
P∩N Bridged Cu(I) Dimers Featuring Both TADF and Phosphorescence. From Overview towards Detailed Case Study of the Excited Singlet and Triplet States †


Abstract
:1. Introduction
2. Results and Discussion
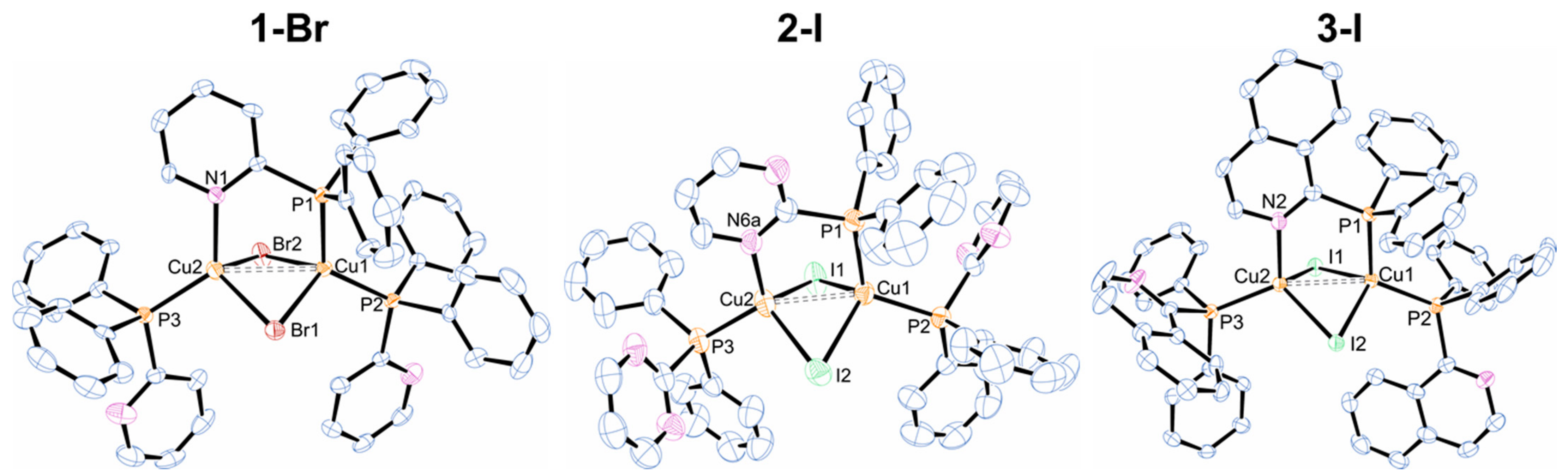
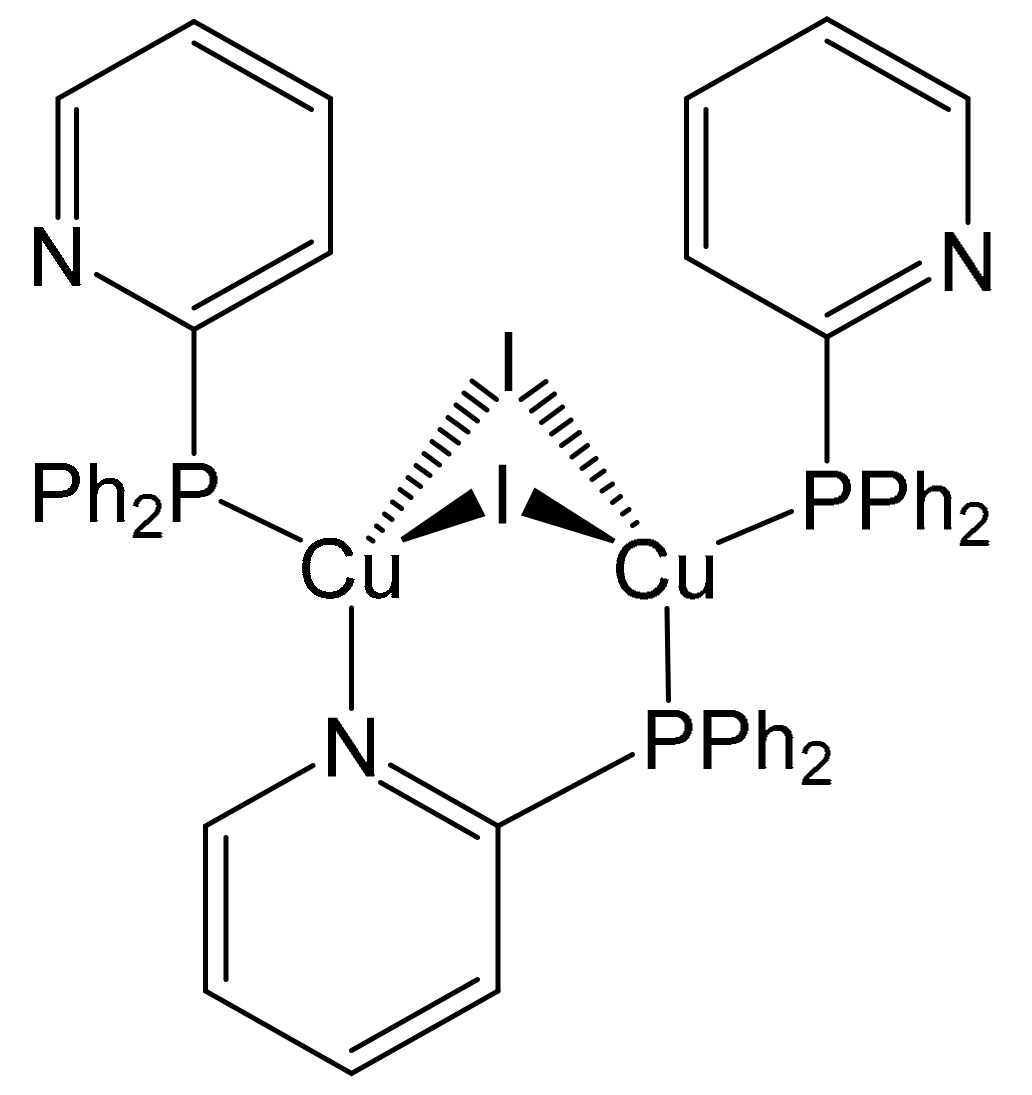
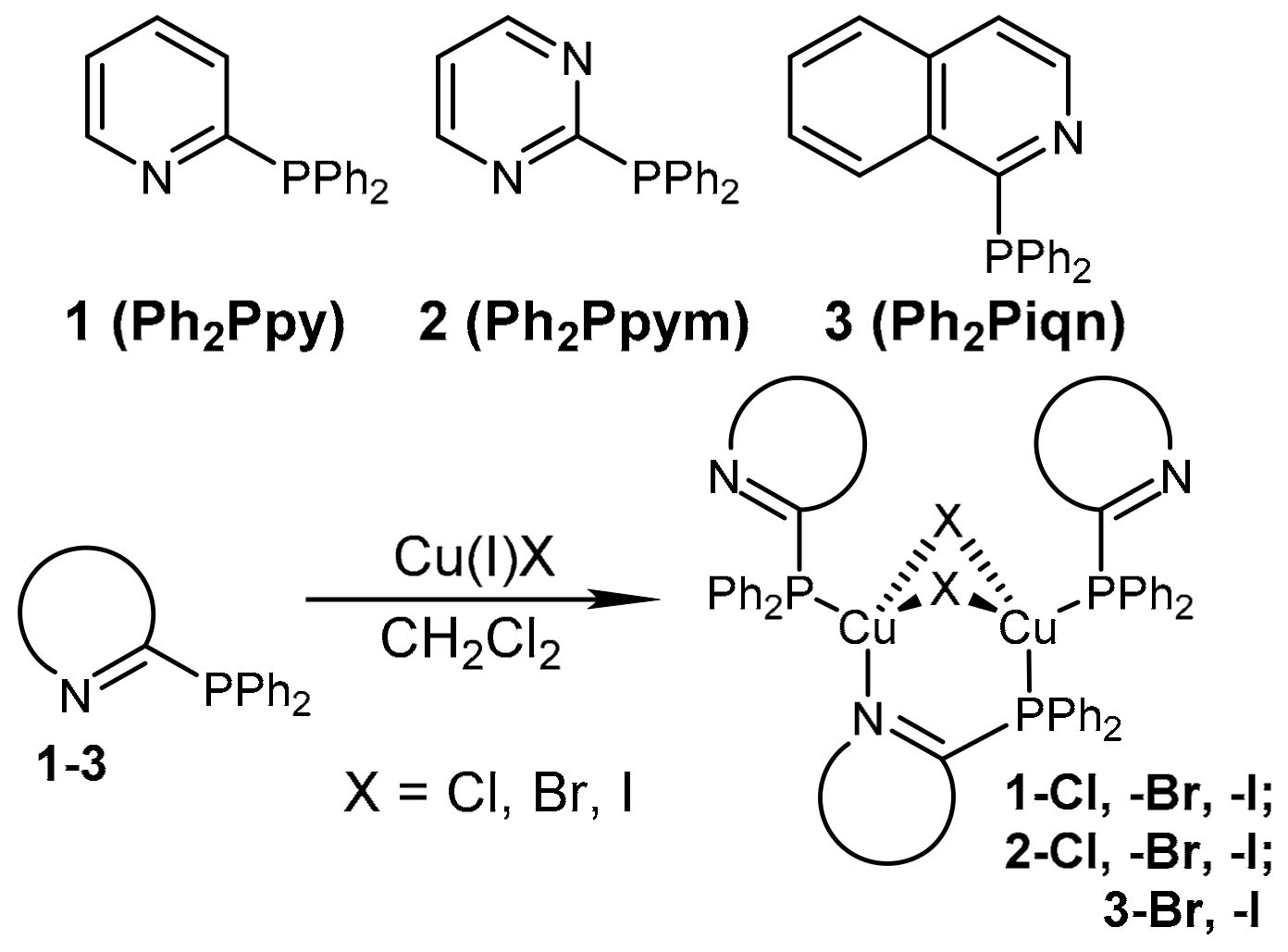
2.1. Syntheses and Structural Characterization
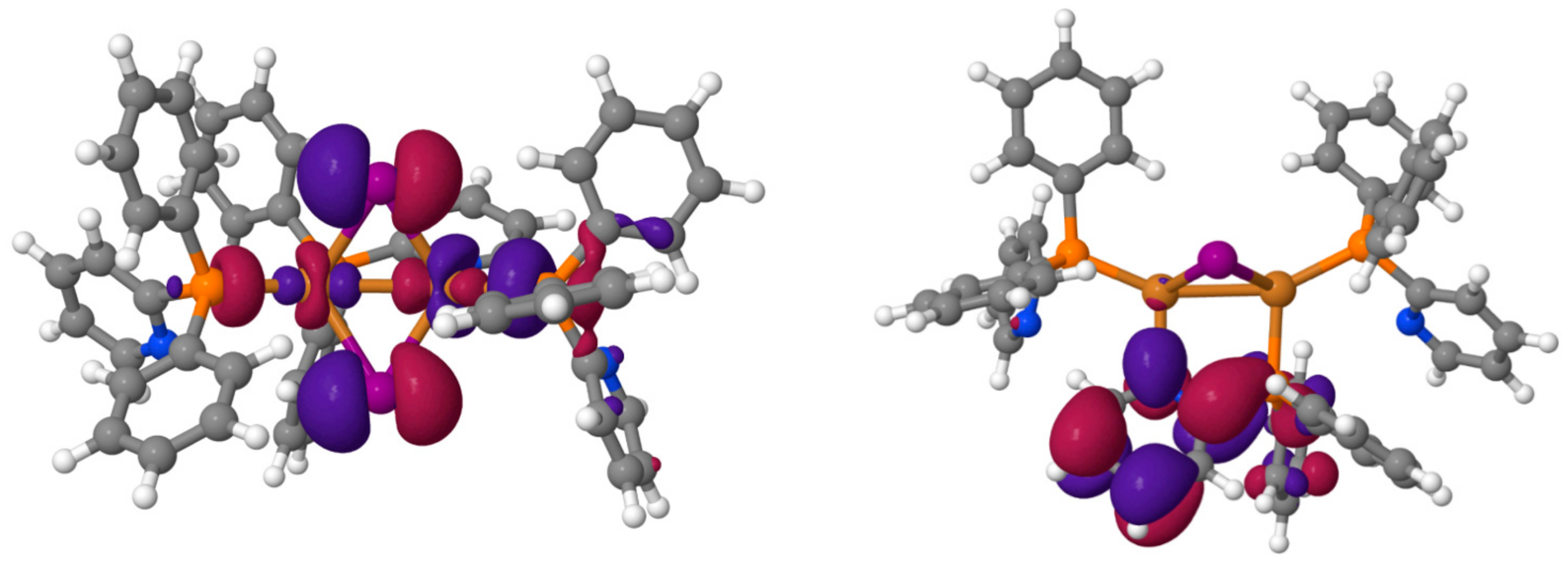
2.2. Computational Investigations
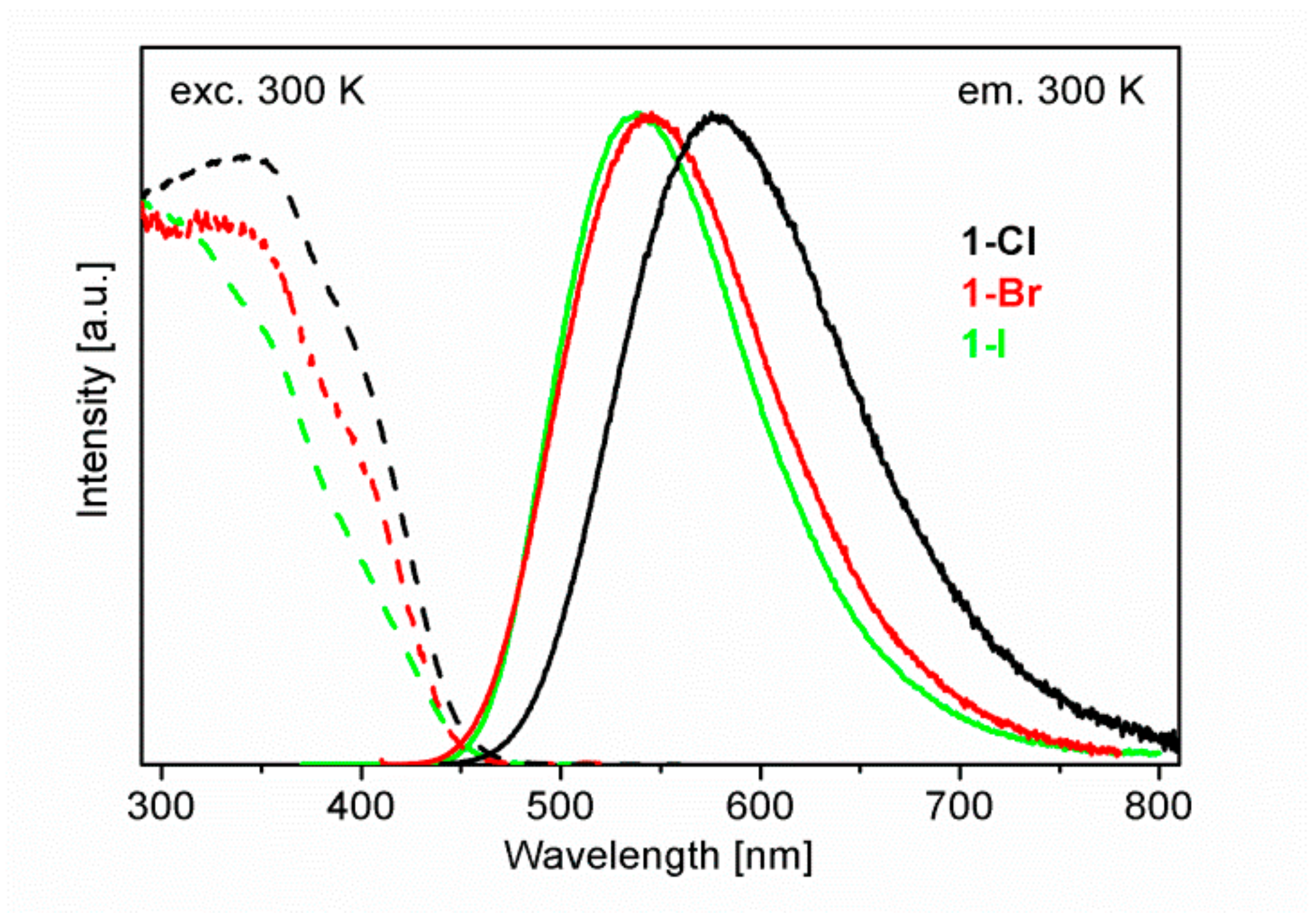
2.3. Luminescence Properties of Cu2X2(P∩N)3 Complexes with X = Cl, Br, I. An Overview
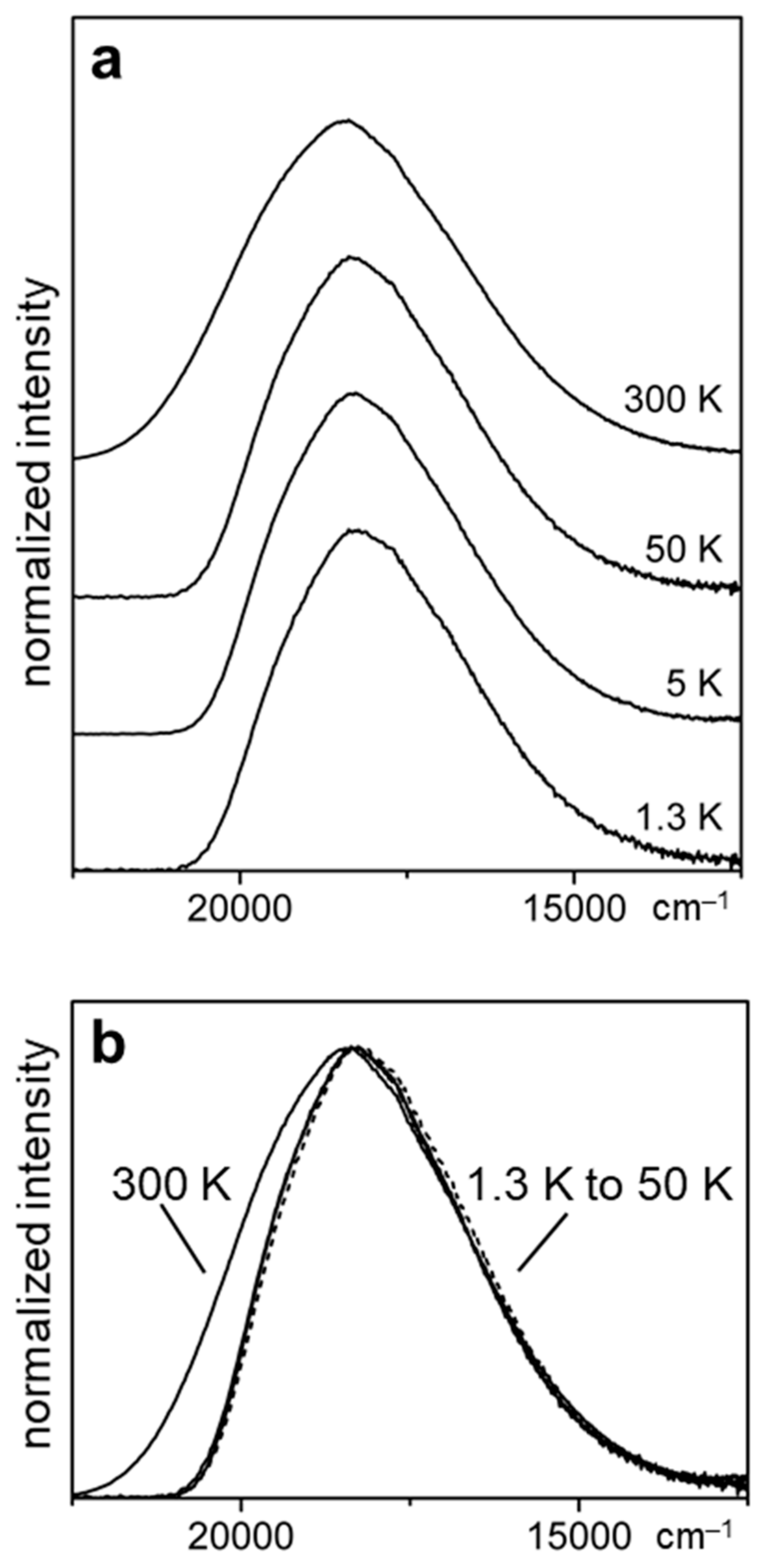
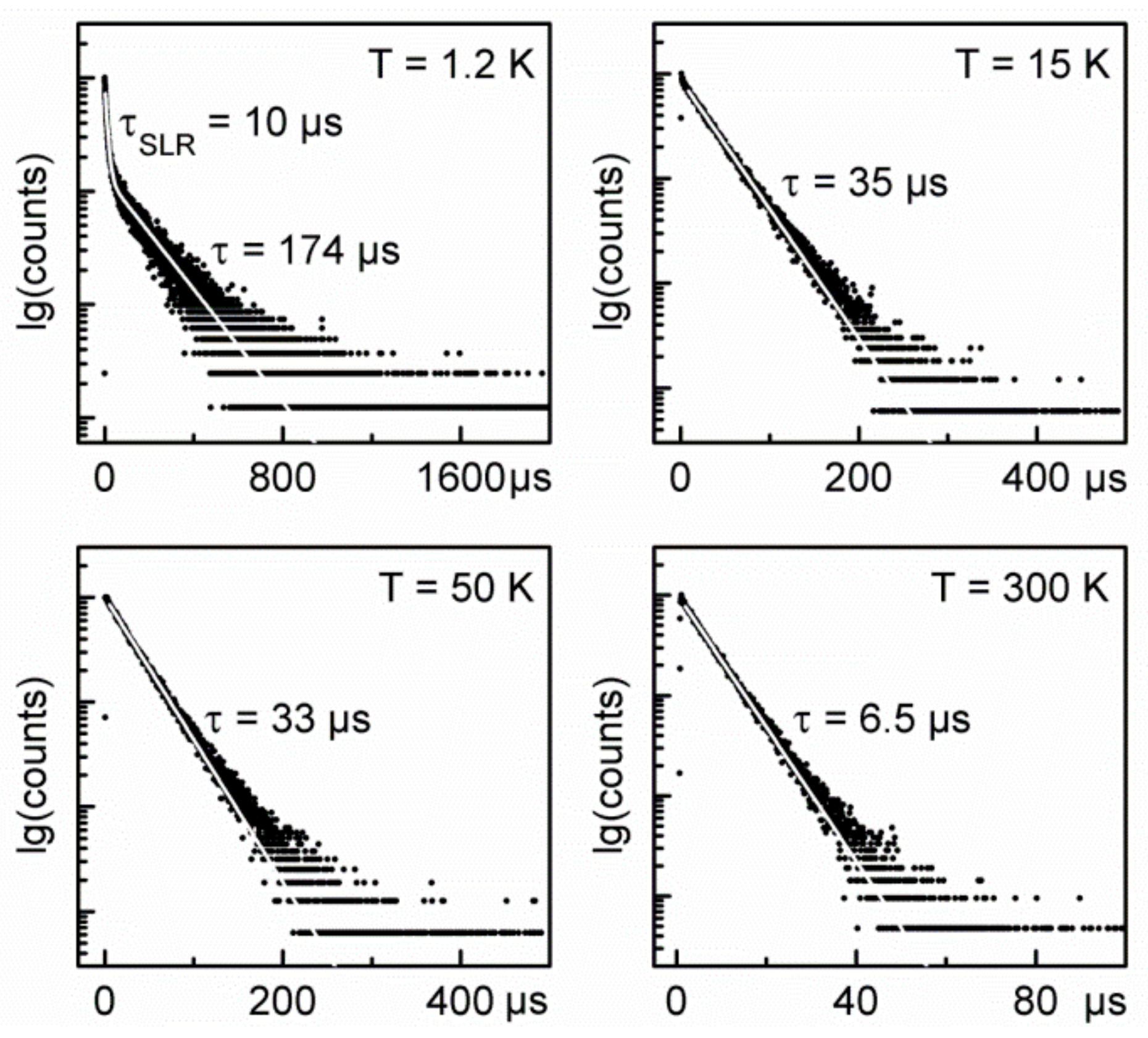
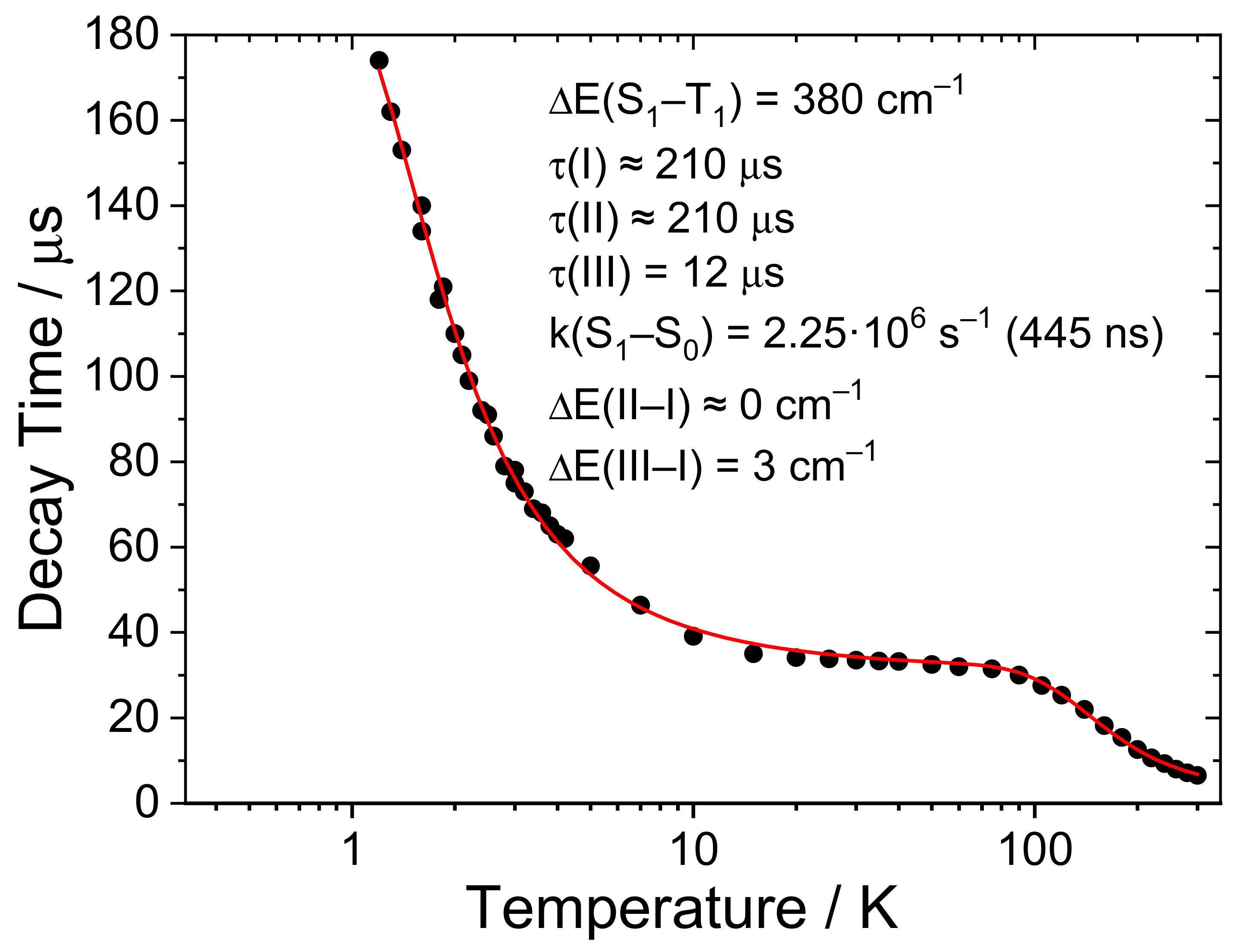
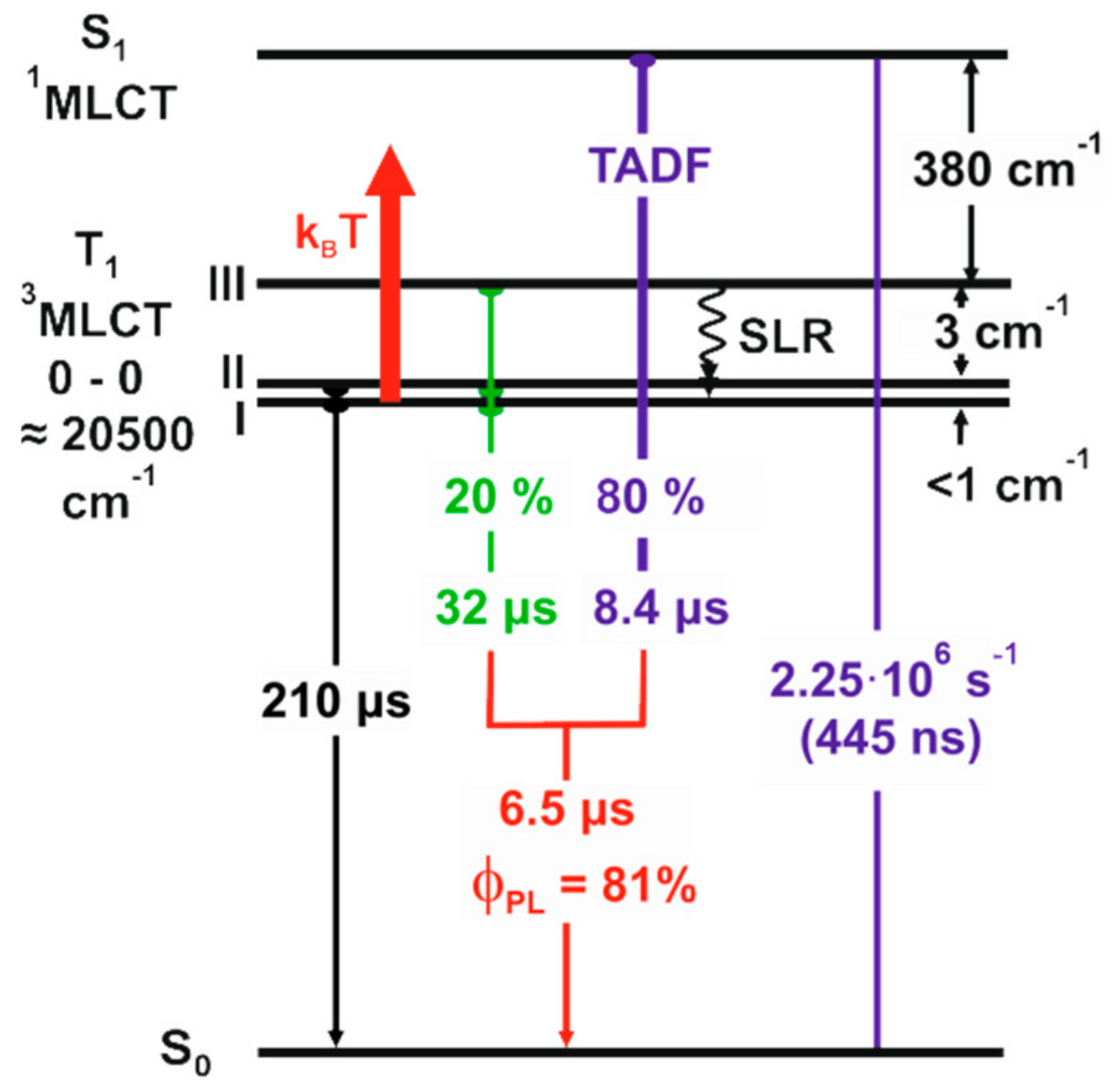
2.4. Detailed Case Study of Cu2I2(Ph2Ppy)3, 1-I. the Lowest Excited Triplet and Singlet States
3. Summarizing Conclusions
4. Materials and Methods
4.1. General
4.2. Photophysical Measurements
4.3. Computational Investigations
4.4. Syntheses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Helfrich, W.; Schneider, W.G. Transients of volume-controlled current and of recombination radiation in anthracene. J. Chem. Phys. 1966, 44, 2902–2909. [Google Scholar] [CrossRef]
- Yersin, H. Highly Efficient OLEDs with Phosphorescent Materials; Wiley-VCH Verlag: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Brütting, W.; Adachi, C. Physics of Organic Semiconductors; Wiley-VCH Verlag: Weinheim, Germany, 2012. [Google Scholar]
- Baldo, M.A.; O’Brien, D.F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.E.; Forrest, S.R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Adachi, C.; Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. [Google Scholar] [CrossRef] [Green Version]
- Yersin, H. Triplet Emitters for OLED applications. Mechanisms of exciton trapping and control of emission properties. Top. Curr. Chem. 2004, 241, 1–26. [Google Scholar] [CrossRef]
- Yersin, H. Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence; Wiley-VCH Verlag: Weinheim, Germany, 2019. [Google Scholar]
- Yersin, H.; Monkowius, U. Komplexe mit Kleinen Singulett-Triplett-Energie-Abständen zur Verwendung in Opto-Elektronischen Bauteilen (Singulett-Harvesting-Effekt). German Patent No. DE102008033563, 21 January 2010. [Google Scholar]
- Czerwieniec, R.; Yu, J.; Yersin, H. Blue-light emission of Cu(I) complexes and singlet harvesting. Inorg. Chem. 2011, 50, 8293–8301. [Google Scholar] [CrossRef]
- Deaton, J.C.; Switalski, S.C.; Kondakov, D.Y.; Young, R.H.; Pawlik, T.D.; Giesen, D.J.; Harkins, S.B.; Miller, A.J.M.; Mickenberg, S.F.; Peters, J.C. E-type delayed fluorescence of a phosphine-supported Cu2(μ-NAr2)2 diamond core: Harvesting singlet and triplet excitons in OLEDs. J. Am. Chem. Soc. 2010, 132, 9499–9508. [Google Scholar] [CrossRef] [Green Version]
- Zink, D.M.; Volz, D.; Baumann, T.; Mydlak, M.; Flügge, H.; Friedrichs, J.; Nieger, M.; Bräse, S. Heteroleptic, dinuclear copper (I) complexes for application in organic light-emitting diodes. Chem. Mater. 2013, 25, 4471–4486. [Google Scholar] [CrossRef]
- Dumur, F. Recent advances in organic light-emitting devices comprising copper complexes: A realistic approach for low-cost and highly emissive devices? Org. Electron. 2015, 21, 27–39. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly phosphorescent bis-cyclometalated iridium complexes: Synthesis, photophysical characterization, and use in organic light emitting diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef]
- Zysman-Colman, E. Iridium (III) in Optoelectronic and Photonics Applications; John Wiley & Sons: Chichester, UK, 2017. [Google Scholar]
- Deaton, J.C.; Castellano, F.N. Archetypal iridium (III) compounds for optoelectronic and photonic applications. In Iridium(III) in Optoelectronic and Photonics Applications; Zysman-Colman, E., Ed.; John Wiley & Sons: Chichester, UK, 2017; pp. 1–69. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.-C.; Chan, A.K.-W.; Chan, M.-Y.; Yam, V.W.-W. Platinum and gold complexes for OLEDs. Top. Curr. Chem. 2016, 374, 1–43. [Google Scholar] [CrossRef]
