] [CrossRef]
- Heyden, M.; Tobias, D.J.; Matyushov, D.V. Terahertz absorption of dilute aqueous solutions. J. Chem. Phys. 2012, 137, 235103(1)–235103(9). [Google Scholar] [CrossRef] [PubMed]
- Doster, W.; Cusack, S.; Petry, W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 1989, 337, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Magazu, S.; Migliardo, F.; Benedetto, A. Mean square displacements from elastic incoherent neutron scattering evaluated by spectrometers working with different energy resolution on dry and hydrated (H2O and D2O) lysozyme. J. Phys. Chem. B 2010, 114, 9268–9274. [Google Scholar] [CrossRef]
- Magazù, S.; Migliardo, F.; Benedetto, A.; La Torre, R.; Hennet, L. Bio-protective effects of homologous disaccharides on biological macromolecules. Eur. Biophys. J. Biophy. 2012, 41, 361–367. [Google Scholar] [CrossRef]
- Zanotti, J.M.; Bellissent-Funel, M.C.; Kolesnikov, A.I. Phase transitions of interfacial water at 165 and 240 K. Connections to bulk water physics and protein dynamics. Eur. Phys. J.-Spec. Top. 2007, 141, 227–233. [Google Scholar] [CrossRef]
- Tarek, M.; Martyna, G.J.; Tobias, D.J. Amplitudes and Frequencies of Protein Dynamics: Analysis of Discrepancies between Neutron Scattering and Molecular Dynamics Simulations. J. Am. Chem. Soc. 2000, 122, 10450–10451. [Google Scholar] [CrossRef]
- Tarek, M.; Tobias, D.J. Role of protein-water hydrogen bond dynamics in the protein dynamical transition. Phys. Rev. Lett. 2002, 88, 138101(1)–138101(4). [Google Scholar] [CrossRef]
- Jia, M.; Yang, J.; Qin, Y.; Wang, D.; Pan, H.; Wang, L.; Xu, J.; Zhong, D. Determination of Protein Surface Hydration by Systematic Charge Mutations. J. Phys. Chem. Lett. 2015, 6, 5100–5105. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Jia, M.H.; Yang, J.; Wang, D.H.; Wang, L.J.; Xu, J.H.; Zhong, D.P. Molecular Origin of Ultrafast Water–Protein Coupled Interactions. J. Phys. Chem. Lett. 2016, 7, 4171–4177. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Kao, Y.T.; Zhang, L.; Yang, Y.; Wang, L.; Stites, W.E.; Zhong, D.; Zewail, A.H. Protein surface hydration mapped by site-specific mutations. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13979–13984. [Google Scholar] [CrossRef] [Green Version]
- Skinner, J.J.; Lim, W.K.; Bédard, S.; Black, B.E.; Englander, S.W. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012, 21, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Skinner, J.J.; Lim, W.K.; Bédard, S.; Black, B.E.; Englander, S.W. Protein hydrogen exchange: Testing current models. Protein Sci. 2012, 21, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Damjanović, A.; Schlessman, J.L.; Fitch, C.A.; García, A.E.; García-Moreno, E.B. Role of Flexibility and Polarity as Determinants of the Hydration of Internal Cavities and Pockets in Proteins. Biophys. J. 2007, 93, 2791–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Cui, Q. Reverse Protonation of Buried Ion-Pairs in Staphylococcal Nuclease Mutants. J. Chem. Theory Comput. 2021, 17, 4550–4563. [Google Scholar] [CrossRef] [PubMed]
- Terazima, M.; Kataoka, M.; Ueoka, R.; Okamoto, Y. Molecular Science of Fluctuations Toward Biological Functions; Springer: Tokyo, Japan, 2016; pp. 151–161. [Google Scholar]
- Hirano, S.; Kamikubo, H.; Yamazaki, Y.; Kataoka, M. Elucidation of information encoded in tryptophan 140 of staphylococcal nuclease. Proteins, structure, function, and bioinformatics 2005, 58, 271–277. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 374–379. [Google Scholar] [CrossRef]
