
An Alternative HIV-1 Non-Nucleoside Reverse Transcriptase Inhibition Mechanism: Targeting the p51 Subunit

 , , , and
, , , and 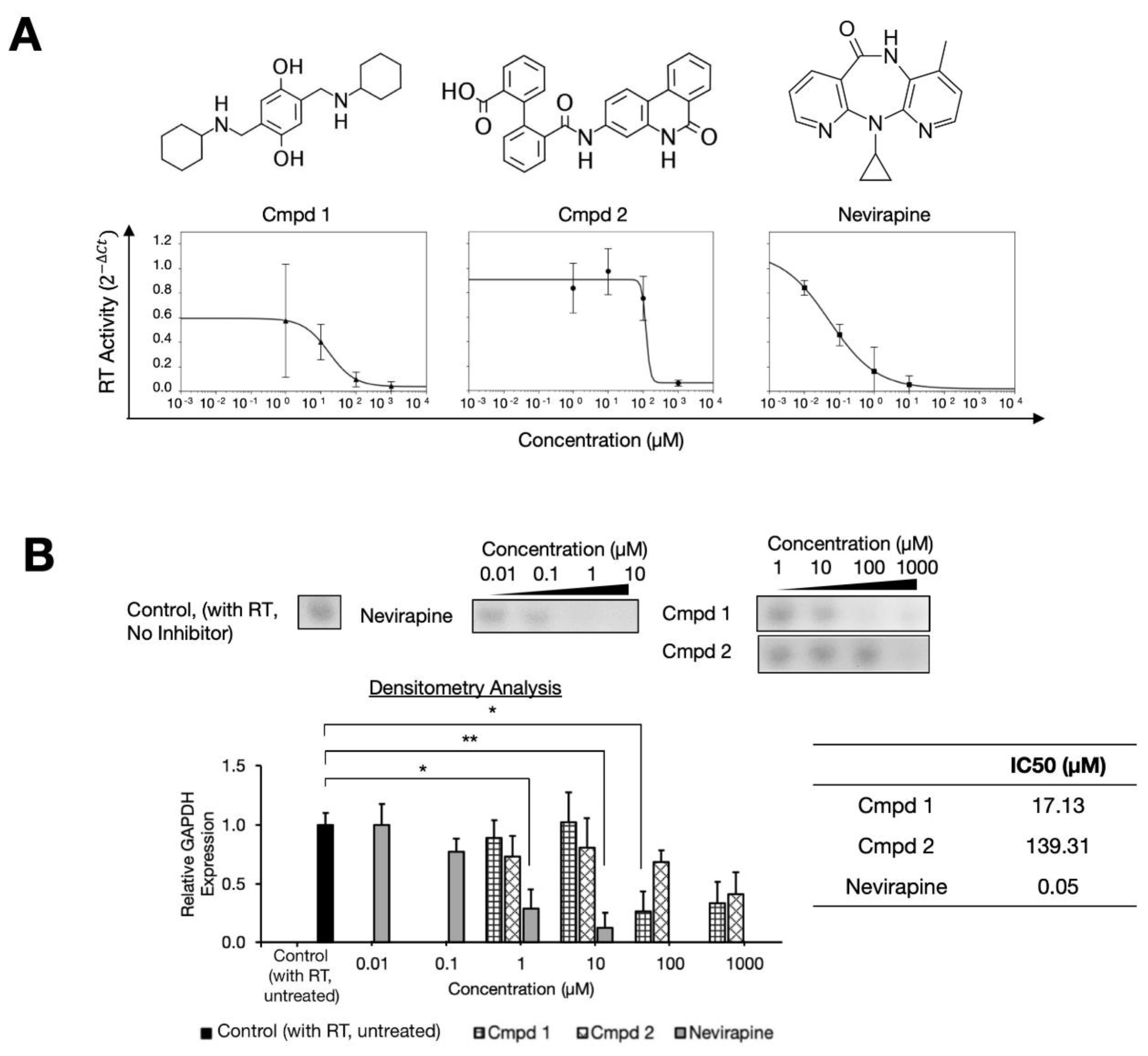{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Two Compound Scaffolds that Inhibit HIV-1 RT Activity