- Li, K.; Ming Tong, G.S.; Wan, Q.; Cheng, G.; Tong, W.-Y.; Ang, W.-H.; Kwong, W.-L.; Che, C.-M. Highly phosphorescent platinum (II) emitters: Photophysics, materials and biological applications. Chem. Sci. 2016, 7, 1653–1673. [Google Scholar] [CrossRef] [Green Version]
- Armaroli, N.; Bolink, H.J. Photoluminescent Materials and Electroluminescent Devices; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Yersin, H.; Rausch, A.F.; Czerwieniec, R.; Hofbeck, T.; Fischer, T. The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs. Coord. Chem. Rev. 2011, 255, 2622–2652. [Google Scholar] [CrossRef]
- Yersin, H.; Rausch, A.F.; Czerwieniec, R. Organometallic emitters for OLEDs: Triplet harvesting, singlet harvesting, case structures, and trends. In Physics of Organic Semiconductors; Brütting, W., Adachi, C., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2012; pp. 371–424. [Google Scholar] [CrossRef]
- Yersin, H.; Finkenzeller, W.J. Triplet emitters for organic light-emitting diodes: Basic properties. In Highly Efficient OLEDs with Phosphorescent Materials; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2007; pp. 1–97. [Google Scholar] [CrossRef]
- Che, C.-M.; Kwok, C.-C.; Lai, S.-W.; Rausch, A.F.; Finkenzeller, W.J.; Zhu, N.; Yersin, H. Photophysical properties and OLED applications of phosphorescent platinum (II) Schiff base complexes. Chem. Eur. J. 2010, 16, 233–247. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Chan, S.-C.; Chan, M.C.W.; Hou, Y.-J.; Zhu, N.; Che, C.-M.; Liu, Y.; Wang, Y. Structural, photophysical, and electrophosphorescent properties of platinum (II) complexes supported by tetradentate N2O2 chelates. Chem. Eur. J. 2003, 9, 1263–1272. [Google Scholar] [CrossRef]
- Cheng, G.; Kui, S.C.F.; Ang, W.-H.; Ko, M.-Y.; Chow, P.-K.; Kwong, C.-L.; Kwok, C.-C.; Ma, C.; Guan, X.; Low, K.-H.; et al. Structurally robust phosphorescent [Pt(O^N^C^N)] emitters for high performance organic light-emitting devices with power efficiency up to 126 lm W−1 and external quantum efficiency over 20%. Chem. Sci. 2014, 5, 4819–4830. [Google Scholar] [CrossRef]
- Mao, M.; Peng, J.; Lam, T.-L.; Ang, W.-H.; Li, H.; Cheng, G.; Che, C.-M. High-performance organic light-emitting diodes with low-efficiency roll-off using bulky tetradentate [Pt(O^N^C^N)] emitters. J. Mater. Chem. C 2019, 7, 7230–7236. [Google Scholar] [CrossRef]
- Cheng, G.; Kwak, Y.; To, W.-P.; Lam, T.-L.; Tong, G.S.M.; Sit, M.-K.; Gong, S.; Choi, B.; Choi, W.I.; Yang, C.; et al. High-efficiency solution-processed organic light-emitting diodes with tetradentate platinum(II) emitters. ACS Appl. Mater. Interfaces 2019, 11, 45161–45170. [Google Scholar] [CrossRef]
- Vezzu, D.A.K.; Deaton, J.C.; Jones, J.S.; Bartolotti, L.; Harris, C.F.; Marchetti, A.P.; Kondakova, M.; Pike, R.D.; Huo, S. Highly luminescent tetradentate bis-cyclometalated platinum complexes: Design, synthesis, structure, photophysics, and electroluminescence application. Inorg. Chem. 2010, 49, 5107–5119. [Google Scholar] [CrossRef]
- Fleetham, T.; Li, G.; Wen, L.; Li, J. Efficient “pure” blue OLEDs employing tetradentate Pt complexes with a narrow spectral bandwidth. Adv. Mater. 2014, 26, 7116–7121. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Yersin, H. Diversity of copper(I) complexes showing thermally activated delayed fluorescence: Basic photophysical analysis. Inorg. Chem. 2015, 54, 4322–4327. [Google Scholar] [CrossRef]
- Osawa, M.; Hoshino, M.; Hashimoto, M.; Kawata, I.; Igawa, S.; Yashima, M. Application of three-coordinate copper(I) complexes with halide ligands in organic light-emitting diodes that exhibit delayed fluorescence. Dalton Trans. 2015, 44, 8369–8378. [Google Scholar] [CrossRef]
- Yu, R.; Lu, C.-Z. Ionic [Cu(NN)(PP)]+ TAD9727 F complexes with pyridine-based diimine chelating ligands and their use in OLEDs. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 177–198. [Google Scholar] [CrossRef]
- Hirtenlehner, C.; Monkowius, U. Syntheses, crystal structures and blue luminescence of Cu2X2(Ph3P)2[(−)-nicotine]2 (X. = Br, I). Inorg. Chem. Commun. 2012, 15, 109–112. [Google Scholar] [CrossRef]
- So, G.K.-M.; Cheng, G.; Wang, J.; Chang, X.; Kwok, C.-C.; Zhang, H.; Che, C.-M. Efficient color-tunable copper(I) complexes and their applications in solution-processed organic light-emitting diodes. Chem. Asian J. 2017, 12, 1490–1498. [Google Scholar] [CrossRef] [Green Version]
