
Novel Tamoxifen Nanoformulations for Improving Breast Cancer Treatment: Old Wine in New Bottles

 ,
,  ,
, 

Abstract
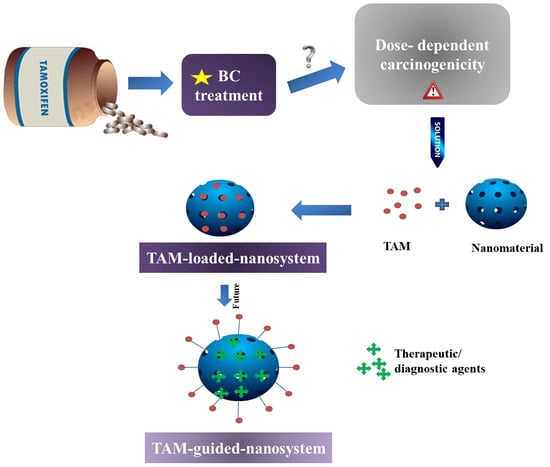
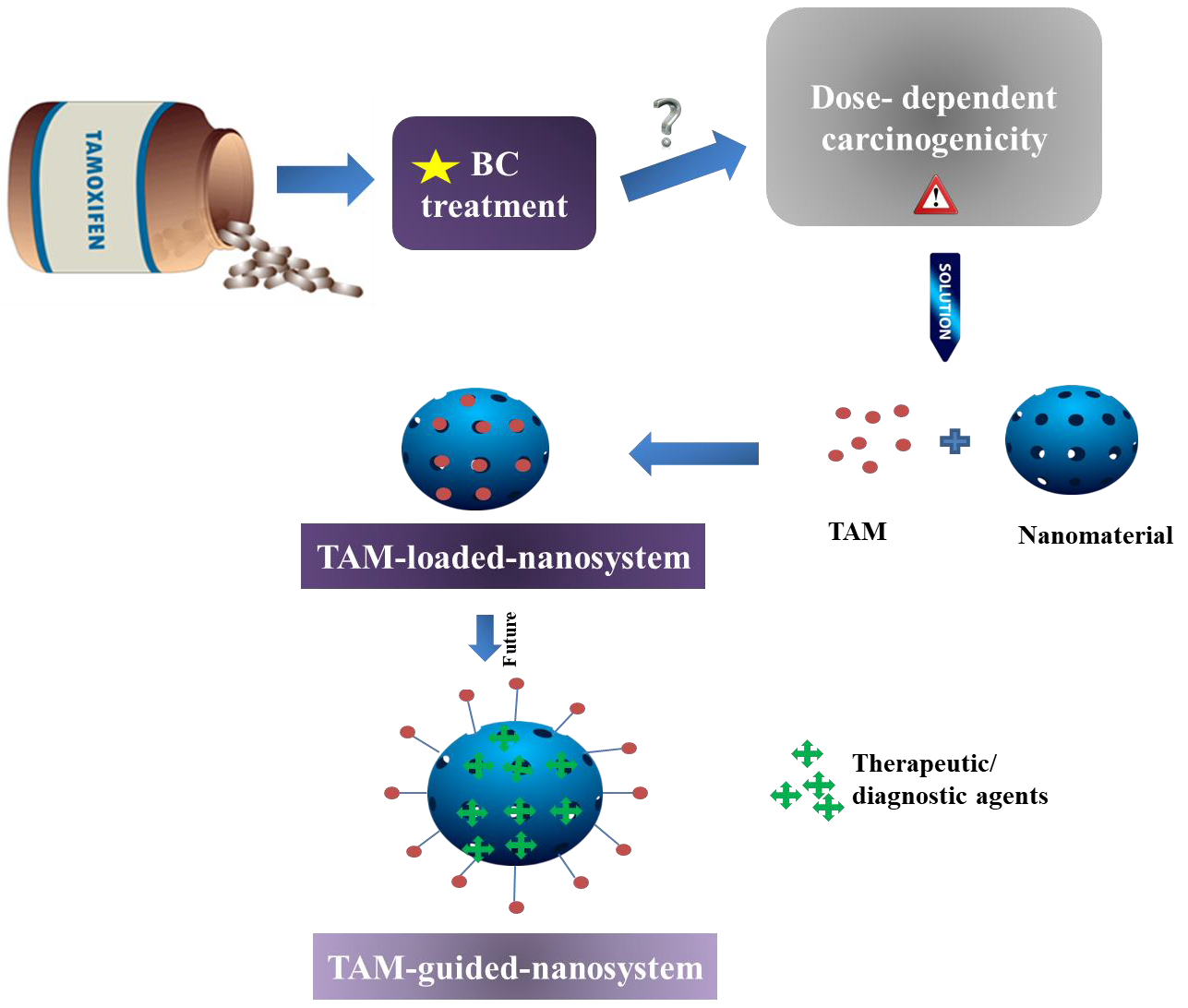
:
1. Introduction
BC Targeting Therapy
2. TAM as a Gold Standard in BC Therapy
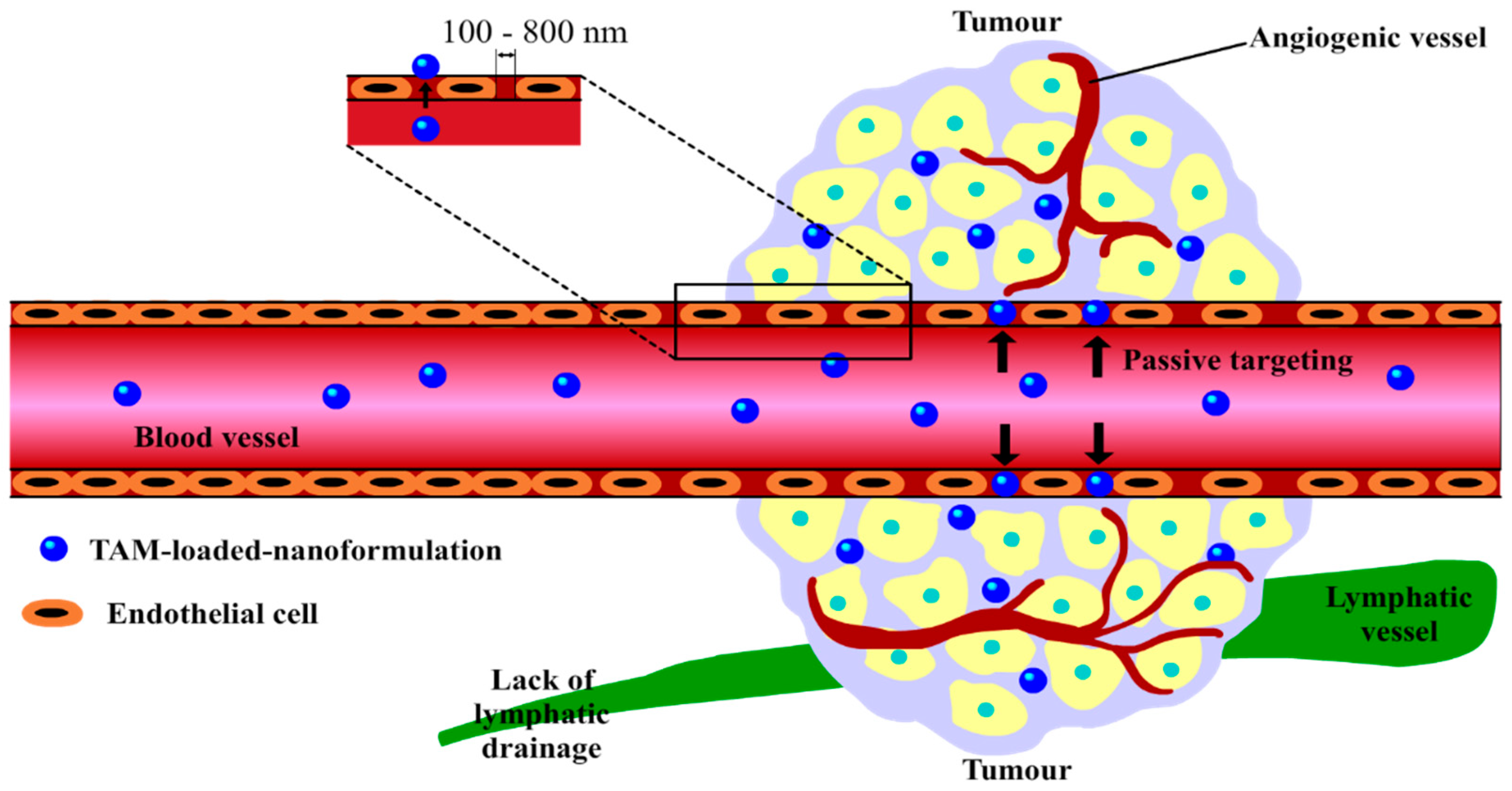
3. Nano-Enabled-Formulations Containing TAM
3.1. Liposomes
3.2. Micelles
3.3. Other Nanoparticles
3.4. Other TAM-Loaded Nanoformulations
3.5. TAM as a Targeting Vector
4. Authors’ Opinion on the Future of Nanoformulated TAM
Author Contributions
Funding
Conflicts of Interest
References
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Gene Funct. Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer. J. Clin. 2016, 66, 7–30. [Google Scholar]
- Tyczynski, J.E.; Plesko, I.; Aareleid, T.; Primic-Zakelj, M.; Dalmas, M.; Kurtinaitis, J.; Stengrevics, A.; Parkin, D.M. Breast cancer mortality patterns and time trends in 10 new EU member states: Mortality declining in young women, but still increasing in the elderly. Int. J. Cancer 2004, 112, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer in Australia Statistics. Available online: www.breast-cancer.canceraustralia.gov.au/statistics (accessed on 1 February 2020).
