Unless otherwise specified, all compounds were purchased from commercial sources (Bide Chemical Co., Ltd., Xiangtan, Hunan, China; Sigma-Aldrich, St. Louis, MO, USA; and J&K Chemical Ltd., Beijing, China), and no further purification was required. 1H-NMR and 13C-NMR spectra were recorded at room temperature on a Bruker AVANCE 400 or 500 spectrometer (Karlsruhe, Germany) with TMS as an internal reference. Peaks were labeled as single (s), doublet (d), two doublets (dd), doublet of triplets (dt), triplet (t), or multiplet (m). A Shimadzu LCMS-IT-TOF high-resolution mass spectrometer (Kyoto, Japan) was used for molecular weight confirmation. The purity of compounds was confirmed to be over 95% and was determined by reversed-phase high performance liquid chromatography (HPLC) with a TC-C18 column (250 mm × 4.6 mm, 5 µm) using acetonitrile/water (70%, v/v) as the mobile phase (1.0 mL/min) at 25 °C.
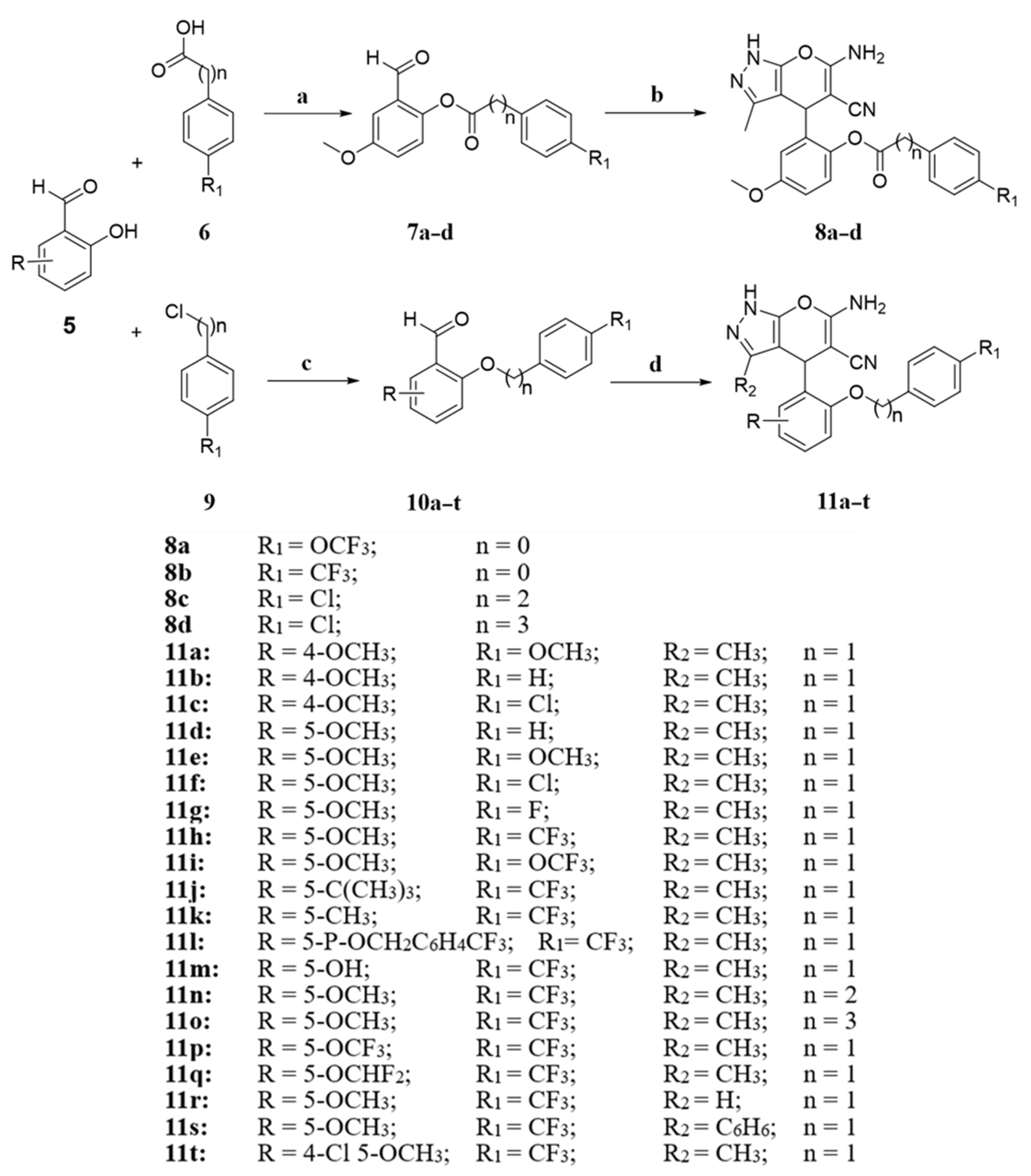
3.1.5. General Procedure for Synthesis of Intermediate Compounds 10a–q and 10t
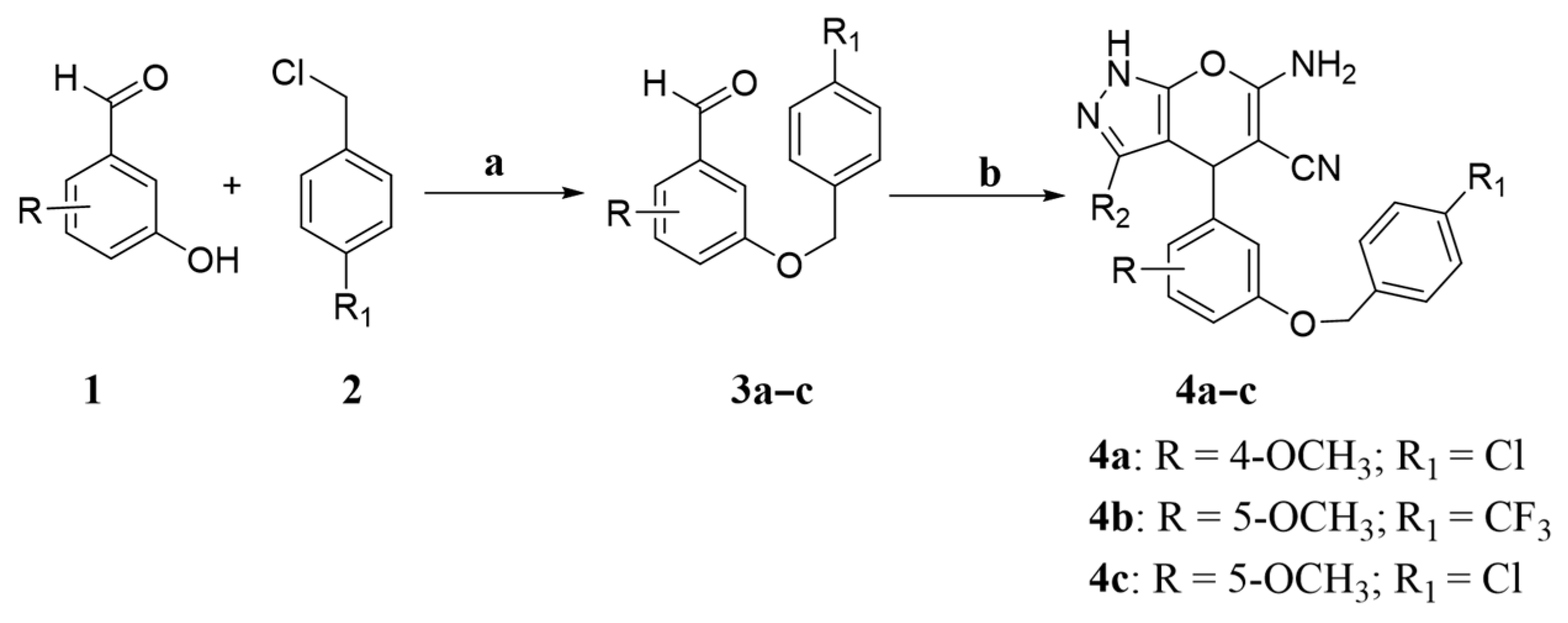
The synthesis method was the same as that used for 3a–c.