- Richards, F.M. Areas, Volumes, Packing, and Protein Structure. Annu. Rev. B. B. 1977, 6, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Petsko, G.A.; Tsernoglou, D. Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature 1979, 280, 558–563. [Google Scholar] [CrossRef]
- Nandi, P.K.; English, N.J.; Futera, Z.; Benedetto, A. Hydrogen-bond dynamics at the bio-water interface in hydrated proteins: A molecular-dynamics study. Phys. Chem. Chem. Phys. 2016, 19, 318–329. [Google Scholar] [CrossRef]
- Roh, J.H.; Novikov, V.N.; Gregory, R.B.; Curtis, J.E.; Chowdhuri, Z.; Sokolov, A.P. Onsets of Anharmonicity in Protein Dynamics. Phys. Rev. Lett. 2005, 95, 38101(1)–38101(4). [Google Scholar] [CrossRef]
- Pizzitutti, F.; Marchi, M.; Sterpone, F.; Rossky, P.J. How Protein Surfaces Induce Anomalous Dynamics of Hydration Water. J. Phys. Chem. B. 2007, 111, 7584–7590. [Google Scholar] [CrossRef] [PubMed]
- Sterpone, F.; Marchetti, G.; Pierleoni, C.; Marchi, M. Molecular modeling and simulation of water near model micelles: Diffusion, rotational dynamics and structure of the hydration interface. J. Phys. Chem. B. 2006, 110, 11504–11510. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. HBonanza: A computer algorithm for molecular-dynamics-trajectory hydrogen-bond analysis. J. Mol. Graph. Model. 2011, 31, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Laage, D. A Molecular Jump Mechanism of Water Reorientation. Science (Am. Assoc. Adv. Sci.) 2006, 311, 832–835. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Daura, X.; Suter, R.; van Gunsteren, W.F. Validation of molecular simulation by comparison with experiment: Rotational reorientation of tryptophan in water. J. Chem. Phys. 1999, 110, 3049–3055. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Starr, F.W.; Nielsen, J.K.; Stanley, H.E. Fast and Slow Dynamics of Hydrogen Bonds in Liquid Water. Phys. Rev. Lett. 1999, 82, 2294–2297. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | SN0 | SN1 | SN2 | SN3 |
|---|---|---|---|---|
| Coil(%) | 17 | 19 | 18 | 20 |
| β-Sheet(%) | 28 | 27 | 28 | 26 |
| β-Bridge(%) | 2 | 3 | 2 | 2 |
| Bend(%) | 14 | 11 | 12 | 12 |
| Turn(%) | 12 | 14 | 13 | 14 |
| α-Helix(%) | 25 | 24 | 25 | 25 |
| 3-Helix(%) | 2 | 2 | 1 | 1 |
| Temperature (K) | 150 | 180 | 210 | 240 | 270 | 300 |
|---|---|---|---|---|---|---|
| SASA (nm2) | 80.2 ± 0.6 | 81.5 ± 1.1 | 82.9 ± 0.8 | 83.9 ± 1.3 | 84.4 ± 1.2 | 85.6 ± 1.6 |
| Mutation | SN0 | SN1 | SN2 | SN3 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Temperature (K) | 150 | 180 | 210 | 240 | 270 | 300 | 300 | 300 | 300 |
| Position (nm) | 0.276 | 0.274 | 0.274 | 0.272 | 0.27 | 0.272 | 0.272 | 0.276 | 0.276 |
| Height | 1.794 | 1.719 | 1.667 | 1.554 | 1.513 | 1.503 | 1.491 | 1.415 | 1.381 |
| Mutation | SN0 | SN1 | SN2 | SN3 |
|---|---|---|---|---|
| τs (ps) | 58.9 ± 0.35 | 57.8 ± 0.30 | 49.4 ± 0.40 | 46.3 ± 0.36 |
| τ1 (ps) | 4.8 ± 0.05 | 4.7 ± 0.05 | 4.3 ± 0.05 | 3.1 ± 0.13 |
| Mutation | SN0 | SN1 | SN2 | SN3 |
|---|---|---|---|---|
| tμ (ps) | 4.8 ± 0.01 | 4.3 ± 0.01 | 4.2 ± 0.01 | 3.8 ± 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Xiang, S.; Zhu, H.; Li, L. The Structural and Dynamical Properties of the Hydration of SNase Based on a Molecular Dynamics Simulation. Molecules 2021, 26, 5403. https://doi.org/10.3390/molecules26175403
Liu H, Xiang S, Zhu H, Li L. The Structural and Dynamical Properties of the Hydration of SNase Based on a Molecular Dynamics Simulation. Molecules. 2021; 26(17):5403. https://doi.org/10.3390/molecules26175403
Chicago/Turabian StyleLiu, Hangxin, Shuqing Xiang, Haomiao Zhu, and Li Li. 2021. "The Structural and Dynamical Properties of the Hydration of SNase Based on a Molecular Dynamics Simulation" Molecules 26, no. 17: 5403. https://doi.org/10.3390/molecules26175403