- Zink, D.M.; Bächle, M.; Baumann, T.; Nieger, M.; Kühn, M.; Wang, C.; Klopper, W.; Monkowius, U.; Hofbeck, T.; Yersin, H.; et al. Synthesis, structure, and characterization of dinuclear copper(I) halide complexes with P^N ligands featuring exciting photoluminescence properties. Inorg. Chem. 2013, 52, 2292–2305. [Google Scholar] [CrossRef]
- Wallesch, M.; Volz, D.; Zink, D.M.; Schepers, U.; Nieger, M.; Baumann, T.; Bräse, S. Bright coppertunities: Multinuclear Cu(I) complexes with N-P ligands and their applications. Chem. Eur. J. 2014, 20, 6578–6590. [Google Scholar] [CrossRef] [PubMed]
- Leitl, M.J.; Zink, D.M.; Schinabeck, A.; Baumann, T.; Volz, D.; Yersin, H. Copper(I) complexes for thermally activated delayed fluorescence: From photophysical to device properties. Top. Curr. Chem. 2016, 374, 25–65. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Leitl, M.J.; Homeier, H.H.H.; Yersin, H. Cu(I) complexes–thermally activated delayed fluorescence. Photophysical approach and material design. Coord. Chem. Rev. 2016, 325, 2–28. [Google Scholar] [CrossRef]
- Hamze, R.; Peltier, J.L.; Sylvinson, D.; Jung, M.; Cardenas, J.; Haiges, R.; Soleilhavoup, M.; Jazzar, R.; Djurovich, P.I.; Bertrand, G.; et al. Eliminating nonradiative decay in Cu(I) emitters: 99% quantum efficiency and microsecond lifetime. Science 2019, 363, 601–606. [Google Scholar] [CrossRef]
- Hamze, R.; Shi, S.; Kapper, S.C.; Muthiah Ravinson, D.S.; Estergreen, L.; Jung, M.-C.; Tadle, A.C.; Haiges, R.; Djurovich, P.I.; Peltier, J.L.; et al. “Quick-silver>” from a systematic study of highly luminescent, two-coordinate, d10 coinage metal complexes. J. Am. Chem. Soc. 2019, 141, 8616–8626. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Jung, M.C.; Coburn, C.; Tadle, A.; Sylvinson, M.R.D.; Djurovich, P.I.; Forrest, S.R.; Thompson, M.E. Highly efficient photo- and electroluminescence from two-coordinate Cu(I) complexes featuring nonconventional N-heterocyclic carbenes. J. Am. Chem. Soc. 2019, 141, 3576–3588. [Google Scholar] [CrossRef] [PubMed]
- Di, D.; Romanov, A.S.; Yang, L.; Richter, J.M.; Rivett, J.P.; Jones, S.; Thomas, T.H.; Jalebi, M.A.; Friend, R.H.; Linnolahti, M.; et al. High-performance light-emitting diodes based on carbene-metal-amides. Science 2017, 356, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Conaghan, P.J.; Menke, S.M.; Romanov, A.S.; Jones, S.T.E.; Pearson, A.J.; Evans, E.W.; Bochmann, M.; Greenham, N.C.; Credgington, D. Efficient vacuum-processed light-emitting diodes based on carbene-metal-amides. Adv. Mater. 2018, 30, e1802285. [Google Scholar] [CrossRef]
- Romanov, A.S.; Jones, S.T.E.; Gu, Q.; Conaghan, P.J.; Drummond, B.H.; Feng, J.; Chotard, F.; Buizza, L.; Foley, M.; Linnolahti, M.; et al. Carbene metal amide photoemitters: Tailoring conformationally flexible amides for full color range emissions including white-emitting OLED. Chem. Sci. 2020, 11, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Föller, J.; Ganter, C.; Steffen, A.; Marian, C.M. Computer-aided design of luminescent linear N-heterocyclic carbene copper(I) pyridine complexes. Inorg. Chem. 2019, 58, 5446–5456. [Google Scholar] [CrossRef]
- Osawa, M.; Hoshino, M. Molecular design and synthesis of metal complexes as emitters for TADF-type OLEDs. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 119–176. [Google Scholar] [CrossRef]
- Nozaki, K.; Iwamura, M. Highly emissive d10 metal complexes as TADF emitters with versatile structures and photophysical properties. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 61–91. [Google Scholar] [CrossRef]
- Tsuboyama, A. Luminescent dinuclear copper(I) complexes with short intramolecular Cu-Cu distances. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 93–118. [Google Scholar] [CrossRef]
- Yersin, H.; Czerwieniec, R.; Shafikov, M.Z.; Suleymanova, A.F. TADF material design: Photophysical background and case studies focusing on CuI and AgI complexes. Chemphyschem 2017, 18, 3508–3535. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Cardinali, F.; Listorti, A. Photochemistry and photophysics of coordination compounds: Copper. In Photochemistry and Photophysics of Coordination Compounds I; Balzani, V., Campagna, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 69–115. [Google Scholar] [CrossRef]