4-Methoxy-2-((4-methoxybenzyl)oxy)benzaldehyde (10a). White solid. Yield, 82%. 1H-NMR (400 MHz, DMSO) δ 10.20 (s, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 6.99–6.95 (m, 2H), 6.81 (d, J = 2.2 Hz, 1H), 6.65 (dd, J = 8.7, 1.8 Hz, 1H), 5.20 (s, 2H), 3.86 (s, 3H), 3.76 (s, 3H).
2-(Benzyloxy)-4-methoxybenzaldehyde (10b). White solid. Yield, 80%. 1H-NMR (400 MHz, CDCl3) δ 10.41 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.48–7.37 (m, 5H), 6.59 (dd, J = 8.7, 1.8 Hz, 1H), 6.54 (d, J = 2.2 Hz, 1H), 5.19 (s, 2H), 3.88 (s, 3H).
2-((4-Chlorobenzyl)oxy)-4-methoxybenzaldehyde (10c). White solid. Yield, 83%. 1H-NMR (400 MHz, CDCl3) δ 10.38 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.40 (s, 4H), 6.60 (dd, J = 8.7, 1.7 Hz, 1H), 6.50 (d, J = 2.2 Hz, 1H), 5.15 (s, 2H), 3.88 (s, 3H).
2-(Benzyloxy)-5-methoxybenzaldehyde (10d). White solid. Yield, 86%. 1H-NMR (400 MHz, CDCl3) δ 10.53 (s, 1H), 7.47–7.34 (m, 6H), 7.14 (dd, J = 9.0, 3.3 Hz, 1H), 7.03 (d, J = 9.1 Hz, 1H), 5.18 (s, 2H), 3.83 (s, 3H).
5-Methoxy-2-((4-methoxybenzyl)oxy)benzaldehyde (10e). White solid. Yield, 79%.1H-NMR (400 MHz, DMSO) δ 10.33 (s, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 9.1 Hz, 1H), 7.25 (dd, J = 9.1, 3.2 Hz, 1H), 7.17 (d, J = 3.1 Hz, 1H), 6.97–6.93 (m, 2H), 5.16 (s, 2H), 3.76 (s, 3H), 3.75 (s, 3H).
2-((4-Chlorobenzyl)oxy)-5-methoxybenzaldehyde (10f). White solid. Yield, 81%. 1H-NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 7.42–7.38 (m, 4H), 7.37 (d, J = 3.2 Hz, 1H), 7.14 (dd, J = 9.0, 3.2 Hz, 1H), 6.99 (d, J = 9.1 Hz, 1H), 5.14 (s, 2H), 3.83 (s, 3H).
2-((4-Fluorobenzyl)oxy)-5-methoxybenzaldehyde (10g). White solid. Yield, 83%. 1H-NMR (400 MHz, CDCl3) δ 10.50 (s, 1H), 7.45–7.39 (m, 2H), 7.37 (d, J = 3.2 Hz, 1H), 7.14 (dd, J = 8.3, 2.5 Hz, 1H), 7.12–7.08 (m, 2H), 7.01 (d, J = 9.1 Hz, 1H), 5.13 (s, 2H), 3.83 (s, 3H).
5-Methoxy-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10h). White solid. Yield, 82%. 1H-NMR (400 MHz, CDCl3) δ 10.55 (s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 3.2 Hz, 1H), 7.14 (dd, J = 9.0, 3.2 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 5.24 (s, 2H), 3.84 (s, 3H).
5-Methoxy-2-((4-(trifluoromethoxy)benzyl)oxy)benzaldehyde (10i). White solid. Yield, 76%. 1H-NMR (400 MHz, CDCl3) δ 10.50 (s, 1H), 7.46 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 3.2 Hz, 1H), 7.25 (d, J = 7.4 Hz, 2H), 7.12 (dd, J = 9.0, 3.3 Hz, 1H), 6.98 (d, J = 9.1 Hz, 1H), 5.15 (s, 2H), 3.81 (s, 3H).
5-(Tert-butyl)-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10j). White solid. Yield, 87%. 1H-NMR (400 MHz, CDCl3) δ 10.59 (s, 1H), 7.92 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.62–7.56 (m, 3H), 6.97 (d, J = 8.7 Hz, 1H), 5.27 (s, 2H), 1.34 (s, 9H).
5-Methyl-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10k). White solid. Yield, 89%. 1H-NMR (400 MHz, CDCl3) δ 10.56 (s, 1H), 7.69 (d, J = 7.8 Hz, 3H), 7.58 (d, J = 8.1 Hz, 2H), 7.36 (dd, J = 8.5, 2.1 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 5.25 (s, 2H), 2.35 (s, 3H).
2,5-Bis((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10l). White solid. Yield, 79%. 1H-NMR (400 MHz, CDCl3) δ 10.54 (s, 1H), 7.72–7.64 (m, 4H), 7.60–7.55 (m, 4H), 7.46 (d, J = 3.2 Hz, 1H), 7.21 (dd, J = 9.0, 3.3 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 5.24 (s, 2H), 5.14 (s, 2H).
