Probing the Protein-Protein Interaction between the ATRXADD Domain and the Histone H3 Tail



Abstract
:1. Introduction
2. Results
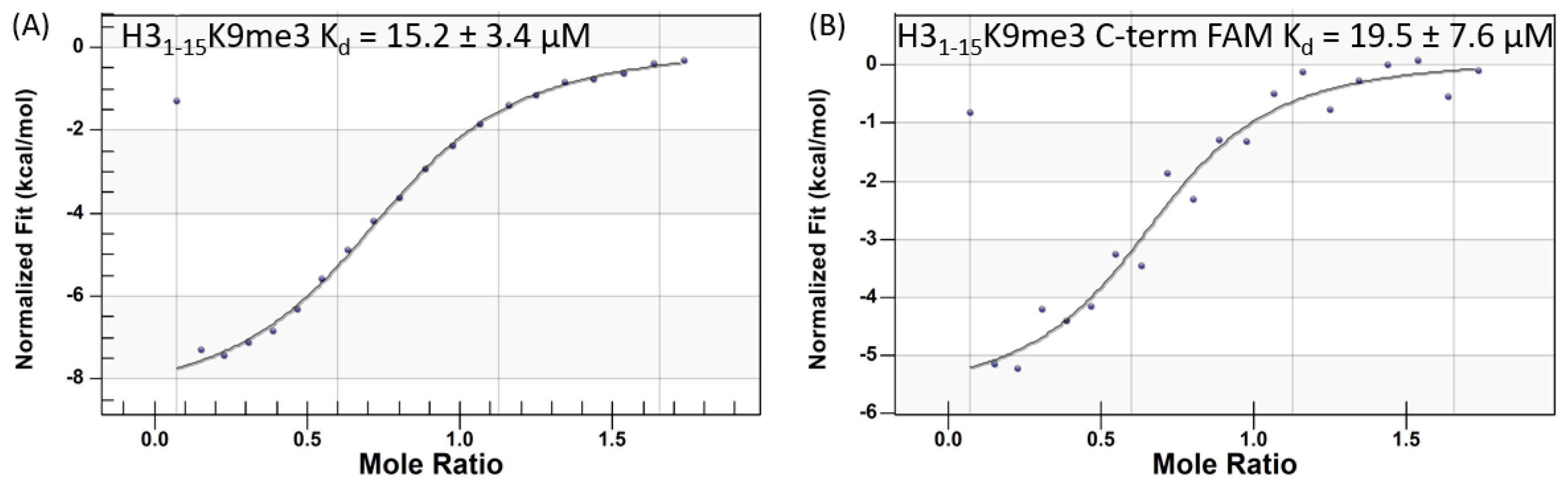
2.1. Isothermal Titration Calorimetry Studies
2.2. Assay Development
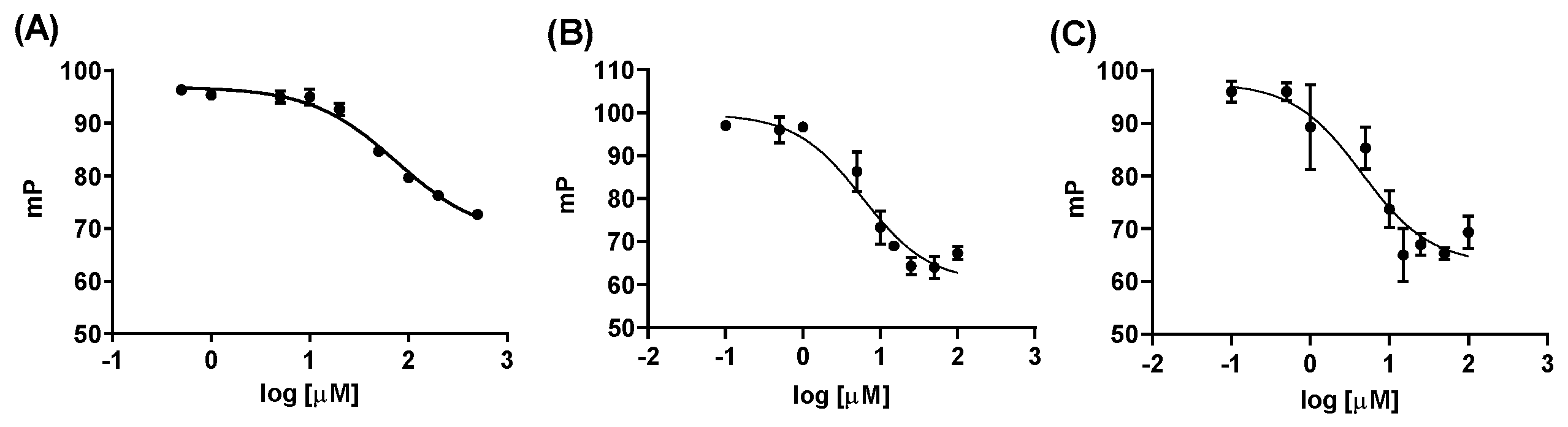
2.2.1. Fluorescence Polarization Assay
2.2.2. AlphaScreen Assay
2.3. Screening Results
2.3.1. Biochemical Screening
2.3.2. Virtual Screening
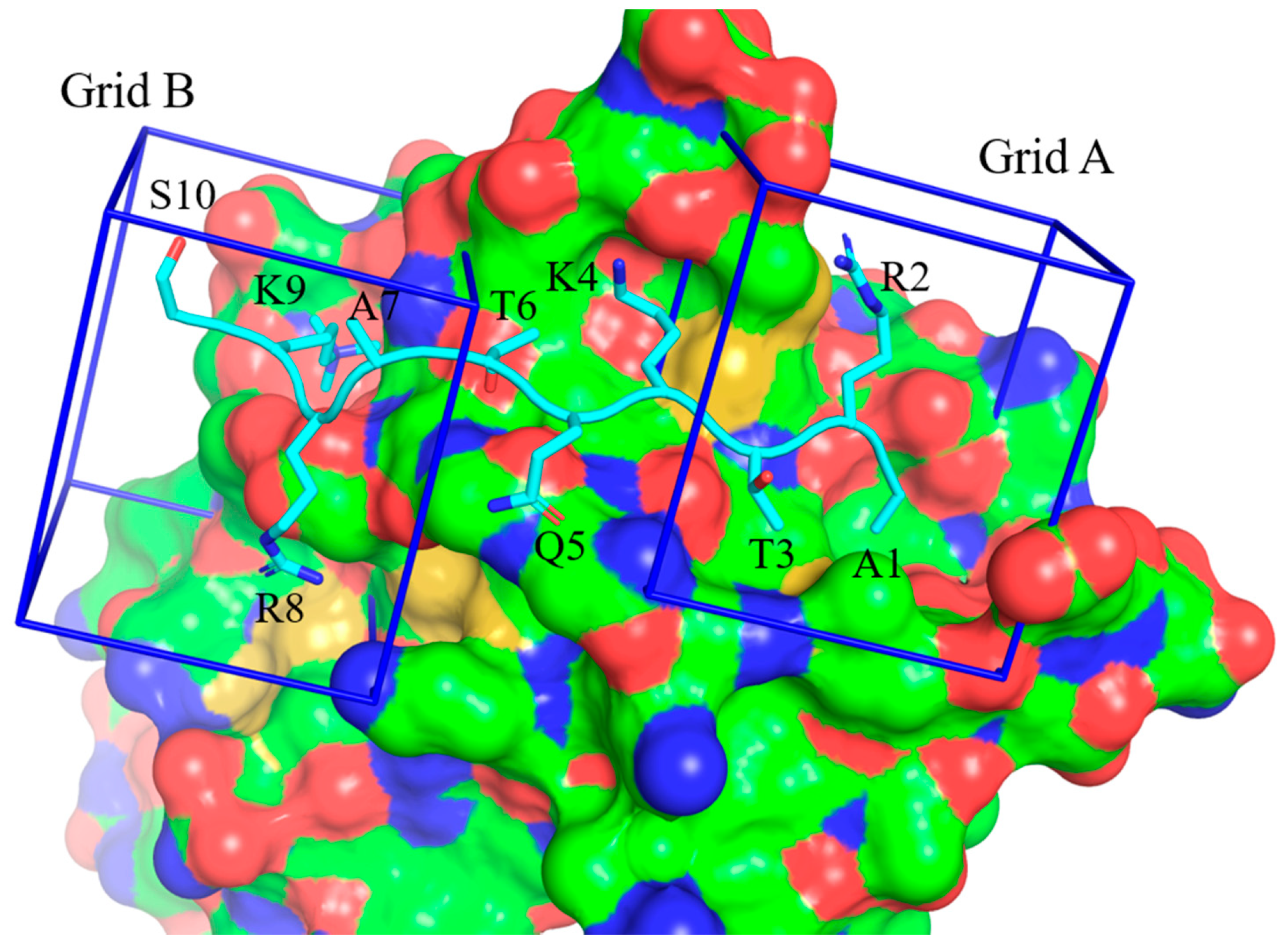
2.4. Truncated H3 Peptide Studies
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Plasmids and Cloning
4.3. Protein Expression and Purification
4.4. Isothermal Titration Calorimetry
4.5. Microscale Thermophoresis
4.6. Fluorescence Polarization Assay
4.7. Alphascreen Assay
4.7.1. Initial Assay Development
4.7.2. Optimized Assay Protocol
4.8. Biochemical Screening
4.9. Virtual Screening
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gibbons, R.J.; Picketts, D.J.; Villard, L.; Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with α-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, D.; Xiang, B.; Li, H. Structural