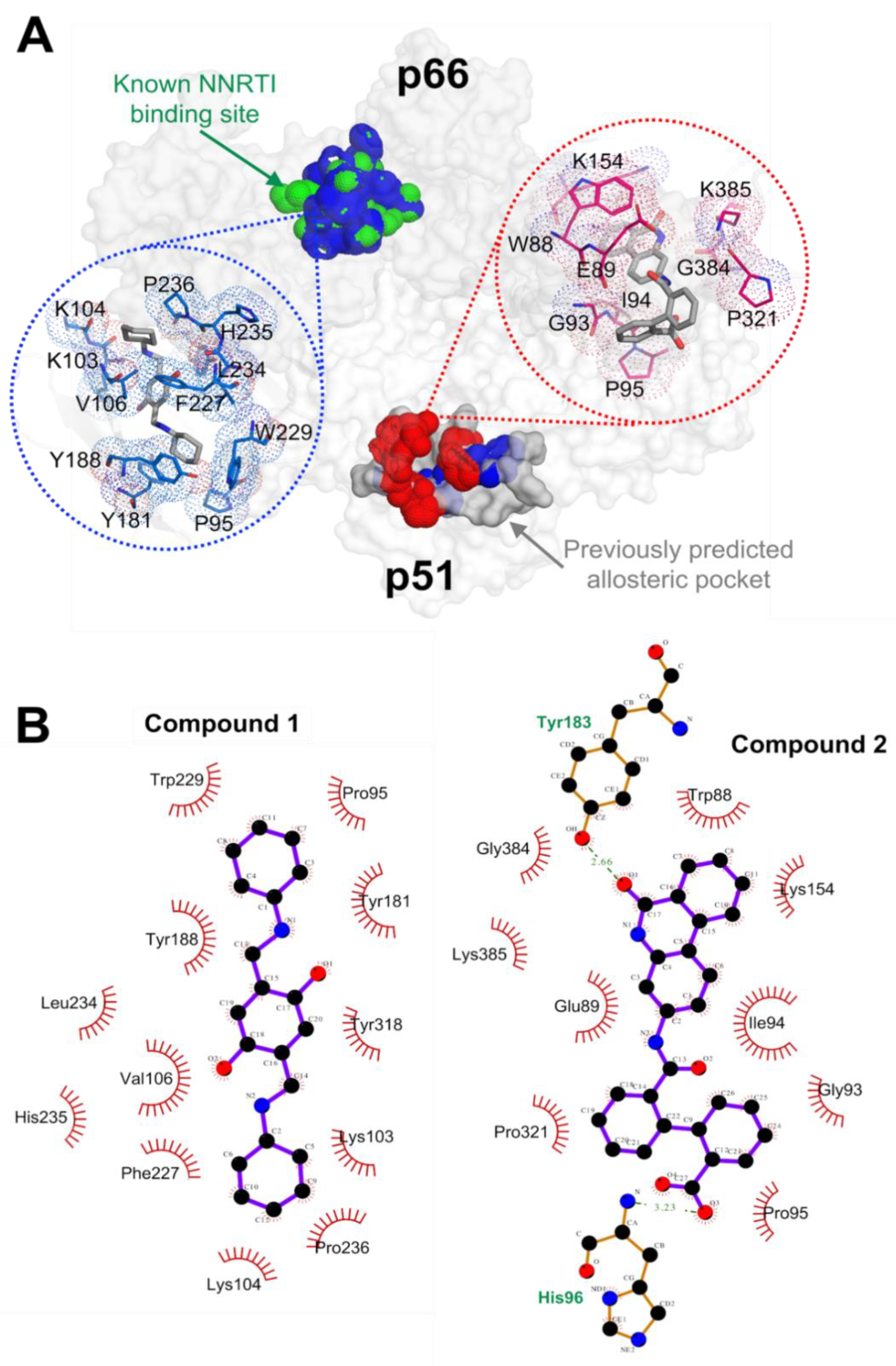
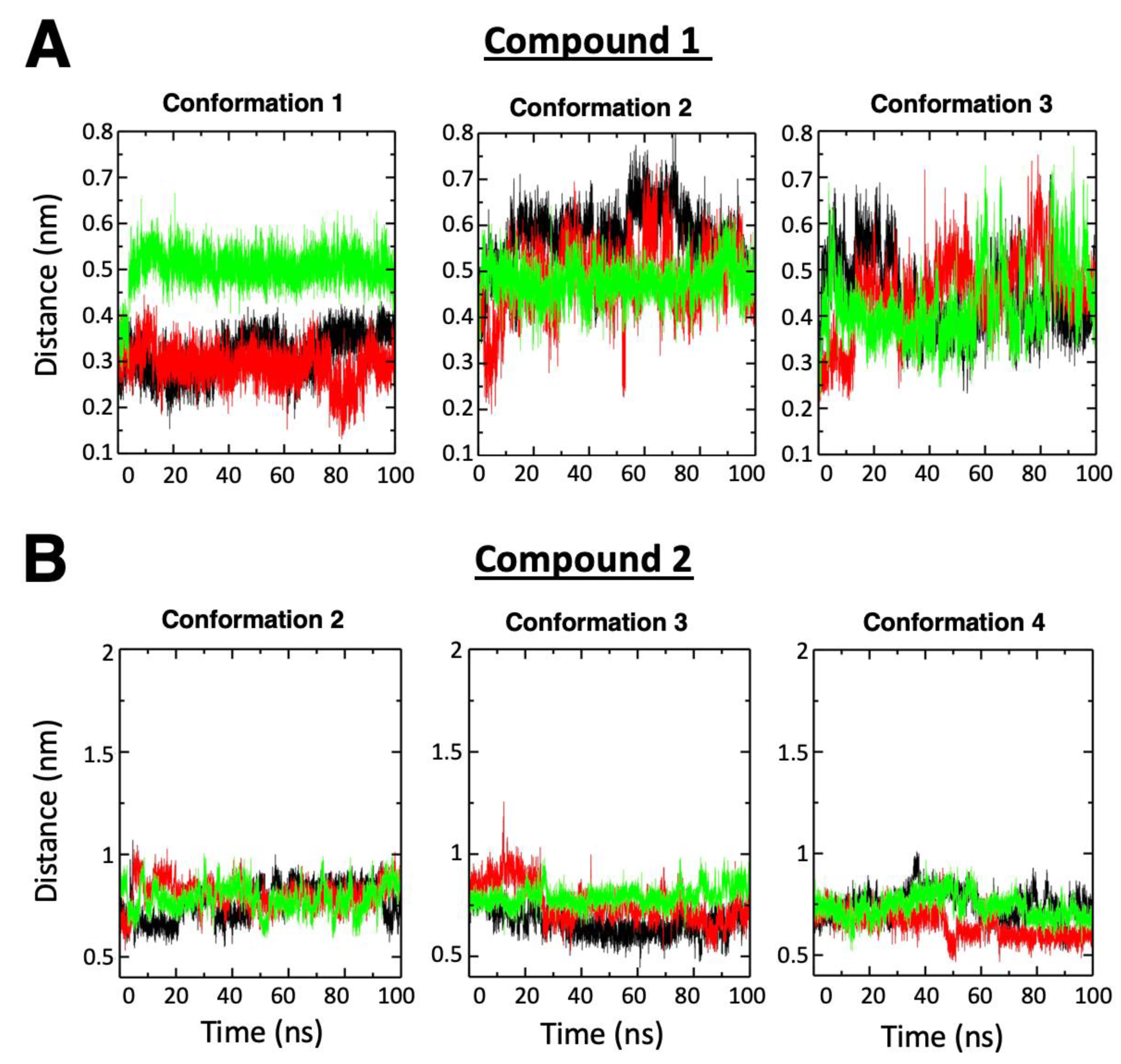
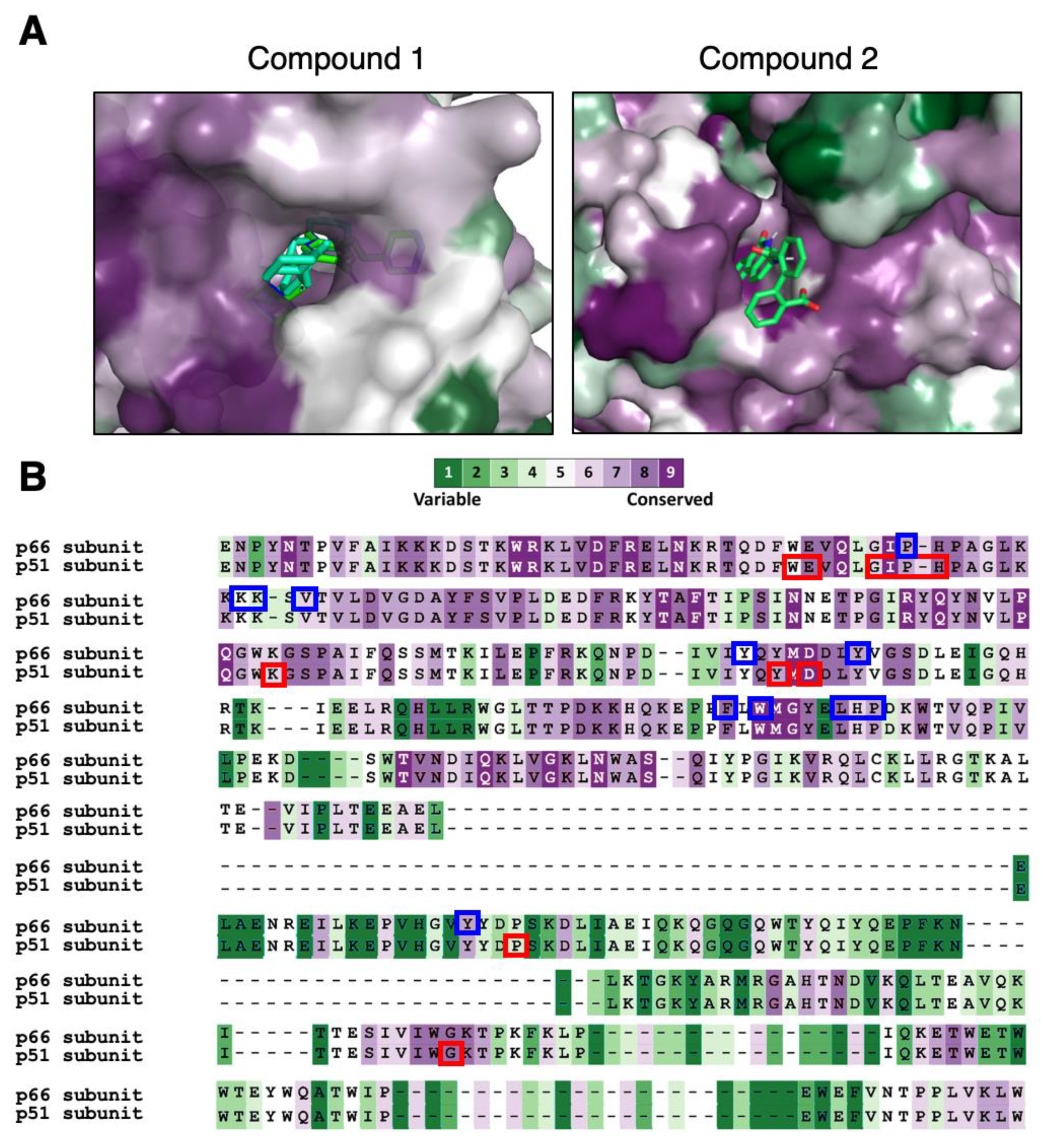
2.2. RT Binding Sites of the Two Compounds
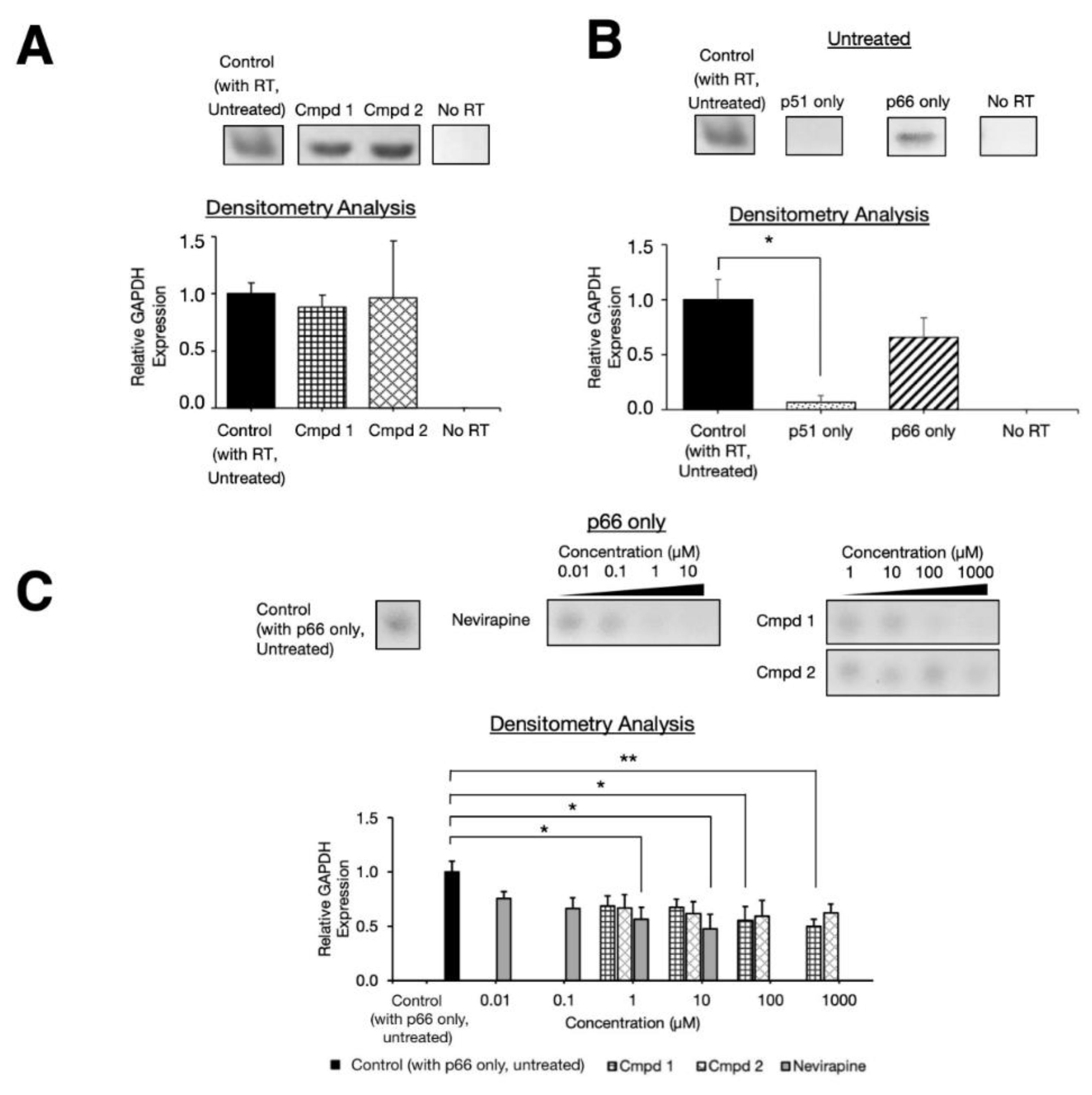
2.3. Experimental Inhibition on Separate HIV-1 RT p66 and p51 Subunits
3. Discussion
4. Materials & Methods
4.1. RNA Extraction
4.2. Library Reagents Preparation
4.3. cDNA Synthesis Reverse Transcription (HIV-1)
4.4. Quantitative Polymerase Chain Reaction (qPCR) Gene Amplification
4.5. Inhibitory Concentration Calculation
4.6. Statistical Analysis
4.7. Structural Docking
4.8. MD Simulations of HIV-1 RT Complexes
4.9. Conventional MD Simulation Analysis
4.10. Binding Energy Calculations
4.11. Analysis of Structural Conservation among RT Proteins
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Accession Codes
References
- World Health Organization (WHO). Number of People (All Ages) Living with HIV Estimates by WHO Region. Available online: http://apps.who.int/gho/data/view.main.22100WHO?lang=en (accessed on 17 November 2020).