5-Hydroxy-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10m). White solid. Yield, 56%. 1H-NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 3.2 Hz, 1H), 7.11 (dd, J = 8.9, 3.2 Hz, 1H), 6.96 (d, J = 8.9 Hz, 1H), 5.23 (s, 2H).
5-Methoxy-2-(4-(trifluoromethyl)phenethoxy)benzaldehyde (10n). White solid. Yield, 69%. 1H-NMR (400 MHz, CDCl3) δ 10.40 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 3.3 Hz, 1H), 7.13 (dd, J = 9.0, 3.3 Hz, 1H), 6.93 (d, J = 9.1 Hz, 1H), 4.30 (t, J = 6.5 Hz, 2H), 3.82 (s, 3H), 3.22 (t, J = 6.5 Hz, 2H).
5-Methoxy-2-(3-(4-(trifluoromethyl)phenyl)propoxy)benzaldehyde (10o). White solid. Yield, 78%. 1H-NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 7.58 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 3.3 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.13 (dd, J = 9.0, 3.3 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 4.07 (t, J = 6.1 Hz, 2H), 3.83 (s, 3H), 2.92 (t, J = 7.6 Hz, 2H), 2.25–2.14 (m, 2H).
5-(Trifluoromethoxy)-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10p). White solid. Yield, 87%. 1H-NMR (400 MHz, CDCl3) δ 10.54 (s, 1H), 7.76 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 7.42 (dd, J = 9.1, 2.6 Hz, 1H), 7.06 (d, J = 9.1 Hz, 1H), 5.29 (s, 2H).
5-(Difluoromethoxy)-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10q). Diethyl bromo difluoromethylphosphonate (2 mmol) was added in one portion to a solution of 14 m (1 mmol) and KOH (20 mmol) in CH3CN–H2O (10 mL, 1:1) at 0 °C, and the reaction mixture was allowed to warm to room tempeature. After 20 min, the reaction mixture was diluted with ethyl acetate (10 mL), and the organic phase was separated. Then, the water phase was washed with another 10 mL of ethyl acetate, and the combined organic fractions were dried over anhydrous Na2SO4. Evaporation of the solvent yielded a crude product that was purified on a silica gel column to obtain compound 10q. White solid. Yield, 82%. 1H-NMR (400 MHz, CDCl3) δ 10.36 (s, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 7.49 (d, J = 1.3 Hz, 1H), 7.27–7.23 (m, 2H), 6.62 (t, J = 72.9 Hz, 1H), 5.18 (s, 2H). 19F-NMR (376 MHz, CDCl3) δ −62.65 (s), −81.41 (d, J = 72.9 Hz).
4-Chloro-5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10t). The synthesis method was the same as that used for 3a–c. White solid. Yield, 67%. 1H-NMR (400 MHz, CDCl3) δ 10.48 (s, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.44 (s, 1H), 7.14 (s, 1H), 5.22 (s, 2H), 3.94 (s, 3H).
3.1.6. General Procedure for Synthesis of Intermediate Compounds 11a–t
The synthesis method was the same as that used for 4a–c.
6-Amino-4-(4-methoxy-2-((4-methoxybenzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11a). White solid. Yield, 41%. 1H-NMR (500 MHz, DMSO-d6) δ 11.98 (s, 1H), 7.33 (d, J = 7.3 Hz, 2H), 6.93 (d, J = 7.6 Hz, 2H), 6.89 (d, J = 8.0 Hz, 1H), 6.72 (s, 2H), 6.63 (s, 1H), 6.48 (d, J = 8.3 Hz, 1H), 5.03 (q, J = 11.8 Hz, 2H), 4.89 (s, 1H), 3.75 (s, 3H), 3.72 (s, 3H), 1.74 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.59, 159.45, 159.33, 156.75, 155.56, 135.49, 130.07, 129.66, 129.41, 124.97, 121.47, 114.26, 105.98, 100.07, 98.41, 69.75, 57.35, 55.55, 55.51 and 10.05. HRMS (ESI) calculated for C23H22N4O4 [M + H]+: 419.1714, found: 419.1708. Purity: 99.77% (by HPLC).
6-Amino-4-(2-(benzyloxy)-4-methoxyphenyl)-3-methyl-1,4-dihydropyrano [2,3-c] pyrazole-5-carbonitrile (11b). White solid. Yield, 36%. 1H-NMR (500 MHz, DMSO-d6) δ 11.99 (s, 1H), 7.44–7.35 (m, 4H), 7.31 (t, J = 6.8 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.74 (s, 2H), 6.62 (s, 1H), 6.49 (d, J = 8.4 Hz, 1H), 5.12 (q, J = 12.6 Hz, 2H), 4.94 (s, 1H), 3.72 (s, 3H), 1.75 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.59, 159.48, 156.68, 155.57, 137.60, 135.51, 130.15, 128.89, 128.15, 127.83, 124.97, 121.46, 106.07, 100.04, 98.40, 69.93, 57.36, 55.56 and 10.05. HRMS (ESI) calculated for C22H20N4O3 [M + H]+: 389.1608, found: 389.1598. Purity: 99.01% (by HPLC).
6-Amino-4-(2-((4-chlorobenzyl)oxy)-4-methoxyphenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11c). white solid. Yield, 52%. 1H-NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 7.44 (m, J = 8.2 Hz, 4H), 6.93 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.62 (s, 1H), 6.50 (d, J = 8.1 Hz, 1H), 5.11 (q, J = 12.6 Hz, 2H), 4.90 (s, 1H), 3.73 (s, 3H), 1.75 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.54, 159.52, 156.53, 155.57, 136.60, 135.49, 132.68, 130.27, 129.68, 128.84, 124.83, 121.47, 106.04, 100.12, 98.29, 69.02, 57.28, 55.57 and 10.05. HRMS (ESI) calculated for C22H19ClN4O3 [M + H]+: 423.1218, found: 423.1219. Purity: 99.91% (by HPLC).
6-Amino-4-(2-(benzyloxy)-5-methoxyphenyl)-3-methyl-1,4-dihydropyrano [2,3-c] pyrazole-5-carbonitrile (11d). White solid. Yield, 44%. 1H-NMR (500 MHz, DMSO-d6) δ 12.03 (s, 1H), 7.43–7.34 (m, 4H), 7.31 (t, J = 6.9 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.81 (s, 2H), 6.76 (d, J = 8.9 Hz, 1H), 6.53 (s, 1H), 5.05 (q, J = 12.3 Hz, 2H), 4.99 (s, 1H), 3.64 (s, 3H), 1.77 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.82, 155.52, 153.96, 150.05, 137.86, 135.62, 134.07, 128.85, 128.09, 127.84, 121.37, 115.61, 114.39, 112.34, 98.00, 70.69, 56.81, 55.69 and 10.04. HRMS (ESI) calculated for C22H20N4O3 [M + H]+: 389.1608, found: 389.1601. Purity: 99.93% (by HPLC).