- Schinabeck, A.; Leitl, M.J.; Yersin, H. Dinuclear Cu(I) complex with combined bright TADF and phosphorescence. Zero-field splitting and spin-lattice relaxation effects of the triplet state. J. Phys. Chem. Lett. 2018, 9, 2848–2856. [Google Scholar] [CrossRef]
- Schinabeck, A.; Rau, N.; Klein, M.; Sundermeyer, J.; Yersin, H. Deep blue emitting Cu(I) tripod complexes. Design of high quantum yield materials showing TADF-assisted phosphorescence. Dalton Trans. 2018, 47, 17067–17076. [Google Scholar] [CrossRef]
- Schinabeck, A.; Chen, J.; Kang, L.; Teng, T.; Homeier, H.H.H.; Suleymanova, A.F.; Shafikov, M.Z.; Yu, R.; Lu, C.-Z.; Yersin, H. Symmetry-based design strategy for unprecedentedly fast decaying thermally activated delayed fluorescence (TADF). Application to dinuclear Cu(I) compounds. Chem. Mater. 2019, 31, 4392–4404. [Google Scholar] [CrossRef]
- Xu, K.; Chen, B.-L.; Yang, F.; Liu, L.; Zhong, X.-X.; Wang, L.; Zhu, X.-J.; Li, F.-B.; Wong, W.-Y.; Qin, H.-M. Largely color-tuning prompt and delayed fluorescence: Dinuclear Cu(I) halide complexes with tert -amines and phosphines. Inorg. Chem. 2021, 60, 4841–4851. [Google Scholar] [CrossRef]
- Gan, X.-M.; Yu, R.; Chen, X.-L.; Yang, M.; Lin, L.; Wu, X.-Y.; Lu, C.-Z. A unique tetranuclear Ag(I) complex emitting efficient thermally activated delayed fluorescence with a remarkably short decay time. Dalton Trans. 2018, 47, 5956–5960. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Suleymanova, A.F.; Czerwieniec, R.; Yersin, H. Design strategy for Ag(I)-based thermally activated delayed fluorescence reaching an efficiency breakthrough. Chem. Mater. 2017, 29, 1708–1715. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Suleymanova, A.F.; Schinabeck, A.; Yersin, H. Dinuclear Ag(I) complex designed for highly efficient thermally activated delayed fluorescence. J. Phys. Chem. Lett. 2018, 9, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Kawata, I.; Ishii, R.; Igawa, S.; Hashimoto, M.; Hoshino, M. Application of neutral d10 coinage metal complexes with an anionic bidentate ligand in delayed fluorescence-type organic light-emitting diodes. J. Mater. Chem. C 2013, 1, 4375–4383. [Google Scholar] [CrossRef]
- Chen, J.; Teng, T.; Kang, L.; Chen, X.-L.; Wu, X.-Y.; Yu, R.; Lu, C.-Z. Highly efficient thermally activated delayed fluorescence in dinuclear Ag(I) complexes with a bis-bidentate tetraphosphane bridging ligand. Inorg. Chem. 2016, 55, 9528–9536. [Google Scholar] [CrossRef]
- Chan, K.-T.; Lam, T.-L.; Yu, D.; Du, L.; Phillips, D.L.; Kwong, C.-L.; Tong, G.S.M.; Cheng, G.; Che, C.-M. Strongly luminescent tungsten emitters with emission quantum yields of up to 84 %: TADF and high-efficiency molecular tungsten OLEDs. Angew. Chem. Int. Ed. Engl. 2019, 58, 14896–14900. [Google Scholar] [CrossRef]
- Fernandez-Cestau, J.; Bertrand, B.; Blaya, M.; Jones, G.A.; Penfold, T.J.; Bochmann, M. Synthesis and luminescence modulation of pyrazine-based gold(III) pincer complexes. Chem. Commun. 2015, 51, 16629–16632. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; To, W.-P.; Kwak, Y.; Cho, Y.; Cheng, G.; Tong, G.S.M.; Che, C.-M. Thermally stable donor-acceptor type (Alkynyl)gold(III) TADF emitters achieved EQEs and luminance of up to 23.4% and 70 300 cd m-2 in vacuum-deposited OLEDs. Adv. Sci. 2019, 6, 1802297. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; To, W.-P.; Tong, G.S.M.; Cheng, G.; Du, L.; Phillips, D.L.; Che, C.-M. Tetradentate gold(III) complexes as thermally activated delayed fluorescence (TADF) emitters: Microwave-assisted synthesis and high-performance OLEDs with long operational lifetime. Angew. Chem. Int. Ed. Engl. 2020, 59, 6375–6382. [Google Scholar] [CrossRef]
- Li, L.-K.; Tang, M.-C.; Lai, S.-L.; Ng, M.; Kwok, W.-K.; Chan, M.-Y.; Yam, V.W.-W. Strategies towards rational design of gold(III) complexes for high-performance organic light-emitting devices. Nat. Photonics 2019, 13, 185–191. [Google Scholar] [CrossRef]