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-hiv drug discovery and development: Current innovations and future trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Huo, Z.; Kang, D.; Wu, G.; Zhou, Z.; Liu, X.; Zhan, P. Current insights into anti-HIV drug discovery and development: A review of recent patent literature (2014–2017). Expert Opin. Ther. Pat. 2018, 28, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.J.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Parniak, M.A.; Sluis-Cremer, N. Inhibitors of HIV-I reverse transcriptase. Adv. Pharmacol. 2000, 49, 67–109. [Google Scholar]
- Gu, S.-X.; Zhu, Y.-Y.; Wang, C.; Wang, H.-F.; Liu, G.-Y.; Cao, S.; Huang, L. Recent discoveries in HIV-1 reverse transcriptase inhibitors. Curr. Opin. Pharmacol. 2020, 54, 166–172. [Google Scholar] [CrossRef]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005, 4, 236. [Google Scholar] [CrossRef]
- Qadir, M.I.; Malik, S.A. HIV fusion inhibitors. Rev. Med. Virol. 2010, 20, 23–33. [Google Scholar] [CrossRef]
- Princen, K.; Schols, D. HIV chemokine receptor inhibitors as novel anti-HIV drugs. Cytokine Growth Factor Rev. 2005, 16, 659–677. [Google Scholar] [CrossRef]
- Qian, K.; Morris-Natschke, S.L.; Lee, K.H. HIV entry inhibitors and their potential in HIV therapy. Med. Res. Rev. 2009, 29, 369–393. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Updated Recommendations on First-Line and Second-Line Antiretroviral Regimens and Post-Exposure Prophylaxis and Recommendations on Early Infant Diagnosis of HIV: Interim Guidelines: Supplement to the 2016 Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Maggiolo, F.; Gianotti, N.; Comi, L.; Di Filippo, E.; Fumagalli, L.; Nozza, S.; Galli, L.; Valenti, D.; Rizzi, M.; Castagna, A. Rilpivirine plus cobicistat-boosted darunavir as a two-drug switch regimen in HIV-infected, virologically suppressed subjects on steady standard three-drug therapy: A randomized, controlled, non-inferiority trial (PROBE 2). J. Antimicrob. Chemother. 2020, 75, 1332–1337. [Google Scholar] [CrossRef]
- Cheng, Y.; Dutschman, G.E.; Bastow, K.F.; Sarngadharan, M.; Ting, R. Human immunodeficiency virus reverse transcriptase. General properties and its interactions with nucleoside triphosphate analogs. J. Biol. Chem. 1987, 262, 2187–2189. [Google Scholar] [PubMed]
- Huang, P.; Farquhar, D.; Plunkett, W. Selective action of 2′, 3′-didehydro-2′, 3′-dideoxythymidine triphosphate on human immunodeficiency virus reverse transcriptase and human DNA polymerases. J. Biol. Chem. 1992, 267, 2817–2822. [Google Scholar] [PubMed]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A., Jr.; Hughes, S.; Arnold, E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 Å resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Radzio, J.; Anderson, K.S.; Nicolas, S.C. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007, 16, 1728–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.J. Off-target effects of drugs that disrupt human mitochondrial DNA maintenance. Front. Mol. Biosci. 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Seckler, J.M.; Barkley, M.D.; Wintrode, P.L. Allosteric suppression of HIV-1 reverse transcriptase structural dynamics upon inhibitor binding. Biophys. J. 2011, 100, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, C.F.; Gago, F.; Santoro, M.; Gori, C.; Svicher, V.; Rodríguez Barrios, F.; d’Arrigo, R.; Ciccozzi, M.; Bertoli, A.; Monforte, A.d.A. High sequence conservation of human immunodeficiency virus type 1 reverse transcriptase under drug pressure despite the continuous appearance of mutations. J. Virol. 2005, 79, 10718–10729. [Google Scholar] [CrossRef] [Green Version]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J.; Van Nieuwenhove, J.; Rodríguez-Barrios, F.; de Castro, S.; Velázquez, S.; Ceccherini-Silberstein, F.; De Clercq, E.; Camarasa, M.-J.; Perno, C.-F.; Gago, F.; et al. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 2005, 579, 2294–2300. [Google Scholar] [CrossRef]