6-Amino-4-(5-methoxy-2-((4-methoxybenzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11e). White solid. Yield, 48%. 1H-NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.33 (d, J = 8.5 Hz, 2H), 7.01 (d, J = 9.0 Hz, 1H), 6.93 (d, J = 8.7 Hz, 2H), 6.79 (s, 2H), 6.76 (dd, J = 8.9, 3.1 Hz, 1H), 6.52 (d, J = 2.9 Hz, 1H), 4.97 (m, 3H), 3.76 (s, 3H), 3.65 (s, 3H), 1.76 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.81, 159.30, 155.51, 153.89, 150.12, 135.61, 134.09, 129.69, 126.66, 121.37, 115.55, 114.53, 114.23, 112.32, 98.01, 70.54, 56.82, 55.68, 55.52 and 10.04. HRMS (ESI) calculated for C23H22N4O4 [M + H]+: 419.1714, found: 419.1723. Purity: 99.35% (by HPLC).
6-Amino-4-(2-((4-chlorobenzyl)oxy)-5-methoxyphenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11f). White solid. Yield, 48%. 1H-NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.47–7.37 (m, 4H), 7.00 (d, J = 9.0 Hz, 1H), 6.80 (s, 2H), 6.77 (dd, J = 9.0, 3.1 Hz, 1H), 6.56 (d, J = 2.6 Hz, 1H), 5.04 (q, J = 12.4 Hz, 2H), 4.96 (s, 1H), 3.65 (s, 3H), 1.77 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.78, 155.53, 153.97, 149.89, 136.86, 135.63, 133.95, 132.63, 129.69, 128.82, 121.39, 115.76, 114.42, 112.36, 97.91, 69.75, 56.76, 55.70 and 10.04. HRMS (ESI) calculated for C22H19ClN4O3 [M + H]+: 423.1218, found: 423.1229. Purity: 99.91% (by HPLC).
6-Amino-4-(2-((4-fluorobenzyl)oxy)-5-methoxyphenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11g). White solid. Yield, 54%. 1H-NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.47–7.40 (m, 2H), 7.19 (t, J = 8.8 Hz, 2H), 7.02 (d, J = 8.9 Hz, 1H), 6.80–6.75 (m, 3H), 6.55 (d, J = 2.8 Hz, 1H), 5.02 (q, J = 12.0 Hz, 2H), 4.96 (s, 1H), 3.65 (s, 3H), 1.76 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 162.10 (d, J = 244.4 Hz), 161.78, 155.53, 153.96, 149.97, 135.62, 134.02, 134.00, 130.13, 130.05, 121.40, 115.70, 115.63 (d, J = 21.4 Hz), 114.46, 112.34, 97.93, 69.94, 56.78, 55.68 and 10.04. HRMS (ESI) calculated for C22H19FN4O3 [M + H]+: 407.1514, found: 407.1521. Purity: 99.85% (by HPLC).
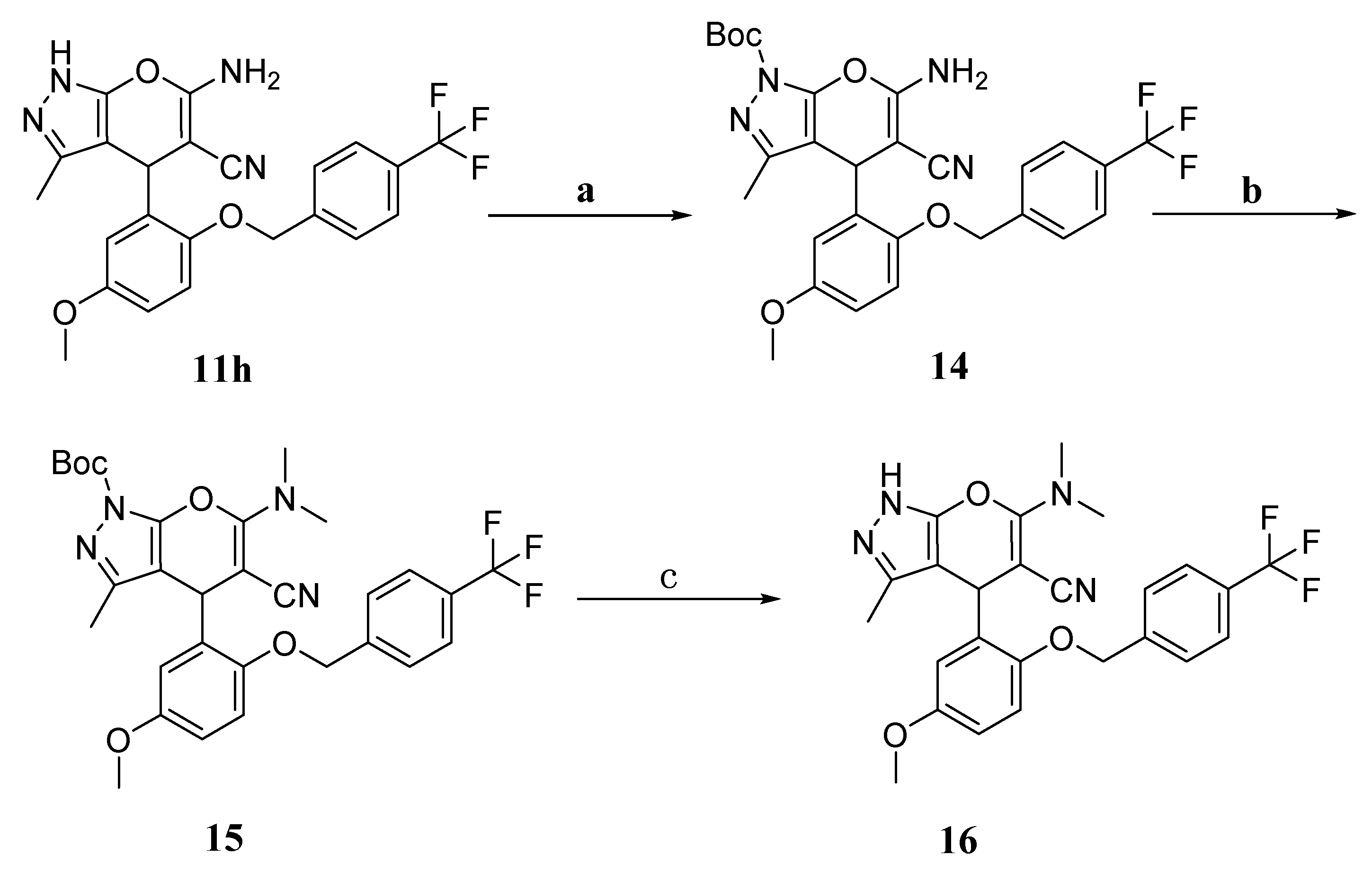
6-Amino-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11h). White solid. Yield, 50%. 1H-NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.01 (d, J = 9.0 Hz, 1H), 6.81 (s, 2H), 6.78 (dd, J = 8.9, 3.1 Hz, 1H), 6.59 (s, 1H), 5.16 (q, J = 13.2 Hz, 2H), 4.99 (s, 1H), 3.66 (s, 3H), 1.77 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.78, 155.55, 154.03, 149.78, 142.74, 135.65, 133.91, 128.57 (q, J = 39.9 Hz), 128.18, 125.70 (q, J = 3.8 Hz), 124.75 (q, J = 253.7 Hz), 121.39, 115.87, 114.34, 112.39, 97.88, 69.62, 56.74, 55.70 and 10.04. HRMS (ESI) calculated for C23H19F3N4O3 [M + H]+: 457.1482, found: 457.1496. Purity: 99.96% (by HPLC).