- Lee, C.-H.; Tang, M.-C.; Kong, F.K.-W.; Cheung, W.-L.; Ng, M.; Chan, M.-Y.; Yam, V.W.-W. Isomeric tetradentate ligand-containing cyclometalated gold(III) complexes. J. Am. Chem. Soc. 2020, 142, 520–529. [Google Scholar] [CrossRef]
- Sakai, Y.; Sagara, Y.; Nomura, H.; Nakamura, N.; Suzuki, Y.; Miyazaki, H.; Adachi, C. Zinc complexes exhibiting highly efficient thermally activated delayed fluorescence and their application to organic light-emitting diodes. Chem. Commun. 2015, 51, 3181–3184. [Google Scholar] [CrossRef]
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. [Google Scholar] [CrossRef]
- Hosokai, T.; Matsuzaki, H.; Nakanotani, H.; Tokumaru, K.; Tsutsui, T.; Furube, A.; Nasu, K.; Nomura, H.; Yahiro, M.; Adachi, C. Evidence and mechanism of efficient thermally activated delayed fluorescence promoted by delocalized excited states. Sci. Adv. 2017, 3, e1603282. [Google Scholar] [CrossRef] [Green Version]
- Noda, H.; Nakanotani, H.; Adachi, C. Excited state engineering for efficient reverse intersystem crossing. Sci. Adv. 2018, 4, eaao6910. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shi, Y.-Z.; Wang, K.; Zhang, M.; Zheng, C.-J.; Sun, D.-M.; Dai, G.-L.; Fan, X.-C.; Wang, D.-Q.; Liu, W.; et al. Thermally activated delayed fluorescence carbonyl derivatives for organic light-emitting diodes with extremely narrow full width at half-maximum. ACS Appl. Mater. Interfaces 2019, 11, 13472–13480. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M.R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 2018, 3, 18020. [Google Scholar] [CrossRef]
- Komatsu, R.; Ohsawa, T.; Sasabe, H.; Nakao, K.; Hayasaka, Y.; Kido, J. Manipulating the electronic excited state energies of pyrimidine-based thermally activated delayed fluorescence emitters to realize efficient deep-blue emission. ACS Appl. Mater. Interfaces 2017, 9, 4742–4749. [Google Scholar] [CrossRef]
- Li, M.; Liu, Y.; Duan, R.; Wei, X.; Yi, Y.; Wang, Y.; Chen, C.-F. Aromatic-imide-based thermally activated delayed fluorescence materials for highly efficient organic light-emitting diodes. Angew. Chem. Int. Ed Engl. 2017, 56, 8818–8822. [Google Scholar] [CrossRef]
- Sharma, N.; Wong, M.Y.; Samuel, I.D.W.; Zysman-Colman, E. Solution-processed TADF materials and devices based on organic emitters. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 501–541. [Google Scholar] [CrossRef]
- Wong, M.Y.; Zysman-Colman, E. Purely organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-K.; Kim, D.; Brédas, J.-L. Thermally activated delayed fluorescence (TADF) path toward efficient electroluminescence in purely organic materials: Molecular level insight. Acc. Chem. Res. 2018, 51, 2215–2224. [Google Scholar] [CrossRef]
- Sommer, G.A.; Mataranga-Popa, L.N.; Czerwieniec, R.; Hofbeck, T.; Homeier, H.H.H.; Müller, T.J.J.; Yersin, H. Design of conformationally distorted donor-acceptor dyads showing efficient thermally activated delayed fluorescence. J. Phys. Chem. Lett. 2018, 9, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.D.; Brütting, W. Efficiency enhancement of organic light-emitting diodes exhibiting delayed fluorescence and nonisotropic emitter orientation. In Highly Efficient OLEDs; Yersin, H., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2018; pp. 199–228. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Su, S.-J. Marching toward highly efficient, pure-blue, and stable thermally activated delayed fluorescent organic light-emitting diodes. Adv. Funct. Mater. 2018, 28, 1802558. [Google Scholar] [CrossRef]
- Yersin, H.; Mataranga-Popa, L.; Czerwieniec, R.; Dovbii, Y. Design of a new mechanism beyond thermally activated delayed fluorescence toward fourth generation organic light emitting diodes. Chem. Mater. 2019, 31, 6110–6116. [Google Scholar] [CrossRef]
- Yersin, H.; Mataranga-Popa, L.; Li, S.-W.; Czerwieniec, R. Design strategies for materials showing thermally activated delayed fluorescence and beyond: Towards the fourth-generation OLED mechanism. J. Soc. Inf. Disp. 2018, 26, 194–199. [Google Scholar] [CrossRef]
- Leitl, M.J.; Küchle, F.-R.; Mayer, H.A.; Wesemann, L.; Yersin, H. Brightly blue and green emitting Cu(I) dimers for singlet harvesting in OLEDs. J. Phys. Chem. A 2013, 117, 11823–11836. [Google Scholar] [CrossRef] [PubMed]