- Sánchez-Murcia, P.A.; de Castro, S.; García-Aparicio, C.; Jiménez, M.A.; Corona, A.; Tramontano, E.; Sluis-Cremer, N.; Menéndez-Arias, L.; Velázquez, S.; Gago, F.; et al. Peptides mimicking the β7/β8 Loop of HIV-1 reverse transcriptase p51 as “hotspot-targeted” dimerization inhibitors. ACS Med. Chem. Lett. 2020, 11, 811–817. [Google Scholar] [CrossRef]
- Suzuki, T.; Higgins, P.J.; Crawford, D.R. Control selection for RNA quantitation. Biotechniques 2000, 29, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grob, P.M.; Wu, J.C.; Cohen, K.A.; Ingraham, R.H.; Shih, C.-K.; Hargrave, K.D.; Mctague, T.L.; Merluzzi, V.J. Nonnucleoside inhibitors of HIV-1 reverse transcriptase: Nevirapine as a prototype drug. AIDS Res. Hum. Retrovir. 1992, 8, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, K.D.; Proudfoot, J.R.; Grozinger, K.G.; Cullen, E.; Kapadia, S.R.; Patel, U.R.; Fuchs, V.U.; Mauldin, S.C.; Vitous, J.; Behnke, M.L.; et al. Novel non-nucleoside inhibitors of HIV-1 reverse transcriptase. 1. Tricyclic pyridobenzo- and dipyridodiazepinones. J. Med. Chem. 1991, 34, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Chiang, R.Z.H.; Gan, S.K.E.; Su, C.T.T. A computational study for rational HIV-1 non-nucleoside reverse transcriptase inhibitor selection and the discovery of novel allosteric pockets for inhibitor design. Biosci. Rep. 2018, 38, BSR20171113. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Starnes, M.C.; Gao, W.Y.; Ting, R.Y.; Cheng, Y.C. Enzyme activity gel analysis of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 1988, 263, 5132–5134. [Google Scholar]
- Hizi, A.; McGill, C.; Hughes, S.H. Expression of soluble, enzymatically active, human immunodeficiency virus reverse transcriptase in Escherichia coli and analysis of mutants. Proc. Natl. Acad. Sci. USA 1988, 85, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, M.; Ertl, P.; Larder, B.A.; Purifoy, D.J.; Darby, G.; Powell, K.L. Characterization of human immunodeficiency virus type 1 reverse transcriptase by using monoclonal antibodies: Role of the C terminus in antibody reactivity and enzyme function. J. Virol. 1988, 62, 3662–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, J.Z.; Nguyen, P.V.; Lee, H.K.; Gan, S.K.E. GelApp: Mobile gel electrophoresis analyser. Nat. Methods 2015, 1–2. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar] [PubMed]
- Yeo, J.Y.; Yap, P.; Goh, G.-R.; Koh, D.W.-S.; Gan, S.K.-E. HIV-1 mutations in HIV-1 Gag, protease, RT p66 and when they appear: Insights from an in vitro BSL2 assay on mutation rates and types. bioRxiv 2020. bioRxiv:679852. Available online: https://www.biorxiv.org/content/10.1101/679852v2 (accessed on 17 November 2020).
- Kang, D.; Song, Y.; Chen, W.; Zhan, P.; Liu, X. “Old dogs with new tricks”: Exploiting alternative mechanisms of action and new drug design strategies for clinically validated HIV targets. Mol. Biosyst. 2014, 10, 1998–2022. [Google Scholar] [CrossRef]
- Su, C.T.T.; Ling, W.L.; Lua, W.H.; Haw, Y.X.; Gan, S.K.E. Structural analyses of 2015-updated drug-resistant mutations in HIV-1 protease: An implication of protease inhibitor cross-resistance. BMC Bioinform. 2016, 17, 500. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Chen, W.; Zhao, T.; Li, Z.; Jiang, X.; Ginex, T.; Vílchez, D.; Luque, F.J.; Kang, D.; Gao, P.; et al. Exploiting the tolerant region I of the Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) binding pocket: Discovery of potent diarylpyrimidine-typed HIV-1 NNRTIs against wild-type and E138K mutant virus with significantly improved water solubility and favorable safety profiles. J. Med. Chem. 2019, 62, 2083–2098. [Google Scholar]
- Phua, S.X.; Chan, K.F.; Su, C.T.; Poh, J.J.; Gan, S.K. Perspective: The promises of a holistic view of proteins—Impact on antibody engineering and drug discovery. Biosci. Rep. 2019, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Su, C.T.T.; Kwoh, C.K.; Verma, C.S.; Gan, S.K.E. Modeling the full length HIV-1 Gag polyprotein reveals the role of its p6 subunit in viral maturation and the effect of non-cleavage site mutations in protease drug resistance. J. Biomol. Struct. Dyn. 2017, 36, 1–12. [Google Scholar] [CrossRef]