6-Amino-4-(5-methoxy-2-((4-(trifluoromethoxy)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11i). White solid. Yield, 47%. 1H-NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.02 (d, J = 8.9 Hz, 1H), 6.80 (s, 2H), 6.78 (dd, J = 9.0, 3.2 Hz, 1H), 6.56 (d, J = 2.2 Hz, 1H), 5.09 (q, J = 12.6 Hz, 2H), 4.98 (s, 1H), 3.65 (s, 3H), 1.76 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.77, 155.55, 154.01, 149.85, 148.17, 137.35, 135.64, 133.98, 129.66, 121.42, 121.39, 120.56 (q, J = 256.6 Hz), 115.75, 114.39, 112.37, 97.90, 69.63, 56.78, 55.69 and 10.02. HRMS (ESI) calculated for C23H19F3N4O4 [M + H]+: 473.1431, found: 473.1444. Purity: 99.82% (by HPLC).
6-Amino-4-(5-(tert-butyl)-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11j). White solid. Yield, 36%. 1H-NMR (400 MHz, DMSO-d6) δ 11.99 (s, 1H), 7.73 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 7.9 Hz, 2H), 7.19 (dd, J = 8.5, 2.4 Hz, 1H), 7.11 (s, 1H), 6.95 (d, J = 8.6 Hz, 1H), 6.77 (s, 2H), 5.18 (q, J = 13.4 Hz, 2H), 4.97 (s, 1H), 1.74 (s, 3H), 1.21 (s, 9H). 13C-NMR (126 MHz, DMSO-d6) δ 161.78, 155.69, 153.66, 143.39, 142.73, 135.55, 131.42, 128.54 (q, J = 31.5 Hz), 128.07, 126.54, 125.69 (q, J = 3.8 Hz), 124.92, 124.35 (q, J = 252.4 Hz), 121.51, 112.54, 98.01, 68.87, 56.90, 34.18, 31.71 and 10.02. HRMS (ESI) calculated for C26H25F3N4O2 [M + H]+: 483.2002, found: 483.1984. Purity: 99.84% (by HPLC).
6-Amino-3-methyl-4-(5-methyl-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11k). Yellow solid. Yield, 40%. 1H-NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.61 (d, J = 7.8 Hz, 2H), 7.02–6.93 (m, 2H), 6.84 (s, 1H), 6.79 (s, 2H), 5.20 (q, J = 13.3 Hz, 2H), 5.01 (s, 1H), 2.19 (s, 3H), 1.76 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 161.72, 155.58, 153.50, 142.67, 135.61, 132.36, 130.29, 130.05, 128.81, 128.58 (q, J = 31.6 Hz), 128.16, 125.72 (q, J = 3.7 Hz), 124.24 (q, J = 249.1 Hz), 121.47, 113.05, 98.12, 69.08, 56.99, 20.67 and 10.04. HRMS (ESI) calculated for C23H19F3N4O2 [M + H]+: 441.1533, found: 441.1538. Purity: 99.80% (by HPLC).
6-Amino-4-(2,5-bis((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11l). White solid. Yield, 31%. 1H-NMR (400 MHz, DMSO-d6) δ 12.03 (s, 1H), 7.76–7.71 (m, 4H), 7.63 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.02 (d, J = 8.9 Hz, 1H), 6.87 (dd, J = 9.0, 3.0 Hz, 1H), 6.82 (s, 2H), 6.70 (s, 1H), 5.23–5.13 (m, 2H), 5.11 (s, 2H), 4.99 (s, 1H), 1.71 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.78, 155.57, 152.74, 150.10, 142.67, 142.51, 135.64, 128.70 (q, J = 32.0 Hz), 128.59 (q, J = 30.7 Hz), 128.55, 128.20, 125.70 (q, J = 3.8 Hz), 124.74 (q, J = 272.2 Hz), 124.71 (q, J = 272.6 Hz), 121.40, 116.82, 114.76, 114.33, 113.66, 97.78, 69.58, 69.05, 56.70 and 9.97. HRMS (ESI) calculated for C30H22F6N4O3 [M + H]+: 601.1669, found: 601.1690. Purity: 99.77% (by HPLC).
6-Amino-4-(5-hydroxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11m). Yellow solid. Yield, 43%. 1H-NMR (400 MHz, DMSO-d6) δ 12.03 (s, 1H), 8.92 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.64 (d, J = 7.7 Hz, 2H), 6.90 (d, J = 8.8 Hz, 1H), 6.81 (s, 2H), 6.56 (dd, J = 8.8, 2.9 Hz, 1H), 6.45 (d, J = 2.7 Hz, 1H), 5.18–5.08 (m, 2H), 4.99 (s, 1H), 1.78 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.70, 155.50, 152.15, 148.48, 142.94, 135.66, 133.94, 128.56 (q, J = 38.2 Hz), 128.20, 125.73 (q, J = 3.4 Hz), 124.76 (q, J = 248.2 Hz), 121.43, 116.07, 114.73, 114.66, 98.21, 69.95, 57.06 and 10.02. HRMS (ESI) calculated for C22H17F3N4O3 [M + H]+: 443.1326, found: 443.1319. Purity: 99.40% (by HPLC).