- Gneuß, T.; Leitl, M.J.; Finger, L.H.; Rau, N.; Yersin, H.; Sundermeyer, J. A new class of luminescent Cu(I) complexes with tripodal ligands–TADF emitters for the yellow to red color range. Dalton Trans. 2015, 44, 8506–8520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-L.; Yu, R.; Wu, X.-Y.; Liang, D.; Jia, J.-H.; Lu, C.-Z. A strongly greenish-blue-emitting Cu4Cl4 cluster with an efficient spin-orbit coupling (SOC): Fast phosphorescence versus thermally activated delayed fluorescence. Chem. Commun. 2016, 52, 6288–6291. [Google Scholar] [CrossRef]
- Bizzarri, C.; Hundemer, F.; Busch, J.; Bräse, S. Triplet emitters versus TADF emitters in OLEDs: A comparative study. Polyhedron 2018, 140, 51–66. [Google Scholar] [CrossRef]
- Baranov, A.Y.; Berezin, A.S.; Samsonenko, D.G.; Mazur, A.S.; Tolstoy, P.M.; Plyusnin, V.F.; Kolesnikov, I.E.; Artem’ev, A.V. New Cu(I) halide complexes showing TADF combined with room temperature phosphorescence: The balance tuned by halogens. Dalton Trans. 2020, 49, 3155–3163. [Google Scholar] [CrossRef]
- Hofbeck, T.; Monkowius, U.; Yersin, H. Highly efficient luminescence of Cu(I) compounds: Thermally activated delayed fluorescence combined with short-lived phosphorescence. J. Am. Chem. Soc. 2015, 137, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, W.; Henwood, A.F.; Hall, D.; Cordes, D.B.; Slawin, A.M.Z.; Lemaur, V.; Olivier, Y.; Samuel, I.D.W.; Zysman-Colman, E. Luminescent dinuclear Copper(I) complexes bearing an imidazolylpyrimidine bridging ligand. Inorg. Chem. 2020, 59, 14772–14784. [Google Scholar] [CrossRef] [PubMed]
- Hobbollahi, E.; List, M.; Hupp, B.; Mohr, F.; Berger, R.J.F.; Steffen, A.; Monkowius, U. Highly efficient cold-white light emission in a Au2CuCl2(P∩N)2PF6 type salt. Dalton Trans. 2017, 46, 3438–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yersin, H.; Fischer, T.; Monkowius, U.; Hofbeck, T. Copper Complexes for Optoelectronic Applications. German Patent DE 102009030475 A1, 5 January 2011. [Google Scholar]
- Chen, K.; Shearer, J.; Catalano, V.J. Subtle modulation of Cu4X4L2 phosphine cluster cores leads to changes in luminescence. Inorg. Chem. 2015, 54, 6245–6256. [Google Scholar] [CrossRef]
- Artemev, A.V.; Shafikov, M.Z.; Schinabeck, A.; Antonova, O.V.; Berezin, A.S.; Bagryanskaya, I.Y.; Plusnin, P.E.; Yersin, H. Sky-blue thermally activated delayed fluorescence (TADF) based on Ag(I) complexes: Strong solvation-induced emission enhancement. Inorg. Chem. Front. 2019, 6, 3168–3176. [Google Scholar] [CrossRef]
- Artem’ev, A.V.; Davydova, M.P.; Berezin, A.S.; Ryzhikov, M.R.; Samsonenko, D.G. Dicopper(I) paddle-wheel complexes with thermally activated delayed fluorescence adjusted by ancillary ligands. Inorg. Chem. 2020, 59, 10699–10706. [Google Scholar] [CrossRef]
- Davydova, M.P.; Rakhmanova, M.I.; Bagryanskaya, I.Y.; Brylev, K.A.; Artemev, A.V. A 1D coordination polymer based on CuI and 2-(Diphenylphosphino)pyrimidine: Synthesis, structure and luminescent properties. J. Struct. Chem. 2020, 61, 894–898. [Google Scholar] [CrossRef]
- Stoïanov, A.; Gourlaouen, C.; Vela, S.; Daniel, C. Luminescent dinuclear Copper(I) complexes as potential thermally activated delayed fluorescence (TADF) emitters: A theoretical study. J. Phys. Chem. A 2018, 122, 1413–1421. [Google Scholar] [CrossRef]
- Busch, J.M.; Koshelev, D.S.; Vashchenko, A.A.; Fuhr, O.; Nieger, M.; Utochnikova, V.V.; Bräse, S. Various structural design modifications: Para-substituted diphenylphosphinopyridine bridged Cu(I) complexes in organic light-emitting diodes. Inorg. Chem. 2021, 60, 2315–2332. [Google Scholar] [CrossRef]
- Monkowius, U.; Zabel, M.; Fleck, M.; Yersin, H. Gold(I) complexes bearing P∩N-ligands: An unprecedented twelve-membered ring structure stabilized by aurophilic interactions. Z. Naturforsch. B 2009, 64, 1513–1524. [Google Scholar] [CrossRef]
- Bondi, A. van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T.; van Lenthe, E.; van Gisbergen, S.; Baerends, E.J. The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry. J. Chem. Phys. 2005, 122, 204103. [Google Scholar] [CrossRef]
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 26 May 2021).