- Lua, W.H.; Su, C.T.T.; Yeo, J.Y.; Poh, J.J.; Ling, W.L.; Phua, S.X.; Gan, S.K.E. Role of the IgE variable heavy chain in FcεRIα and superantigen binding in allergy and immunotherapy. J. Allergy Clin. Immunol. 2019, 144, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Ling, W.L.; Lua, W.H.; Poh, J.J.; Yeo, J.Y.; Lane, D.P.; Gan, S.K.E. Effect of VH–VL Families in pertuzumab and trastuzumab recombinant production, Her2 and FcγIIA binding. Front. Immunol. 2018, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Lua, W.H.; Ling, W.L.; Yeo, J.Y.; Poh, J.J.; Lane, D.P.S.; Gan, S.K.E. The effects of antibody engineering CH and CL in trastuzumab and pertuzumab recombinant models: Impact on antibody production and antigen-binding. Sci. Rep. 2018, 8, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.T.T.; Ling, W.L.; Lua, W.H.; Poh, J.J.; Gan, S.K.E. The role of Antibody Vκ Framework 3 region towards Antigen binding: Effects on recombinant production and Protein L binding. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Su, C.T.T.; Lua, W.H.; Ling, W.L.; Gan, S.K.E. Allosteric effects between the antibody constant and variable regions: A study of IgA Fc mutations on antigen binding. Antibodies 2018, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, W.-L.; Lua, W.-H.; Gan, S.K.-E. Sagacity in antibody humanization for therapeutics, diagnostics and research purposes: Considerations of antibody elements and their roles. Antib. Ther. 2020, 3, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Poh, J.J.; Phua, S.X.; Chan, K.F.; Gan, S.K.-E. Commentary: Augmented reality scientific phone apps—Making the APD AR holistic review app and using existing AR apps for scientific publications. Sci. Phone Apps Mob. Devices 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Chan, K.-F.; Poh, J.-J.; Wu, W.-L.; Gan, S.K.-E. Augmented reality in scientific visualization and communications: A new dawn of looking at antibody interactions. Antib. Ther. 2020, 3, 221–226. [Google Scholar]
- Goutelle, S.; Maurin, M.; Rougier, F.; Barbaut, X.; Bourguignon, L.; Ducher, M.; Maire, P. The Hill equation: A review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 2008, 22, 633–648. [Google Scholar] [CrossRef]
- AAT Bioquest Inc. Quest Graph™ IC50 Calculator. Available online: https://www.aatbio.com/tools/IC50-calculator (accessed on 17 November 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; Version 3.6.2; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. Comparison of end-point continuum-solvation methods for the calculation of protein-ligand binding free energies. Proteins 2012, 80, 1326–1342. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins 2011, 79, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Schutz, C.N.; Warshel, A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [Green Version]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]





Sample Availability: Samples of the compounds are available from National Cancer Institute (NCI) Developmental Therapeutic Program’s Open Compound Repository, NIH. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, K.-F.; Su, C.T.-T.; Krah, A.; Phua, S.-X.; Yeo, J.Y.; Ling, W.-L.; Bond, P.J.; Gan, S.K.-E. An Alternative HIV-1 Non-Nucleoside Reverse Transcriptase Inhibition Mechanism: Targeting the p51 Subunit. Molecules 2020, 25, 5902. https://doi.org/10.3390/molecules25245902
Chan K-F, Su CT-T, Krah A, Phua S-X, Yeo JY, Ling W-L, Bond PJ, Gan SK-E. An Alternative HIV-1 Non-Nucleoside Reverse Transcriptase Inhibition Mechanism: Targeting the p51 Subunit. Molecules. 2020; 25(24):5902. https://doi.org/10.3390/molecules25245902
Chicago/Turabian StyleChan, Kwok-Fong, Chinh Tran-To Su, Alexander Krah, Ser-Xian Phua, Joshua Yi Yeo, Wei-Li Ling, Peter J. Bond, and Samuel Ken-En Gan. 2020. "An Alternative HIV-1 Non-Nucleoside Reverse Transcriptase Inhibition Mechanism: Targeting the p51 Subunit" Molecules 25, no. 24: 5902. https://doi.org/10.3390/molecules25245902