6-Amino-4-(5-methoxy-2-(4-(trifluoromethyl)phenethoxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11n). White solid. Yield, 37%. 1H-NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.1 Hz, 2H), 6.95 (d, J = 8.9 Hz, 1H), 6.81 (s, 2H), 6.75 (dd, J = 8.9, 3.1 Hz, 1H), 6.54 (s, 1H), 4.80 (s, 1H), 4.18–4.06 (m, 2H), 3.64 (s, 3H), 3.09 (t, J = 6.4 Hz, 2H), 1.63 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.86, 155.41, 153.91, 150.28, 144.23, 135.55, 134.09, 130.31, 127.50 (q, J = 31.9 Hz), 125.54 (q, J = 3.8 Hz), 124.90 (q, J = 272.2 Hz), 121.35, 115.58, 114.40, 112.36, 98.06, 69.65, 56.76, 55.68, 35.41 and 9.69. HRMS (ESI) calculated for C24H21F3N4O3 [M + H]+: 471.1639, found: 471.1631. Purity: 98.11% (by HPLC).
6-Amino-4-(5-methoxy-2-(3-(4-(trifluoromethyl)phenyl)propoxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11o). White solid. Yield, 48%. 1H-NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 6.87 (d, J = 8.9 Hz, 1H), 6.77 (s, 2H), 6.76–6.73 (m, 1H), 6.63 (s, 1H), 4.81 (s, 1H), 3.95–3.73 (m, 2H), 3.66 (s, 3H), 2.77 (t, J = 7.5 Hz, 2H), 2.02–1.92 (m, 2H), 1.80 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.82, 155.58, 153.51, 150.74, 147.17, 135.50, 133.40, 129.73, 127.06 (q, J = 34.4 Hz), 125.55 (q, J = 3.8 Hz), 123.86 (q, J = 260.4 Hz), 121.48, 115.95, 113.67, 112.30, 98.01, 67.93, 56.50, 55.69, 31.97, 30.71, 19.01 and 10.01. HRMS (ESI) calculated for C25H23F3N4O3 [M + H]+: 485.1495, found: 485.1788. Purity: 98.23% (by HPLC).
6-Amino-3-methyl-4-(5-(trifluoromethoxy)-2-((4-(trifluoromethyl) benzyl) oxy) phenyl)-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11p). White solid. Yield, 55%. 1H-NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.58 (d, J = 7.7 Hz, 2H), 7.23 (dd, J = 8.9, 2.2 Hz, 1H), 7.17 (d, J = 9.0 Hz, 1H), 7.05 (s, 1H), 6.89 (s, 2H), 5.26 (q, J = 13.2 Hz, 2H), 5.04 (s, 1H), 1.76 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.91, 155.60, 154.64, 142.41, 142.00, 135.71, 134.25, 128.74 (q, J = 35.7 Hz), 128.24, 125.78 (q, J = 3.4 Hz), 123.08 (q, J = 235.6 Hz), 121.60 (q, J = 265.0 Hz), 121.24, 121.18, 114.36, 97.15, 69.39, 55.95 and 9.96. HRMS (ESI) calculated for C23H16F6N4O3 [M + H]+: 511.1199, found: 511.1196. Purity: 99.87% (by HPLC).
6-Amino-4-(5-(difluoromethoxy)-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11q). Yellow solid. Yield, 47%. 1H-NMR (400 MHz, DMSO-d6) δ 12.09 (s, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.64 (d, J = 8.2 Hz, 2H), 7.14 (d, J = 8.8 Hz, 1H), 6.98 (dd, J = 9.0, 3.1 Hz, 1H), 6.88 (s, 2H), 6.75 (s, 1H), 5.16 (s, 2H), 4.87 (s, 1H), 1.73 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.80, 155.84, 155.44, 142.88, 142.09, 137.02, 135.76, 128.80 (q, J = 31.9 Hz), 128.63, 125.75 (q, J = 3.78 Hz), 124.70 (q, J = 273.0 Hz), 120.99, 120.66, 117.30 (t, J = 25.8 Hz), 116.60, 114.37, 97.15, 69.05, 56.21 and 9.87. HRMS (ESI) calculated for C23H17F5N4O3 [M + H]+: 493.1294, found: 493.1279. Purity: 95.59% (by HPLC).
6-Amino-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11r). White solid. Yield, 57%. 1H-NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 8.1 Hz, 2H), 7.29 (s, 1H), 7.02 (d, J = 8.9 Hz, 1H), 6.94 (s, 2H), 6.78 (dd, J = 8.9, 3.1 Hz, 1H), 6.64 (d, J = 3.1 Hz, 1H), 5.27–5.17 (m, 2H), 5.07 (s, 1H), 3.67 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 162.39, 155.21, 153.95, 149.70, 142.76, 134.77, 128.62 (q, J = 32.8 Hz), 128.21, 126.58, 125.76 (q, J = 3.8 Hz), 124.75 (q, J = 272.6 Hz), 121.32, 115.01, 114.04, 112.06, 100.61, 69.52, 55.74, 55.16 and 31.13. HRMS (ESI) calculated for C22H17F3N4O3 [M + H]+: 443.1326, found: 443.1323. Purity: 99.17% (by HPLC).
6-Amino-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11s). Yellow solid. Yield, 42%. 1H-NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H), 7.73 (d, J = 8.1 Hz, 2H), 7.59 (d, J = 7.8 Hz, 2H), 7.45–7.41 (m, 2H), 7.30–7.21 (m, 3H), 6.89 (s, 1H), 6.87 (s, 2H), 6.66 (dd, J = 8.9, 3.1 Hz, 1H), 6.53 (s, 1H), 5.32 (s, 1H), 5.19–5.09 (m, 2H), 3.58 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ 161.06, 156.89, 153.77, 149.66, 142.88, 137.87, 134.06, 129.20, 128.97, 128.72, 128.54 (q, J = 31.9 Hz), 127.94, 126.39, 125.68 (q, J = 3.4 Hz), 124.74 (q, J = 261.7 Hz), 121.16, 115.99, 113.98, 112.51, 97.79, 69.55, 57.68, and 55.65. HRMS (ESI) calculated for C28H21F3N4O3 [M + H]+: 519.1639, found: 519.1627. Purity: 99.66% (by HPLC).
