
First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer’s Disease

 , and
, and
Abstract
: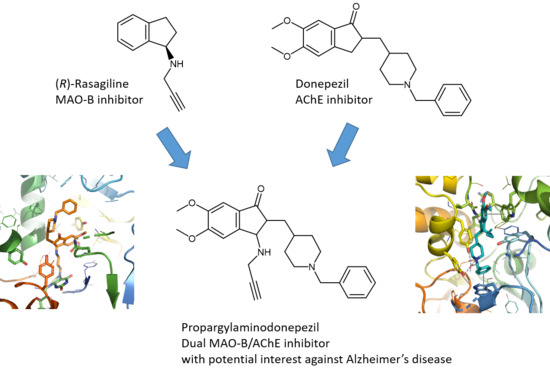
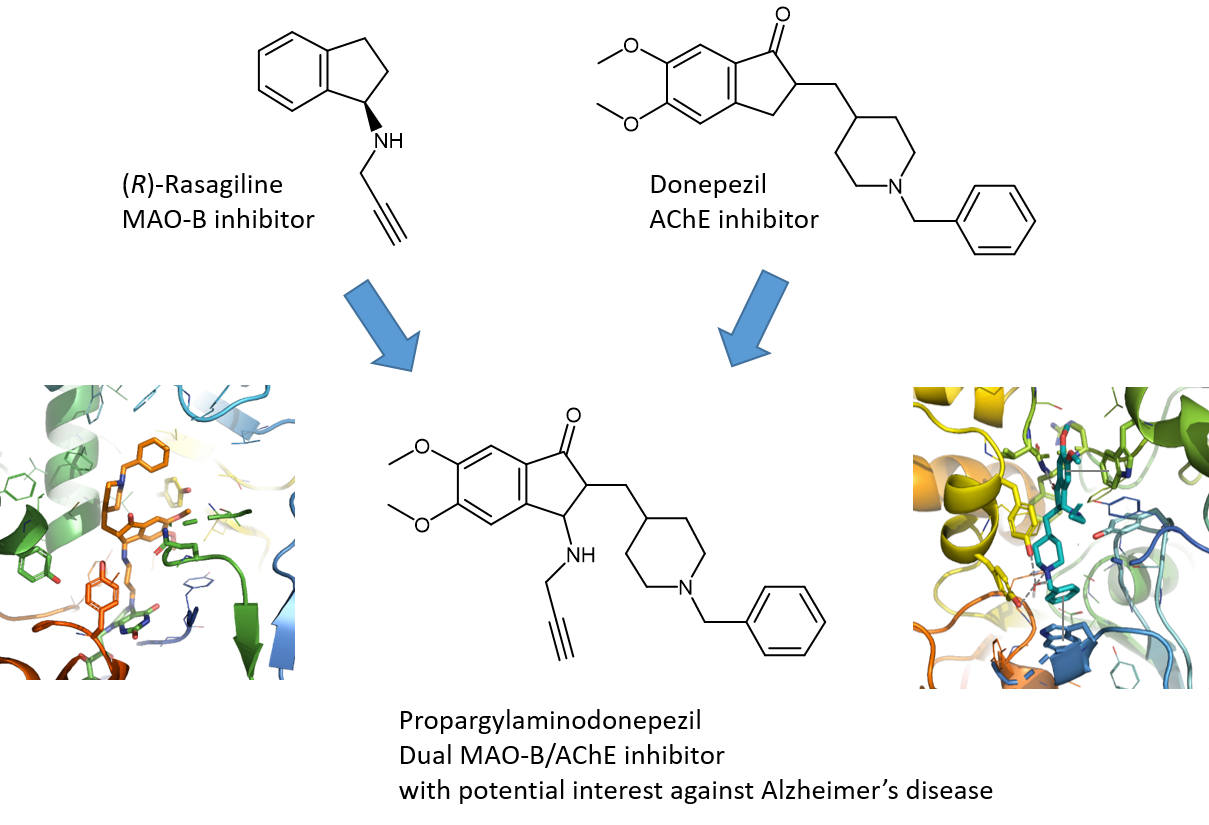
1. Introduction
2. Results
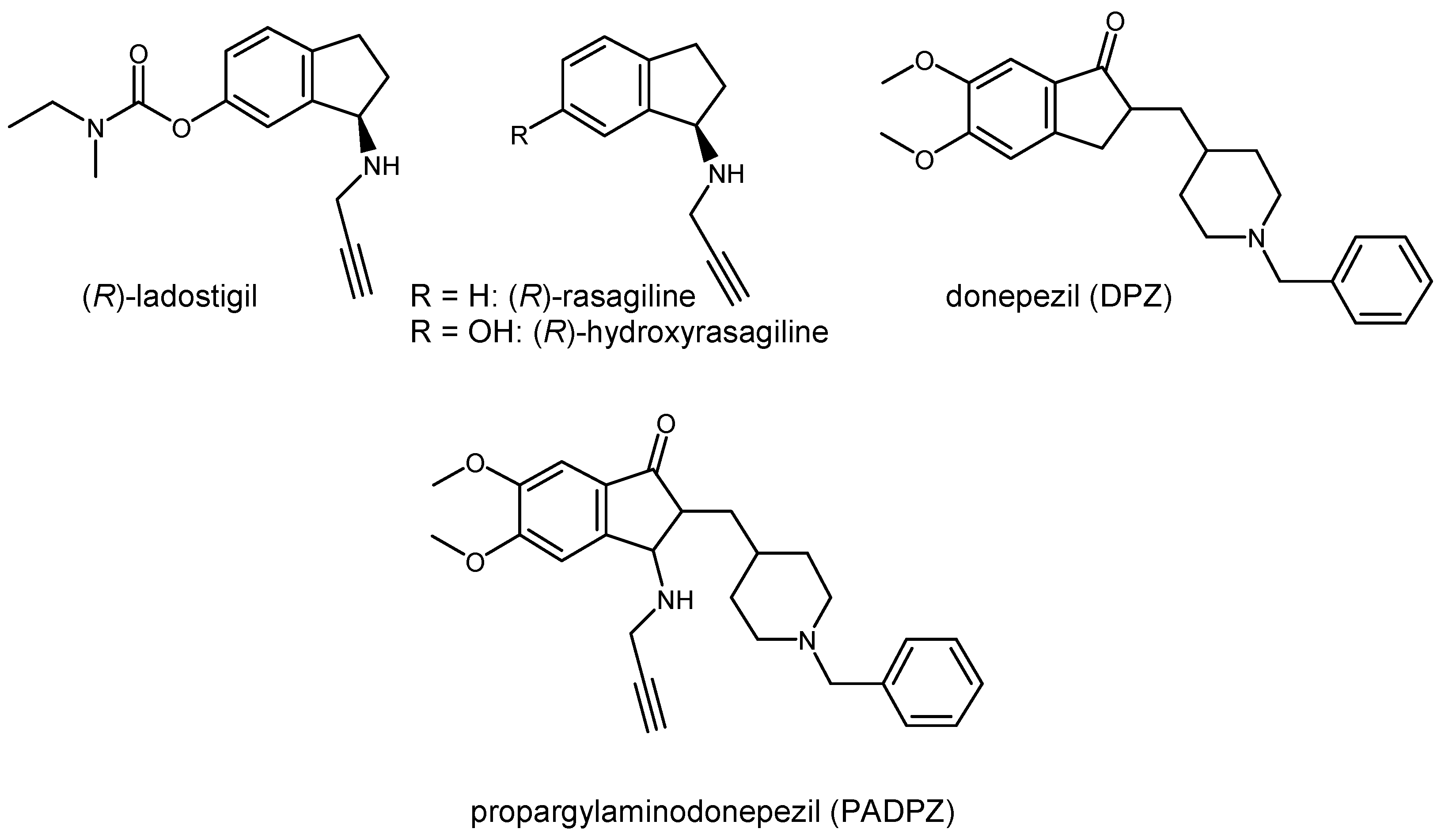
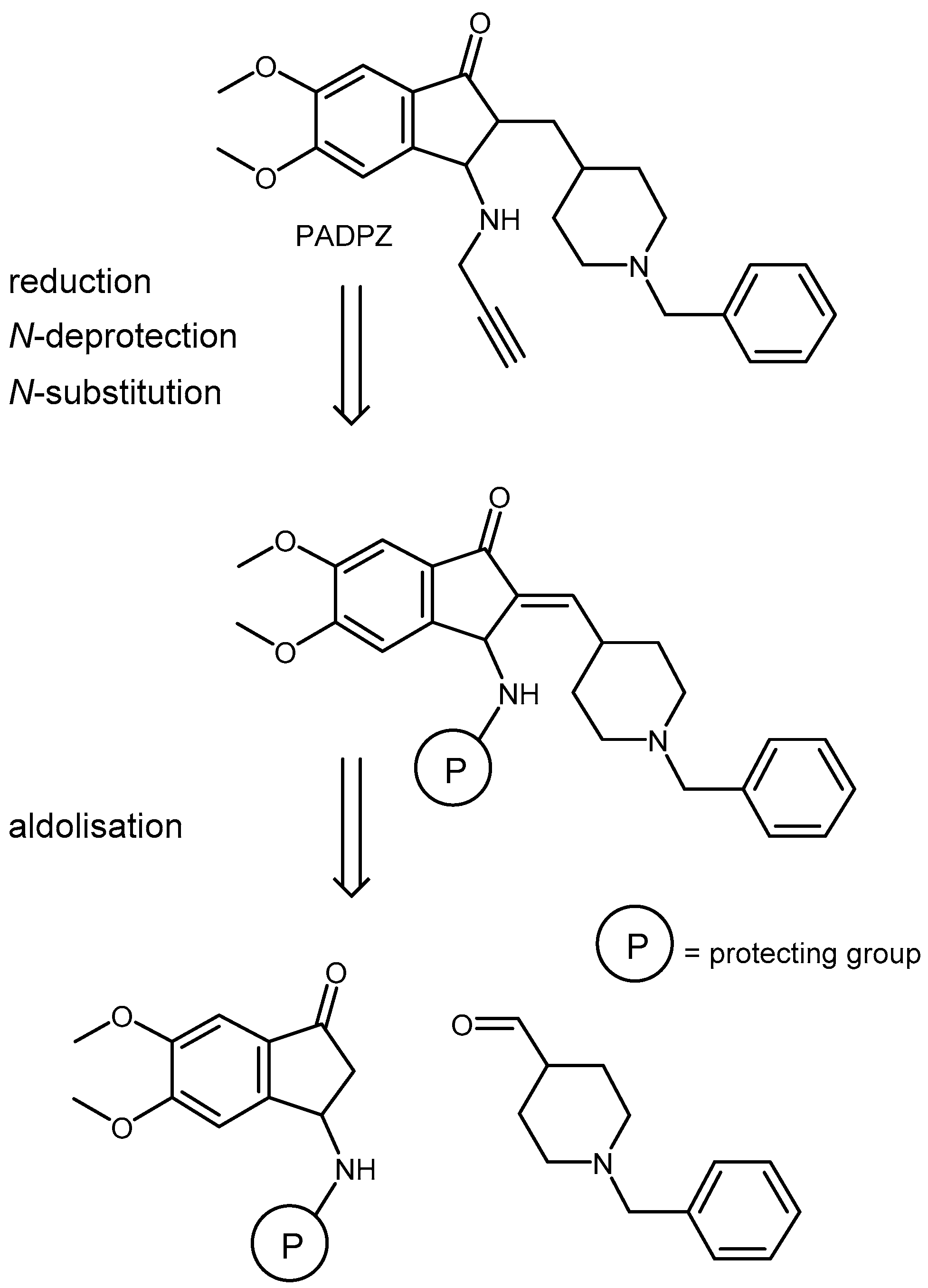
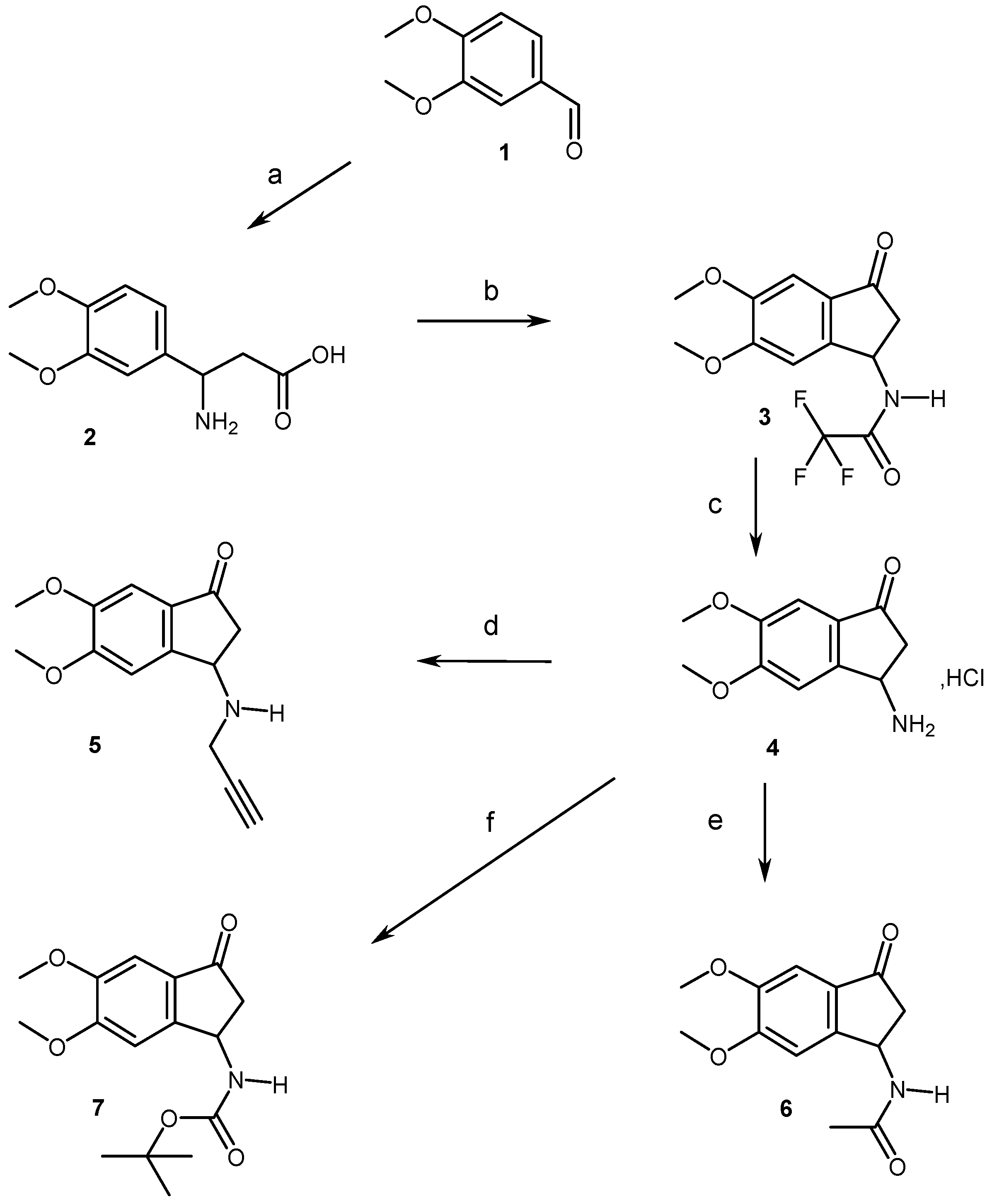
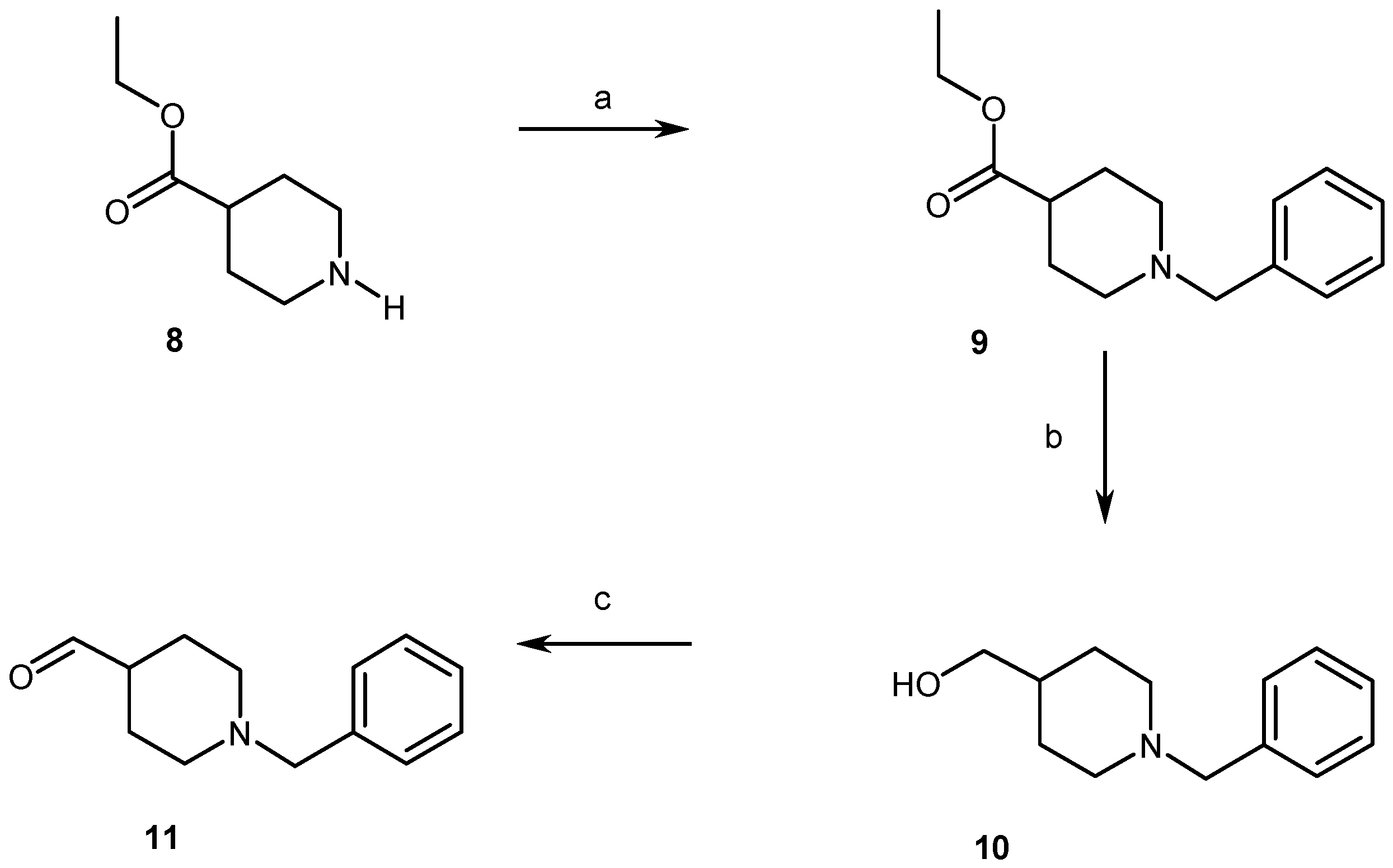
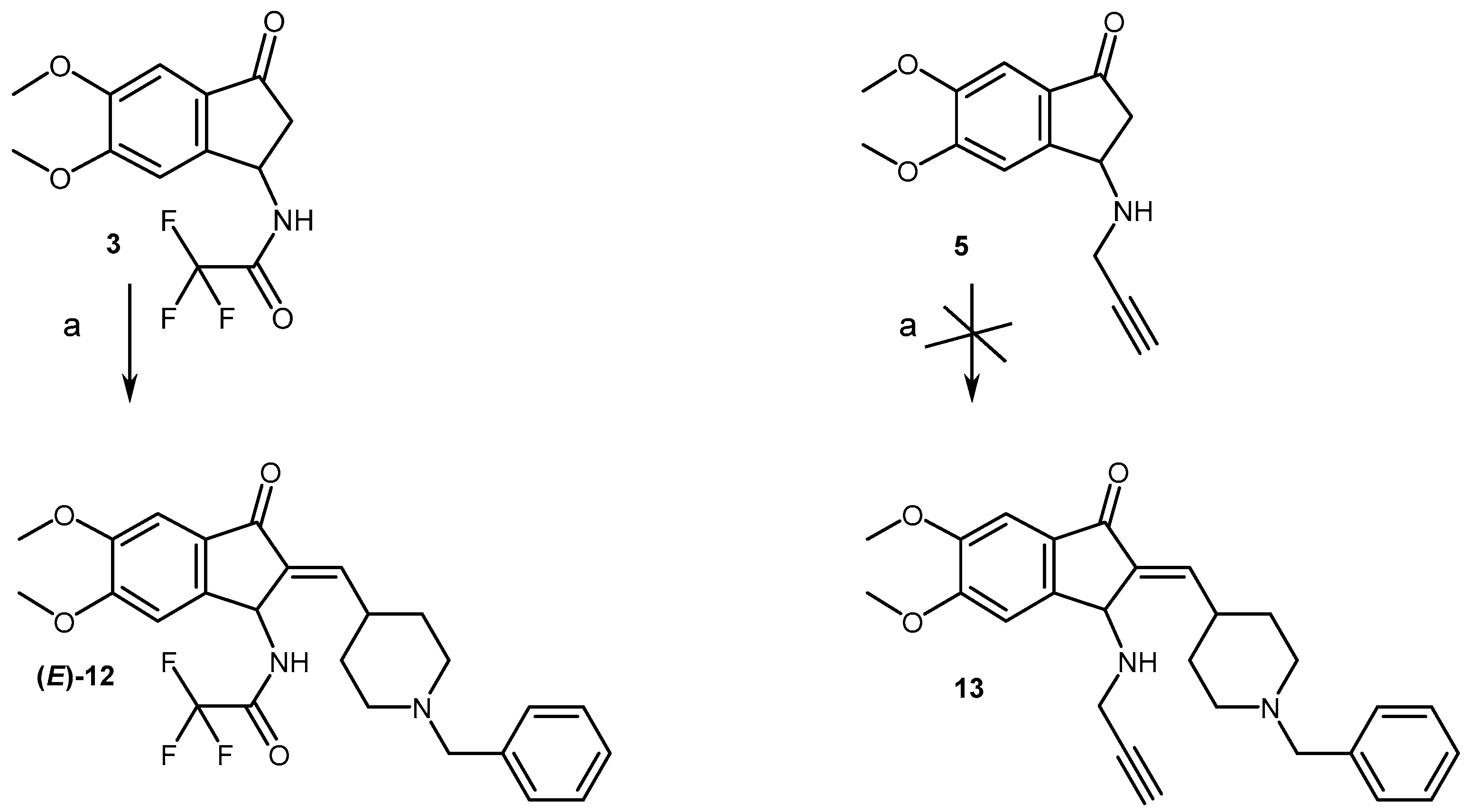
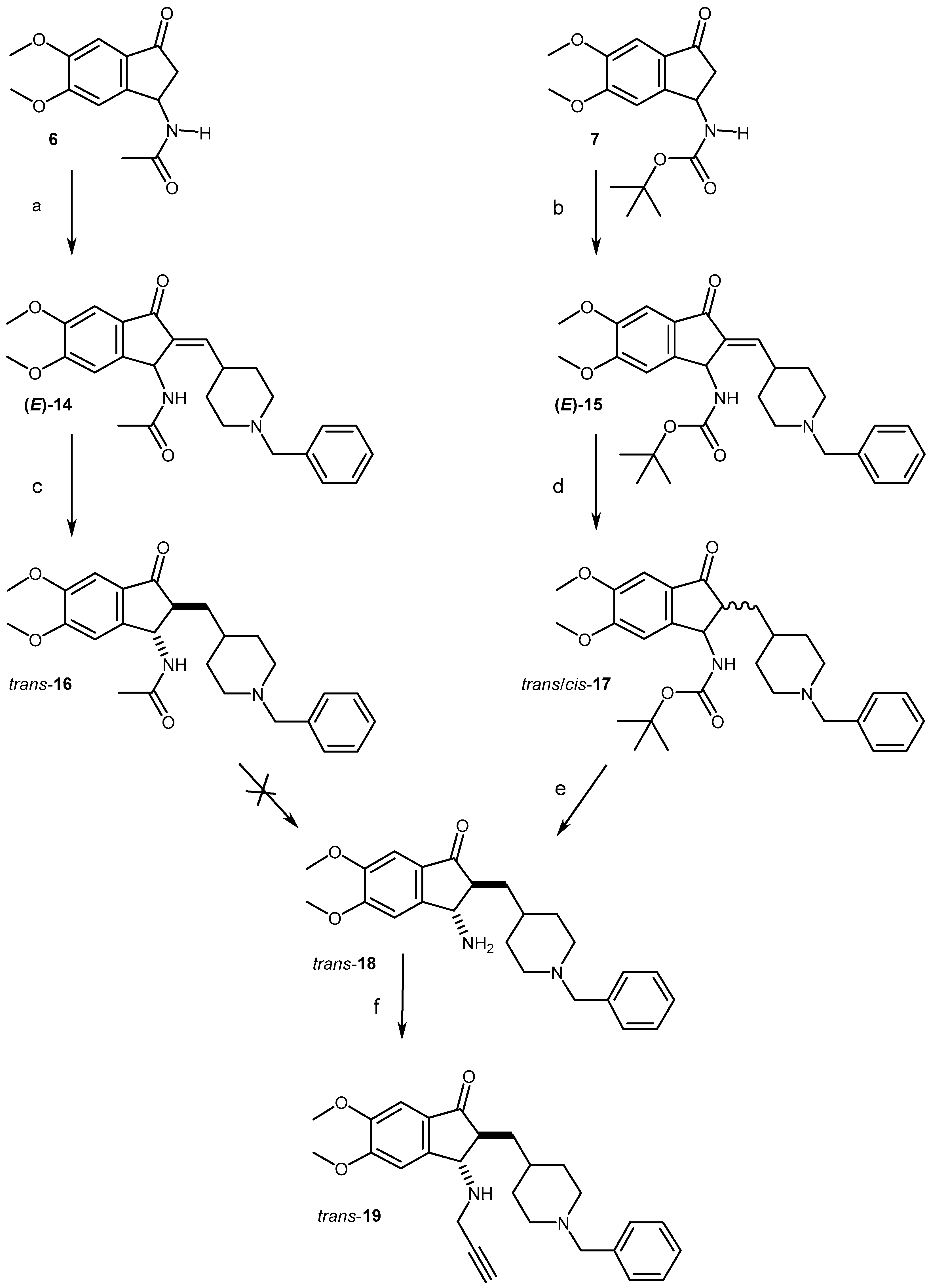
2.1. Chemistry
2.2. In Silico Results
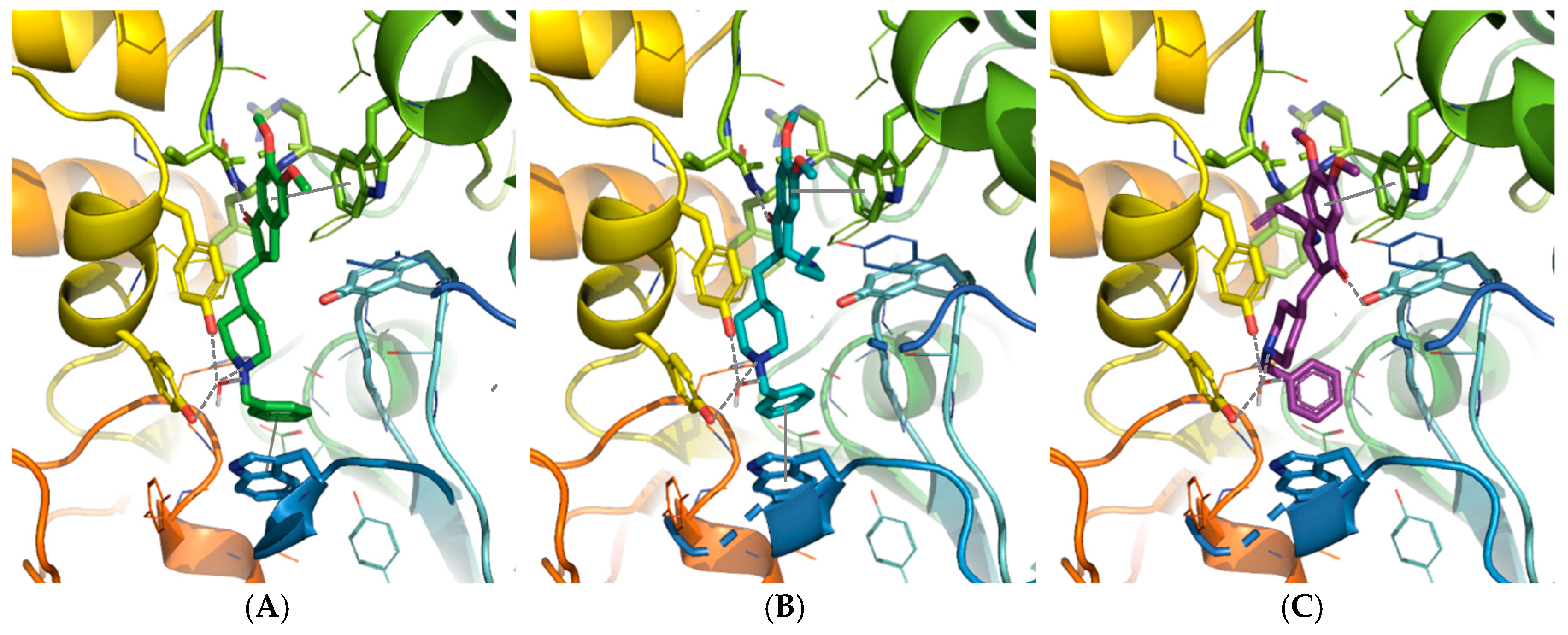
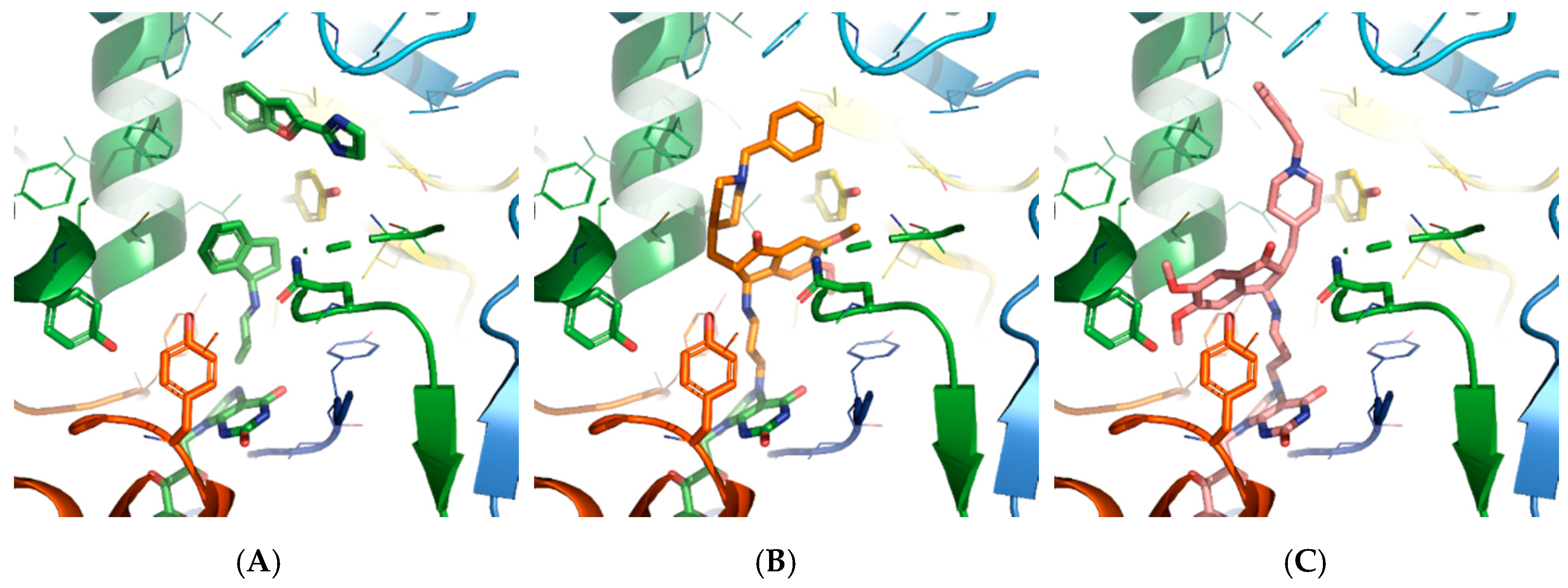
2.2.1. Molecular Modeling Study
2.2.2. In Silico ADME Parameters
2.3. In Vitro Results
Cholinesterases and Monoamine Oxidases Inhibition
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. Synthesis of Compounds (2, 3, 5–11)
4.1.3. General Procedure for the Aldolization Reaction and Preparation of Compounds (12, 15)
4.1.4. Synthesis of Compounds (14, 16–19)
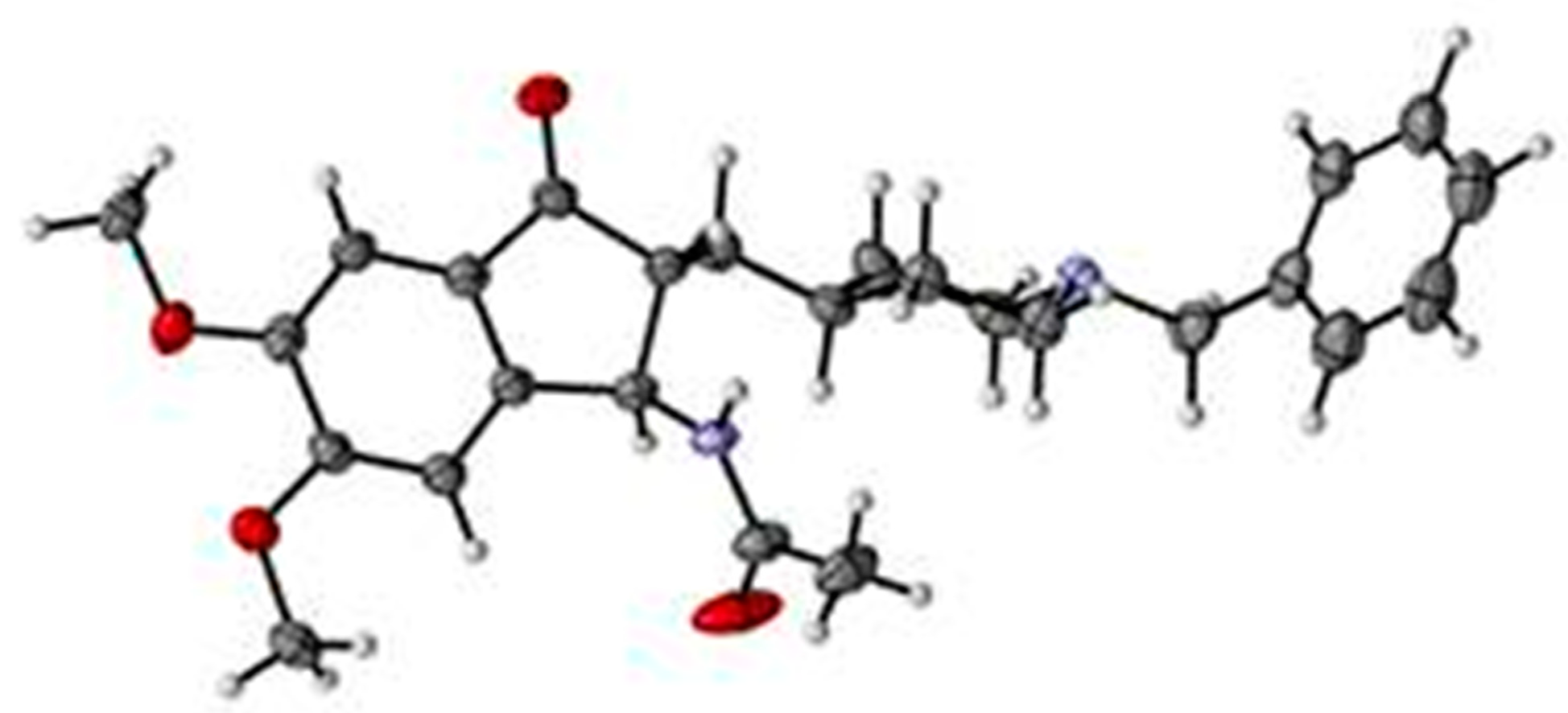
4.2. X-ray Diffractometry