- van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Chen, L.X.; Shaw, G.B.; Novozhilova, I.; Liu, T.; Jennings, G.; Attenkofer, K.; Meyer, G.J.; Coppens, P. MLCT state structure and dynamics of a Copper(I) diimine complex characterized by pump-probe X-ray and laser spectroscopies and DFT calculations. J. Am. Chem. Soc. 2003, 125, 7022–7034. [Google Scholar] [CrossRef]
- Siddique, Z.A.; Yamamoto, Y.; Ohno, T.; Nozaki, K. Structure-dependent photophysical properties of singlet and triplet metal-to-ligand charge transfer states in Copper(I) bis(diimine) compounds. Inorg. Chem. 2003, 42, 6366–6378. [Google Scholar] [CrossRef]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Real-time observation of the photoinduced structural change of bis(2,9-dimethyl-1,10-phenanthroline) Copper(I) by femtosecond fluorescence spectroscopy: A realistic potential curve of the Jahn-Teller distortion. J. Am. Chem. Soc. 2007, 129, 5248–5256. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999, 101, 396–408. [Google Scholar] [CrossRef]
- Mori, K.; Goumans, T.P.M.; van Lenthe, E.; Wang, F. Predicting phosphorescent lifetimes and zero-field splitting of organometallic complexes with time-dependent density functional theory including spin-orbit coupling. Phys. Chem. Chem. Phys. 2014, 16, 14523–14530. [Google Scholar] [CrossRef]
- Toptygin, D. Effects of the solvent refractive index and its dispersion on the radiative decay rate and extinction coefficient of a fluorescent solute. J. Fluoresc. 2003, 13, 201–219. [Google Scholar] [CrossRef]
- Niehaus, T.A.; Hofbeck, T.; Yersin, H. Charge-transfer excited states in phosphorescent organo-transition metal compounds: A difficult case for time dependent density functional theory? RSC Adv. 2015, 5, 63318–63329. [Google Scholar] [CrossRef] [Green Version]
- Rössler, U.; Yersin, H. Destabilization of a self-trapped exciton in a quasi-one-dimensional semiconductor: Mg[Pt(CN)4]7 H2O with hydrostatic pressure. Phys. Rev. B 1982, 26, 3187–3191. [Google Scholar] [CrossRef] [Green Version]
- Gliemann, G.; Yersin, H. Spectroscopic properties of the quasi one-dimensional tetracyanoplatinate(II) compounds. In Clusters; Springer: Berlin/Heidelberg, Germany, 1985; pp. 87–153. [Google Scholar] [CrossRef]
- Tsuboyama, A.; Kuge, K.; Furugori, M.; Okada, S.; Hoshino, M.; Ueno, K. Photophysical properties of highly luminescent Copper(I) halide complexes chelated with 1,2-bis(diphenylphosphino)benzene. Inorg. Chem. 2007, 46, 1992–2001. [Google Scholar] [CrossRef]
- Turro, N.J. Modern Molecular Photochemistry of Organic Molecules; Benjamin/Cummings: Melon Park, CA, USA, 1978. [Google Scholar]
- Leitl, M.J.; Krylova, V.A.; Djurovich, P.I.; Thompson, M.E.; Yersin, H. Phosphorescence versus thermally activated delayed fluorescence. Controlling singlet-triplet splitting in brightly emitting and sublimable Cu(I) compounds. J. Am. Chem. Soc. 2014, 136, 16032–16038. [Google Scholar] [CrossRef]
- Murov, S.L.; Carmichael, J.; Hug, G.L. Handbook of Photochemistry, 2nd ed.; Marcel Dekker: New York, NY, USA, 1993; p. 340. [Google Scholar]
- Shafikov, M.Z.; Zaytsev, A.V.; Kozhevnikov, V.N. Halide-enhanced spin-orbit coupling and the phosphorescence rate in Ir(III) complexes. Inorg. Chem. 2021, 60, 642–650. [Google Scholar] [CrossRef]
- Yersin, H.; Strasser, J. Triplets in metal–organic compounds. Chemical tunability of relaxation dynamics. Coord. Chem. Rev. 2000, 208, 331–364. [Google Scholar] [CrossRef]
- Bergmann, L.; Hedley, G.J.; Baumann, T.; Bräse, S.; Samuel, I.D.W. Direct observation of intersystem crossing in a thermally activated delayed fluorescence Copper complex in the solid state. Sci. Adv. 2016, 2, e1500889. [Google Scholar] [CrossRef] [Green Version]
- Tschierlei, S.; Karnahl, M.; Rockstroh, N.; Junge, H.; Beller, M.; Lochbrunner, S. Substitution-controlled excited state processes in heteroleptic Copper(I) photosensitizers used in hydrogen evolving systems. Chem. Phys. Chem. 2014, 15, 3709–3713. [Google Scholar] [CrossRef]
- Garakyaraghi, S.; Danilov, E.O.; McCusker, C.E.; Castellano, F.N. Transient absorption dynamics of sterically congested Cu(I) MLCT excited states. J. Phys. Chem. A 2015, 119, 3181–3193. [Google Scholar] [CrossRef]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Ultrafast excited-state dynamics of Copper(I) complexes. Acc. Chem. Res. 2015, 48, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Penfold, T.J. Understanding and designing thermally activated delayed fluorescence emitters: Beyond the energy gap approximation. Chem. Rec. 2020, 20, 831–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penfold, T.J.; Gindensperger, E.; Daniel, C.; Marian, C.M. Spin-vibronic mechanism for intersystem crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef] [Green Version]
- Striplin, D.R.; Crosby, G.A. Nature of the emitting 3MLCT manifold of rhenium(I)(diimine)(CO)3Cl complexes. Chem. Phys. Lett. 1994, 221, 426–430. [Google Scholar] [CrossRef]
- Finkenzeller, W.J.; Thompson, M.E.; Yersin, H. Phosphorescence dynamics and spin-lattice relaxation of the OLED emitter Ir(btp)2(acac). Chem. Phys. Lett. 2007, 444, 273–279. [Google Scholar] [CrossRef]
- Finkenzeller, W.J.; Hofbeck, T.; Thompson, M.E.; Yersin, H. Triplet state properties of the OLED emitter Ir(btp)2(acac): Characterization by site-selective spectroscopy and application of high magnetic fields. Inorg. Chem. 2007, 46, 5076–5083. [Google Scholar] [CrossRef]
- Tinti, D.S.; El-Sayed, M.A. New techniques in triplet state phosphorescence spectroscopy: Application to the emission of 2,3-dichloroquinoxaline. J. Chem. Phys. 1971, 54, 2529–2549. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97, Program for the Solution of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. 1990, A46, 467–473. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR 92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Turbomole V6.4 2012. A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 26 May 2021).