6-Amino-4-(4-chloro-5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (11t). White solid. Yield, 44%. 1H-NMR (500 MHz, DMSO-d6) δ 12.05 (s, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.55 (d, J = 6.5 Hz, 2H), 7.23 (s, 1H), 6.87 (s, 3H), 5.30–5.09 (m, 2H), 5.00 (s, 1H), 3.73 (s, 3H), 1.79 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ161.81, 155.56, 149.91, 149.41, 142.20, 135.76, 132.29, 128.67 (q, J = 28.1 Hz), 128.24, 125.72 (q, J = 3.8 Hz), 124.73 (q, J = 272.6 Hz), 121.30, 120.18, 115.64, 114.21, 97.26, 69.79, 56.95, 56.25, and 10.08. HRMS (ESI) calculated for C23H18ClF3N4O3 [M + H]+: 491.1092, found: 491.1089. Purity: 98.50% (by HPLC).
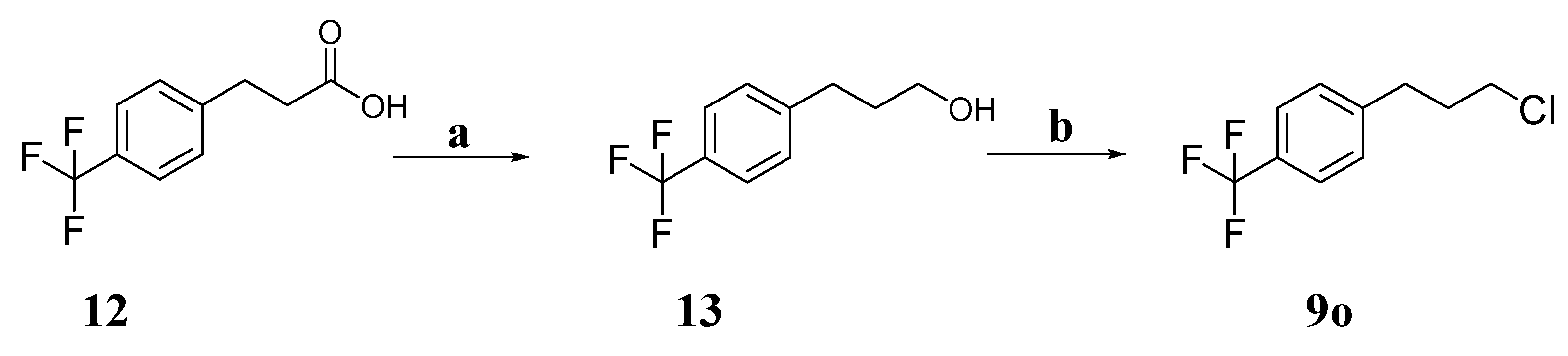
3-(4-(Trifluoromethyl)phenyl)propan-1-ol (13). Lithium aluminum (6.9 mmol) was slowly added to a solution of 12 (4.6 mmol) in tetrahydrofuran at 0 °C. Then, the mixture was refluxed at 85 °C for 6 h. After the addition was complete, water/10% sodium hydroxide/water = 1:1:3 (mL:mL:mL) was added. Then, the solution was filtered and the filtrate was collected. After evaporation of the solvent under reduced pressure, the crude product was purified on a silica gel column to obtain compound 13. White solid. Yield, 56%. 1H-NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.1 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 3.71 (dd, J = 10.3, 5.9 Hz, 2H), 2.83–2.75 (m, 2H), 1.97–1.87 (m, 2H).
1-(3-Chloropropyl)-4-(trifluoromethyl)benzene (9o). Triethylamine (2.5 mmol) was added to a solution of 13 (1.0 mmol) in anhydrous dichloromethane (10 mL) at 0 °C, followed by triphosgene (0.5 mmol) in one portion. The mixture was stirred at 0 °C for 5 min and then allowed to warm to room temperature. After the reaction was complete, the reaction mixture was poured into an aqueous solution containing saturated sodium bicarbonate. After separation of the layers, the aqueous layer was extracted again with dichloromethane (2 × 30 mL). Then, the organic layers were combined, dried with MgSO4, filtered, and concentrated in vacuo, and the crude product was purified on a silica gel column to obtain compound 9o. White solid. Yield, 71%. 1H-NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 3.55 (t, J = 6.4 Hz, 2H), 2.88 (t, J = 7.5 Hz, 2H), 2.17–2.06 (m, 2H).
Tert-butyl-6-amino-5-cyano-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methylpyrano[2,3-c]pyrazole-1(4H)-carboxylate (14). (Boc)2O (1.2 mmol) was added to a solution of 11h (1 mmol) and DMAP (0.2 mmol) in tetrahydrofuran (15 mL) at 0 °C, and then, the mixture was stirred at room temperature for 8 h. After completion, the mixture was extracted with water (30 mL) and the organic layer was separated. After evaporation of the organic layer under reduced pressure, the crude product was purified on a silica gel column to obtain compound 14. White solid. Yield, 60%. 1H-NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 7.9 Hz, 2H), 7.46 (d, J = 6.7 Hz, 2H), 7.07 (s, 2H), 6.97 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.7 Hz, 2H), 5.19–5.09 (m, 2H), 4.89 (s, 1H), 3.68 (s, 3H), 2.08 (s, 3H), 1.52 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 161.03, 156.82, 153.78, 149.63, 148.24, 142.52, 140.27, 132.29, 128.73, 128.38(q, J = 33.3 Hz), 128.14, 125.59 (q, J = 3.7 Hz), 124.70 (q, J = 245.8 Hz), 120.72, 116.47, 114.32, 112.78, 105.48, 85.13, 69.23, 56.45, 55.74, 27.87, 27.79 and 13.19. LC/MS (ESI): 557.20 [M + H]+.
Tert-butyl-5-cyano-6-(dimethylamino)-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl) oxy)phenyl)-3-methylpyrano[2,3-c]pyrazole-1(4H)-carboxylate (15). CH3I (2 mmol) was added to a solution of K2CO3 (2.1 mmol) and 14 (1 mmol) in acetonitrile (20 mL), and the mixture was stirred at 60 °C for 5 h. After the reaction was complete, the solvent was spin-dried under reduced pressure. The mixture was diluted with water (20 mL) and extracted with ethyl acetate (60 mL), and then, the organic layer was separated. After evaporation of the solvent under reduced pressure, the crude product was purified on a silica gel column to obtain compound 15. White solid. Yield, 53%. 1H-NMR (400 MHz, DMSO-d6) δ 7.75 (d, J = 7.9 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 1.9 Hz, 1H), 7.08 (d, J = 8.8 Hz, 1H), 6.91 (dd, J = 9.1, 2.2 Hz, 1H), 5.19 (s, 2H), 4.93 (s, 1H), 3.84 (s, 3H), 3.73 (s, 3H), 2.20 (s, 3H), 1.87 (s, 3H), 1.52 (s, 9H). 13C-NMR (126 MHz, DMSO-d6) δ 162.03, 153.33, 149.52, 148.14, 144.10, 142.20, 128.85 (q, J = 31.9 Hz), 128.66, 125.95, 125.70 (q, J = 3.8 Hz), 124.69 (q, J = 291.9 Hz), 117.01, 116.99, 115.75, 114.14, 113.98, 106.59, 84.88, 69.82, 56.23, 55.75, 35.83, 27.90, 24.80 and 13.24. LC/MS (ESI): 585.19 [M + H] +.