4.3. In Silico Study
4.4. Biological Evaluation
4.4.1. In Vitro Tests of AChE and BuChE Biological Activity
4.4.2. In Vitro Tests of MAO-A and MAO-B Inhibitory Activity
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Multi-Target Directed Ligands. Available online: https://www.clinicaltrials.gov (accessed on 10 November 2020).
- Cavalli, A.; Bolognesi, M.L.; Mìnarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Freon, A.; et al. Design of Donecopride, a Dual Serotonin Subtype 4 Receptor Agonist/Acetylcholinesterase Inhibitor with Potential Interest for Alzheimer’s Disease Treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef] [PubMed]
- Rochais, C.; Lecoutey, C.; Hamidouche, K.; Giannoni, P.; Gaven, F.; Cem, E.; Mignani, S.; Baranger, K.; Freret, T.; Bockaert, J.; et al. Donecopride, a Swiss Army Knife with Potential against Alzheimer’s Disease. Br. J. Pharmacol. 2020, 177, 1988–2005. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M. The Path from Anti Parkinson Drug Selegiline and Rasagiline to Multifunctional Neuroprotective Anti Alzheimer Drugs Ladostigil and M30. Curr. Alzheimer Res. 2006, 3, 541–550. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The Therapeutic Potential of Monoamine Oxidase Inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Bautista-Aguilera, O.M.; Samadi, A.; Chioua, M.; Nikolic, K.; Filipic, S.; Agbaba, D.; Soriano, E.; de Andrés, L.; Rodríguez-Franco, M.I.; Alcaro, S.; et al. N-Methyl-N-((1-Methyl-5-(3-(1-(2-Methylbenzyl)Piperidin-4-Yl)Propoxy)-1H-Indol-2-Yl)Methyl)Prop-2-Yn-1-Amine, a New Cholinesterase and Monoamine Oxidase Dual Inhibitor. J. Med. Chem. 2014, 57, 10455–10463. [Google Scholar] [CrossRef] [Green Version]
- Dallemagne, P.; Rault, S.; Pilo, J.C.; Foloppe, M.P.; Robba, M. One-Pot Cyclization of Alkoxy-3-Aminoindan-1-Ones. Tetrahedron Lett. 1991, 32, 6327–6328. [Google Scholar] [CrossRef]
- Dallemagne, P.; Rault, S.; Pilo, J.C.; Robba, M. A Convenient Route to Indan-1,3-Diones and 3-Hydroxyindan-1-Ones. Bull. Soc. Chim. Fr. 1993, 130, 121–124. [Google Scholar]