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post- d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1-Cl a | 1-Br | 1-I a | 2-I | 3-I·CH2Cl2 | 3-I a |
|---|---|---|---|---|---|---|
| Cu1–Cu2 | 2.878(1) | 2.883(1) | 2.7694(5) | 2.693(1) | 2.799(1) | 2.7204(6) |
| Cu1–P1 | 2.242(1) | 2.240(1) | 2.2514(6) | 2.263(1) | 2.292(1) | 2.2555(8) |
| Cu1–P2 | 2.248(1) | 2.258(1) | 2.2522(7) | 2.249(1) | 2.291(1) | 2.2404(9) |
| Cu2–P3 | 2.224(1) | 2.237(1) | 2.2507(7) | 2.244(1) | 2.285(1) | 2.2468(9) |
| Cu2–N | 2.106(3) | 2.098(4) | 2.104(1) | 2.101(3) | 2.140(3) | 2.076(2) |
| Cu1–X1 | 2.395(1) | 2.570(1) | 2.6733(7) | 2.684(1) | 2.699(1) | 2.6930(5) |
| Cu1–X2 | 2.426 (1) | 2.522(1) | 2.6803(5) | 2.718(1) | 2.641(1) | 2.6954(5) |
| Cu2–X1 | 2.436(1) | 2.509(1) | 2.7280(6) | 2.647(1) | 2.714(1) | 2.6277(5) |
| Cu2–X2 | 2.390(1) | 2.543(1) | 2.6446(5) | 2.702(1) | 2.687(1) | 2.6802(5) |
| X1–Cu1–X2 | 98.38(4) | 101.89(3) | 107.63(2) | 106.85(2) | 107.02(2) | 108.86(2) |
| X1–Cu2–X2 | 98.25(3) | 103.00(3) | 107.07(1) | 108.41(2) | 105.31(2) | 111.32(2) |
| N–Cu2–P3 | 123.23(8) | 123.35(9) | 117.47(2) | 115.9(1) | 111.8(1) | 120.35(2) |
| P1–Cu1–P2 | 123.80(4) | 123.90(5) | 119.87(2) | 118.40(4) | 118.8(1) | 118.16(3) |
| Cu1–X1–Cu2 | 73.42(3) | 69.17(2) | 61.68(1) | 60.69(1) | 62.27(2) | 61.48(1) |
| Cu1–X2–Cu2 | 73.13(3) | 69.40(2) | 62.67(1) | 59.59(1) | 63.38(2) | 60.80(1) |
| Exp a | Calculations Geometry S0 | T1 | |
|---|---|---|---|
| Cu1–Cu2 | 2.77 | 2.86 | 2.59 |
| Cu2–I1 | 2.73 | 2.71 | 2.67 |
| Cu1–I1 | 2.67 | 2.76 | 2.76 |
| Cu1–P2 | 2.25 | 2.36 | 2.36 |
| Cu1–P1 | 2.25 | 2.36 | 2.35 |
| Cu2–P3 | 2.25 | 2.33 | 2.37 |
| Cu2–N1 | 2.10 | 2.22 | 2.03 |
| I1–Cu1–I2 | 107.6 | 106.8 | 102.5 |
| I1–Cu2–I2 | 107.1 | 108.9 | 103.4 |
| Cu1–I1–Cu2 | 61.7 | 63.2 | 56.9 |
| Cu1–I2–Cu2 | 62.7 | 62.3 | 55.9 |
| P1–Cu1–Cu2 | 87.1 | 85.5 | 85.0 |
| P2–Cu1–P1 | 119.9 | 119.7 | 117.3 |
| Geometry | I(T1) | II(T1) | III(T1) | S1 | Δ(S1−T1) | ZFS |
|---|---|---|---|---|---|---|
| ∆E(III−I) | ||||||
| S0 | 2.418 (752.4) | 2.420 (95.1) | 2.424 (6.5) | 2.437 (1.6) | 129 | 52 |
| S0(solvent) | 2.796 (92.5) | 2.797 (8.1) | 2.801 (1.3) | 2.816 (0.2) | 144 | 39 |
| T1 | 1.452 (>103) | 1.453 (>103) | 1.456 (54.4) | 1.507 (2.8) | 411 | 32 |
| Exp a | 2.54 (≈210) | 2.54 (≈210) | 2.54 (12) | ≈2.588 b (0.445) | 380 | 3 |
| Compound | λmax(300 K) a [nm] | ΦPL(300 K) b [%] | τ(300 K) a [µs] | kr(300 K) c [s−1] | knr(300K) d [s−1] | λmax(77 K) a [nm] | ΦPL(77 K) b [%] | τ(77 K) a [µs] | kr(77 K) c [s−1] | knr(77 K) d [s−1] | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cu2Cl2(Ph2Ppy)3 | 1-Cl | 577 | 37 | 7.9 | 4.7 × 104 | 8.0 × 104 | 592 | 71 | 65 | 1.1 × 104 | 4.5 × 103 |
| Cu2Br2(Ph2Ppy)3 | 1-Br | 545 | 53 | 8.8 | 6.0 × 104 | 5.3 × 104 | 567 | 89 | 110 | 8.1 × 103 | 1.0 × 103 |
| Cu2I2(Ph2Ppy)3 | 1-I | 539 | 81 | 6.5 | 1.25 × 105 | 2.92 × 104 | 552 | 92 | 32 | 2.88 × 104 | 2.5 × 103 |
| Cu2Cl2(Ph2Ppym)3 | 2-Cl | 616 | 9 | 1.2 | 7.5 × 104 | 7.6 × 105 | 626 | 14 | 30 | 4.7 × 103 | 2.9 × 104 |
| Cu2Br2(Ph2Ppym)3 | 2-Br | 583 | 33 | 2.5 | 1.3 × 105 | 2.7 × 105 | 584 | 56 | 29 | 1.9 × 104 | 1.5 × 104 |
| Cu2I2(Ph2Ppym)3 | 2-I | 565 | 13 | 1.7 e (2.7 f) | 7.6 × 104 | 5.1 × 105 | 575 | 67 | 17.4 e | 3.9 × 104 | 1.9 × 104 |
| Cu2Br2(Ph2Piqn)3 | 3-Br | 660 | 11 | 2.0 e | 5.5 × 104 | 4.5 × 105 | 668 | 24 | 42 e | 5.7 × 103 | 1.8 × 104 |
| Cu2I2(Ph2Piqn)3 | 3-I | 636 | 38 | 3.3 e | 1.2 × 105 | 1.9 × 105 | 645 | 59 | 22 | 2.7 × 104 | 1.9 × 104 |
| Property | Value |
|---|---|
| E0-0(T1–S0) a | 20,500 cm−1 |
| 2.541 eV | |
| E0-0(S1–S0) b | 20,880 cm−1 |
| 2.588 eV | |
| ΦPL(300 K) | 81% |
| ΦPL(77 K) | 92% |
| k(S1–S0) | 2.25 × 106 s−1 |
| (445 ns) | |
| k(T1–S0), plateau | 3.1 × 104 s−1 |
| (32 µs) | |
| kr(T1–S0) | 2.88 × 104 s−1 |
| (35 µs) | |
| k(TADF + phos), 300 K observed | 15.4 × 104 s−1 |
| 6.5 µs | |
| k(TADF-only), 300 K | 11.9 × 104 s−1 |
| (8.4 µs) | |
| ∆E(S1–T1) | 380 cm−1 |
| (47 meV) | |
| ∆E(II–I) | <1 cm−1 |
| ∆E(III–I,II) | 3 cm−1 |
| (0.37 meV) | |
| k(III–S0) | 8.3 × 104 s−1 |
| (12 µs) | |
| k(I–S0) ≈ k(II–S0) | ≈4.76 × 103 s−1 |
| (≈210 µs) | |
| k(SLR) c at 1.2 K | 1.7 × 104 s−1 |
| (59 μs) |
| 1-Br | 2-I | 3-I·CH2Cl2 | |
|---|---|---|---|
| Empirical formula | C51H42Br2Cu2N3P3 | C48H39Cu2I2N6P3·CH2Cl2 | C63H48Cu2I2N3P3·CH2Cl2 |
| Mr, g mol−1 | 1076.69 | 1258.57 | 1405.78 |
| Size, mm3 | 0.40 × 0.25 × 0.11 | 0.28 × 0.18 × 0.08 | 0.20 × 0.18 × 0.15 |
| Crystal system | monoclinic | monoclinic | triclinic |
| Space group | P21/n | C2/c | |
| a, Å | 14.202(2) | 42.494(5) | 12.4000(13) |
| b, Å | 18.139(3) | 11.0172(14) | 15.5224(17) |
| c, Å | 17.628(3) | 22.311(3) | 16.3296(17) |
| α, deg | 90 | 90 | 97.462(12) |
| β, deg | 98.223(5) | 103.076(3) | 106.923(12) |
| γ, deg | 90 | 90 | 92.752(13) |
| V, Å3 | 4494.5(11) | 10174(2) | 2969.3(6) |
| ρcalcd., g cm−1 | 1.591 | 1.643 | 1.572 |
| Z | 4 | 8 | 2 |
| μ(Mo-Kα), mm−1 | 2.87 | 2.29 | 1.97 |
| T, K | 300 | 293 | 123 |
| Θ range, deg | 1.6–25.1 | 3.0–28.5 | 2.2–27.0 |
| Measured reflections | 27,865 | 14,027 | 42,450 |
| Independent reflections | 7896 | 9516 | 11,932 |
| Reflections with I > 2σ(I)) | 5151 | 7611 | 7253 |
| Absorption correction | multi-scan | multi-scan | analytical |
| Tmin/Tmax | 0.39, 0.74 | 0.894, 0.914 | 0.725, 0.769 |
| Restraints/refined param. | 0/550 | 0/617 | 0/685 |
| R1 (I ≥ 2σ(I)) | 0.047 | 0.031 | 0.034 |
| wR2 | 0.092 | 0.083 | 0.074 |
| ρfin (max/min), e Å−3 | 1.15/−0.65 | 0.60/−0.54 | 1.81/−1.45 |
| CCDC no. | 2,034,780 | 2,034,779 | 2,034,781 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofbeck, T.; Niehaus, T.A.; Fleck, M.; Monkowius, U.; Yersin, H. P∩N Bridged Cu(I) Dimers Featuring Both TADF and Phosphorescence. From Overview towards Detailed Case Study of the Excited Singlet and Triplet States. Molecules 2021, 26, 3415. https://doi.org/10.3390/molecules26113415
Hofbeck T, Niehaus TA, Fleck M, Monkowius U, Yersin H. P∩N Bridged Cu(I) Dimers Featuring Both TADF and Phosphorescence. From Overview towards Detailed Case Study of the Excited Singlet and Triplet States. Molecules. 2021; 26(11):3415. https://doi.org/10.3390/molecules26113415
Chicago/Turabian StyleHofbeck, Thomas, Thomas A. Niehaus, Michel Fleck, Uwe Monkowius, and Hartmut Yersin. 2021. "P∩N Bridged Cu(I) Dimers Featuring Both TADF and Phosphorescence. From Overview towards Detailed Case Study of the Excited Singlet and Triplet States" Molecules 26, no. 11: 3415. https://doi.org/10.3390/molecules26113415