6-(Dimethylamino)-4-(5-methoxy-2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (16). Compound 15 (1 mmol) was added to the solution of trifluoroacetic acid/water = 1:3 (mL:mL), and the mixture was stirred at room temperature for 1 h. After the reaction was complete, the mixture was diluted with saturated sodium carbonate, and then, the organic layer was separated and the solvent was spin-dried under reduced pressure. The crude product was purified on a silica gel column to obtain compound 16. White solid. Yield, 77%. 1H-NMR (400 MHz, DMSO-d6) δ 11.74 (s, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 8.1 Hz, 2H), 7.49 (d, J = 3.0 Hz, 1H), 7.05 (d, J = 9.1 Hz, 1H), 6.88 (dd, J = 9.0, 3.0 Hz, 1H), 5.17 (s, 2H), 4.83 (s, 1H), 3.77 (s, 3H), 3.71 (s, 3H), 1.95 (s, 3H), 1.81 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 161.57, 153.34, 149.44, 142.44, 139.50, 128.72 (q, J = 34.4 Hz), 128.29, 127.35, 125.72 (q, J = 4.2 Hz), 124.72 (q, J = 252.4 Hz), 117.46, 117.27, 115.68, 114.04, 113.68, 97.81, 69.78, 55.91, 55.72, 36.51, 24.78 and 10.53. HRMS (ESI) calculated for C25H23F3N4O3[M + H]+: 485.1795, found: 485.1783. Purity: 96.81% (by HPLC).


 ,
, 
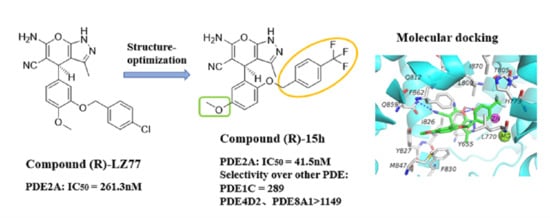









{kind=link}
{kind=link}
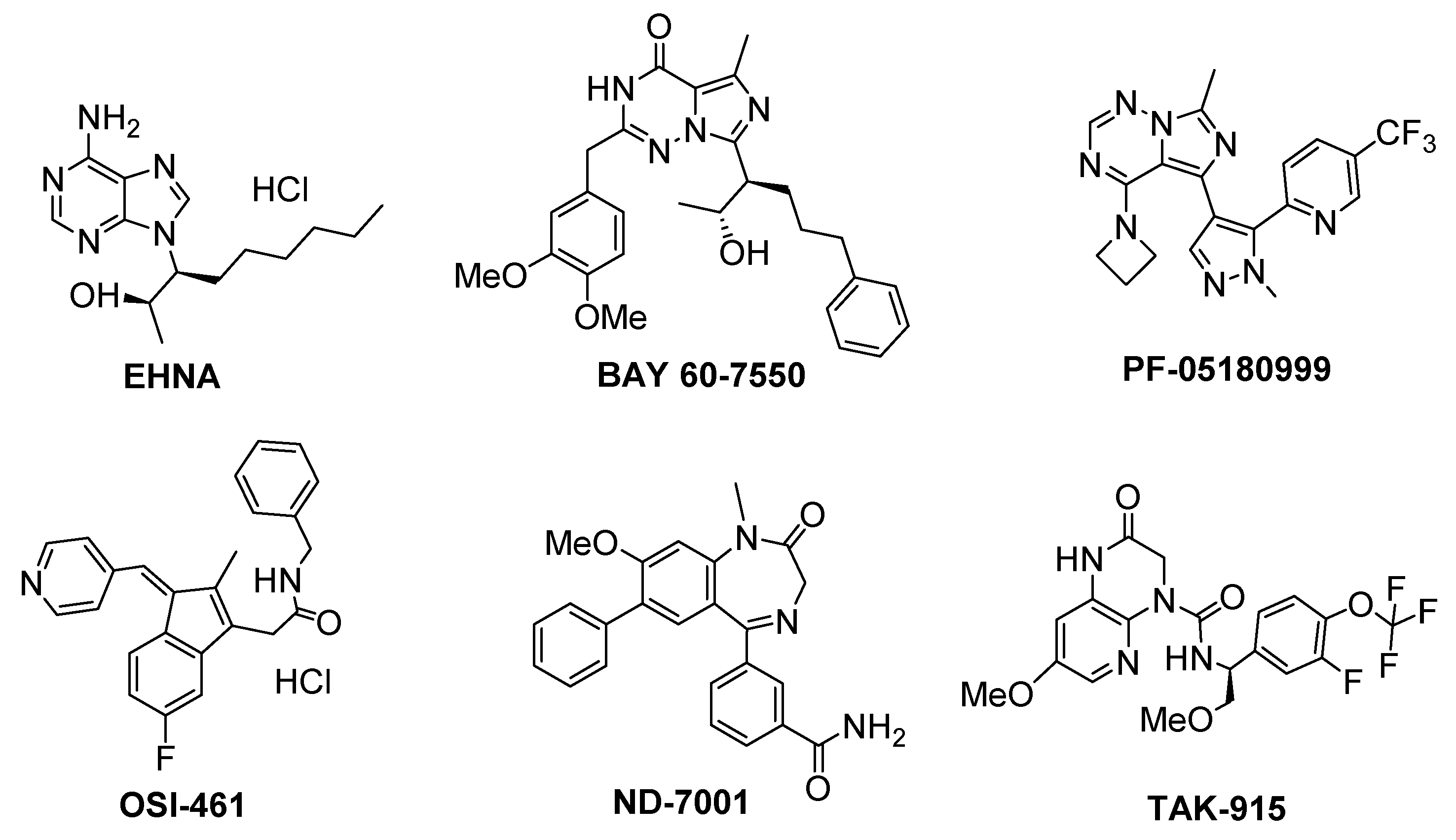
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