- van Greunen, D.G.; Johan van der Westhuizen, C.; Cordier, W.; Nell, M.; Stander, A.; Steenkamp, V.; Panayides, J.-L.; Riley, D.L. Novel N-Benzylpiperidine Carboxamide Derivatives as Potential Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2019, 179, 680–693. [Google Scholar] [CrossRef]
- Diouf, O.; Depreux, P.; Chavatte, P.; Poupaert, J.H. Synthesis and Preliminary Pharmacological Results on New Naphthalene Derivatives as 5-HT4 Receptor Ligands. Eur. J. Med. Chem. 2000, 35, 699–706. [Google Scholar] [CrossRef]
- Punchi Hewage, A.N.D.; Yao, H.; Nammalwar, B.; Gnanasekaran, K.K.; Lovell, S.; Bunce, R.A.; Eshelman, K.; Phaniraj, S.M.; Lee, M.M.; Peterson, B.R.; et al. Small Molecule Inhibitors of the BfrB–Bfd Interaction Decrease Pseudomonas Aeruginosa Fitness and Potentiate Fluoroquinolone Activity. J. Am. Chem. Soc. 2019, 141, 8171–8184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auvray, P.; Moslemi, S.; Sourdaine, P.; Galopin, S.; Séralini, G.-E.; Enguehard, C.; Dallemagne, P.; Bureau, R.; Sonnet, P.; Rault, S. Evidence for new non-steroidal human aromatase inhibitors and comparison with equine aromatase inhibition for an understanding of the mammalian active site. Eur. J. Med. Chem. 1998, 33, 451–462. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.H.; Romero, A.; et al. Donepezil + Propargylamine + 8-Hydroxyquinoline Hybrids as New Multifunctional Metal-Chelators, ChE and MAO Inhibitors for the Potential Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognino, I.; Giangregorio, N.; Pisani, L.; de Candia, M.; Purgatorio, R.; Tonazzi, A.; Altomare, C.D.; Cellamare, S.; Catto, M. A Prospective Repurposing of Dantrolene as a Multitarget Agent for Alzheimer’s Disease. Molecules 2019, 24, 4298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubálek, F.; Binda, C.; Li, M.; Herzig, Y.; Sterling, J.; Youdim, M.B.H.; Mattevi, A.; Edmondson, D.E. Inactivation of Purified Human Recombinant Monoamine Oxidases A and B by Rasagiline and Its Analogues. J. Med. Chem. 2004, 47, 1760–1766. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Patil, S.A. Recent developments in biological activities of indanones. Eur. J. Med. Chem. 2017, 138, 182–198. [Google Scholar] [CrossRef]
- Huang, L.; Miao, H.; Sun, Y.; Meng, F.; Li, X. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 87, 429–439. [Google Scholar] [CrossRef]
- Rampa, A.; Mancini, F.; de Simone, A.; Falchi, F.; Belluti, F.; di Martino, R.M.C.; Gobbi, S.; Andrisano, V.; Tarozzi, A.; Bartolini, M.; et al. From AChE to BACE1 inhibitors: The role of the amine on the indanone scaffold. Bioorg. Med. Chem. Lett. 2015, 25, 2804–2808. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonivento, D.; Milczek, E.M.; McDonald, G.R.; Binda, C.; Holt, A.; Edmondson, D.E.; Mattevi, A. Potentiation of ligand binding through cooperative effects in monoamine oxidase B. J. Biol. Chem. 2010, 285, 36849–36856. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Solubility | |
| Log S (ESOL) | −4.50 |
| Solubility | 1.38 × 10−2 mg/mL; 3.20 × 10−2 mol/L |
| Class | Moderately soluble |
| Pharmacokinetics | |
| GI absorption | High |
| BBB permeant | Yes |
| P-gp substrate | Yes |
| CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No |
| CYP2C9 inhibitor | No |
| CYP2D6 inhibitor | Yes |
| CYP3A4 inhibitor | Yes |
| Lipinski | Yes; 0 violation |
| (h)AChE | (eq)BuChE | |||
|---|---|---|---|---|
| Compound | % Inhibition at 10−6 M | IC50 (µM) (n = 3) | % Inhibition at 10−6 M | IC50 (µM) (n = 3) |
| DPZ | 98% | 0.014 ± 0.0016 | - | 7.4 ± 0.1 1 |
| 16 | 4% | ND | 39% | ND |
| Trans-PADPZ (19) | 71% | 0.442 ± 0.03 | 74% | 4.226 ± 0.375 |
| (h)MAO-B | (h)MAO-A | |||
|---|---|---|---|---|
| Compound | % Inhibition at 10−5 M | IC50 (µM) (n = 3) | % Inhibition at 10−5 M | IC50 (µM) (n = 3) |
| Rasagiline | - | 0.014 1 | - | 0.7 1 |
| Pargiline | - | 2.69 ± 0.48 | - | 10.9 ± 0.6 |
| 5 | 45% | 13.4 ± 3.4 | <10% | ND |
| Trans-PADPZ (19) | 52% | 6.43 ± 0.62 | 13% | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guieu, B.; Lecoutey, C.; Legay, R.; Davis, A.; Sopkova de Oliveira Santos, J.; Altomare, C.D.; Catto, M.; Rochais, C.; Dallemagne, P. First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer’s Disease. Molecules 2021, 26, 80. https://doi.org/10.3390/molecules26010080
Guieu B, Lecoutey C, Legay R, Davis A, Sopkova de Oliveira Santos J, Altomare CD, Catto M, Rochais C, Dallemagne P. First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer’s Disease. Molecules. 2021; 26(1):80. https://doi.org/10.3390/molecules26010080
Chicago/Turabian StyleGuieu, Benjamin, Cedric Lecoutey, Rémi Legay, Audrey Davis, Jana Sopkova de Oliveira Santos, Cosimo Damiano Altomare, Marco Catto, Christophe Rochais, and Patrick Dallemagne. 2021. "First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer’s Disease" Molecules 26, no. 1: 80. https://doi.org/10.3390/molecules26010080