and biochemical characterization of DAXX-ATRX interaction. Protein Cell 2017, 8, 762–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelper, D.; Huang, H.; Jain, A.Y.; Patel, D.J.; Lewis, P.W. Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat. Commun 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnakumar, K.; Bernstein, E. Atrx. Epigenetics 2013, 8, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, ra28Cra328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, R.J.; Wada, T.; Fisher, C.A.; Malik, N.; Mitson, M.J.; Steensma, D.P.; Fryer, A.; Goudie, D.R.; Krantz, I.D.; Traeger-Synodinos, J. Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 2008, 29, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Higgs, D.R.; Gibbons, R.J. The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Nagahashi Marie, S.K.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, L.A.; Goldberg, H.; Bérubé, N.G. Emerging roles of ATRX in cancer. Epigenomics 2015, 7, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Yang, J.-C.; Law, M.J.; Amos, R.; Chapman, L.M.; Jelinska, C.; Garrick, D.; Clynes, D.; Gibbons, R.J.; Rhodes, D.; et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 2011, 18, 777. [Google Scholar] [CrossRef] [PubMed]
- Teske, K.A.; Hadden, M.K. Methyllysine binding domains: Structural insight and small molecule probe development. Eur. J. Med. Chem. 2017, 136, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Tamas, R.; Bock, I.; Tattermusch, A.; Dimitrova, E.; Kudithipudi, S.; Ragozin, S.; Jeltsch, A. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 2011, 20, 2195–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, R.C.; Zaino, A.M.; Hadden, M.K. A metadynamic approach to understand the recognition mechanism of the histone H3 tail with the ATRXADD domain. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2018, 1861, 594–602. [Google Scholar] [CrossRef] [PubMed]
- AlphaScreenTM: Sensitive Homogeneous Assay Technology; PerkinElmer Life and Analytical Sciences: Shelton, CT, USA, 2004.
- Jeltsch, A. Addgene: pGEX-GP-2-ATRX ADD. Available online: https://www.addgene.org/59698/#how-to-cite (accessed on 12 March 2019).
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| H3 Peptide | Kd (µM) 1, ITC | Kd (µM) 2, ITC | Kd (µM) 3, MST | IC50 (µM) 4, FP |
|---|---|---|---|---|
| H31–15K9me0 | 7.9 | 369 ± 47 | 23.4 ± 11 | 76.8 ± 1.3 |
| H31–15K9me1 | 2.5–2.9 | 289 ± 2.4 | 3.4 ± 2.2 | 63.3 ± 9.5 |
| H31–15K9me2 | 0.38–1.3 | 49.5 ± 8.6 | 4.9 ± 2.6 | 10.2 ± 1.0 |
| H31–15K9me3 | 0.27–0.5 | 15.2 ± 3.4 | 1.8 ± 0.2 | 5.5 ± 0.2 |
| H31–15K9me3-FAM | ND 5 | 19.5 ± 7.6 | ND | ND |
| Tagged ATRXADD | H3 Peptide | Kd (μM) 1 | IC50 (μM) 2 |
|---|---|---|---|
| ATRXADD-GST | H31–15K9me3-biotin | 37.6 ± 0.1 | 5.3 ± 0.1 |
| ATRXADD-His | H31–15K9me3-biotin | 14.3 ± 2.7 | 2.8 ± 0.8 |
| Peptide | Protein | Beads | Signal 1 |
|---|---|---|---|
| H31–15K9me3-biotin | ATRXADD-GST | Anti-GST | 722400 ± 33000 |
| H31–15K9me3-biotin | --- | Anti-GST | 224400 ± 14000 |
| H31–15K9me3-biotin | ATRXADD-GST | Ni-NTA | 5218 ± 80 |
| H31–15K9me3-biotin | --- | Ni-NTA | 5778 ± 220 |
| H31–15K9me3-biotin | ATRXADD-His | Ni-NTA | 1246000 ± 58000 |
| H31–15K9me3-biotin | --- | Ni-NTA | 1260 ± 430 |
| Peptide | Beads | IC50 (μM) |
|---|---|---|
| H31–15K9me0 | Ni-NTA | > 10.000 1 |
| H31–15K9me1 | Ni-NTA | > 10.000 |
| H31–15K9me2 | Ni-NTA | 3.9 ± 2.5 |
| H31–15K9me3 | Ni-NTA | 11.6 ± 0.3 |
| Peptide | Truncated Peptide Sequence | FP Assay IC50 |
|---|---|---|
| H31-16 | ARTKQTARKSTGGKAY | 85.8 ± 19.3 μM |
| H31-10 | ARTKQTARKS | 94.8 ± 4.6 μM |
| H31-5 | ARTKQ | ND 1 |
| H33-5 | TKQ | ND |
| H32-6 | RTKQT | ND |
| H33-8 | TKQTAR | ND |
| H31-11K9me3 | ARTKQTARK(me3)ST | 7.6 ± 1.4 μM |
| H37-11K9me3 | ARK(me3)ST | ND |
| H31-5 + H37-11K9me3 | ARTKQ + ARK(me3)ST | ND |
| H31-7K4me3 | ARTK(me3)QTA | ND |
| H33-10K4me3K9me3 | TK(me3)QTARK(me3)S | ND |
| H31-11 A1V | VRTKQTARK(me3)ST | 4.3 ± 0.8 μM |
| H31-11 A1L | LRTKQTARK(me3)ST | 5.05 ± 0.7 μM |
| H31-8 | ARTKQTAR | ≥ 100 μM 2 |
| H33-11K9me3 | TKQTARK(me3)ST | ND |
| H31-11K4me3K9me3 | ARTK(me3)QTARK(me3)ST | ≥ 100 μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaino, A.M.; Dash, R.C.; Hadden, M.K. Probing the Protein-Protein Interaction between the ATRXADD Domain and the Histone H3 Tail. Molecules 2020, 25, 1500. https://doi.org/10.3390/molecules25071500
Zaino AM, Dash RC, Hadden MK. Probing the Protein-Protein Interaction between the ATRXADD Domain and the Histone H3 Tail. Molecules. 2020; 25(7):1500. https://doi.org/10.3390/molecules25071500
Chicago/Turabian StyleZaino, Angela M., Radha Charan Dash, and M. Kyle Hadden. 2020. "Probing the Protein-Protein Interaction between the ATRXADD Domain and the Histone H3 Tail" Molecules 25, no. 7: 1500. https://doi.org/10.3390/molecules25071500