- Dixon, J.M.; Macaskill, E.J. Management of Benign Breast Disease. In Breast Disease; Springer: Berlin, Germany, 2014; Volume 2015, pp. 51–77. [Google Scholar]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various Types And Management Of Breast Cancer: An Overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Cancer Australia. Tests for Breast Cancer. Available online: www.breast-cancer.canceraustralia.gov.au/diagnosis/tests (accessed on 1 February 2020).
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Acharya, S.; Hsieh, S.; Michalski, J.; Shinohara, E.T.; Perkins, S.M.; Information, P.E.K.F.C. Distance to Radiation Facility and Treatment Choice in Early-Stage Breast Cancer. Int. J. Radiat. Oncol. 2016, 94, 691–699. [Google Scholar] [CrossRef]
- Sun, L.; Legood, R.; Dos-Santos-Silva, I.; Gaiha, S.; Sadique, Z. Global treatment costs of breast cancer by stage: A systematic review. PLoS ONE 2018, 13, e0207993. [Google Scholar] [CrossRef] [Green Version]
- Sugerman, D.T. Chemotherapy. JAMA 2013, 310, 218. [Google Scholar] [CrossRef] [Green Version]
- Lancet, T. Breast cancer targeted therapy: Successes and challenges. Lancet 2017, 389, 2350. [Google Scholar] [CrossRef]
- Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [Google Scholar] [CrossRef]
- Tierney, A.J.; Leonard, R.C.; Taylor, J.; Closs, S.J.; Chetty, U.; Rodger, A. Side effects expected and experienced by women receiving chemotherapy for breast cancer. BMJ 1991, 302, 272. [Google Scholar] [CrossRef] [Green Version]
- Hung, M.-H.; Liu, C.-J.; Teng, C.-J.; Hu, Y.-W.; Yeh, C.-M.; Chen, S.-C.; Chien, S.-H.; Hung, Y.-P.; Shen, C.-C.; Chen, T.-J.; et al. Risk of Second Non-Breast Primary Cancer in Male and Female Breast Cancer Patients: A Population-Based Cohort Study. PLoS ONE 2016, 11, e0148597. [Google Scholar] [CrossRef]
- Paterni, I.; Bertini, S.; Granchi, C.; Macchia, M.; Minutolo, F. Estrogen receptor ligands: A patent review update. Expert Opin. Ther. Patents 2013, 23, 1247–1271. [Google Scholar] [CrossRef]
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef]
- Omoto, Y.; Iwase, H. Clinical significance of estrogen receptor β in breast and prostate cancer from biological aspects. Cancer Sci. 2015, 106, 337–343. [Google Scholar] [CrossRef]
- Yip, C.-H.; Rhodes, A. Estrogen and progesterone receptors in breast cancer. Futur. Oncol. 2014, 10, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Elledge, R.M.; Green, S.; Pugh, R.; Allred, D.C.; Clark, G.M.; Hill, J.; Ravdin, P.; Martino, S.; Osborne, C.K. Estrogen receptor (ER) and progesterone receptor (PgR), by ligand-binding assay compared with ER, PgR and pS2, by immuno-histochemistry in predicting response to tamoxifen in metastatic breast cancer: A Southwest Oncology Group Study. Int. J. Cancer. 2000, 89, 111–117. [Google Scholar] [CrossRef]
- Jung, J.; Lee, S.H.; Park, M.; Youn, J.H.; Shin, S.H.; Gwak, H.S.; Yoo, H. Discordances in ER, PR, and HER2 between primary breast cancer and brain metastasis. J. Neuro-Oncology 2017, 137, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Vaz-Luis, I.; Winer, E.P.; Lin, N.U. Human epidermal growth factor receptor-2-positive breast cancer: Does estrogen receptor status define two distinct subtypes? Ann. Oncol. 2013, 24, 283–291. [Google Scholar] [CrossRef]
- Buonomo, O.C.; Caredda, E.; Portarena, I.; Vanni, G.; Orlandi, A.; Bagni, C.; Petrella, G.; Palombi, L.; Orsaria, P. New insights into the metastatic behavior after breast cancer surgery, according to well-established clinicopathological variables and molecular subtypes. PLoS ONE 2017, 12, e0184680. [Google Scholar] [CrossRef] [Green Version]
- Steiman, J.; Peralta, E.A.; Louis, S.; Kamel, O. Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am. J. Surg. 2013, 206, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H. Tamoxifen: Molecular basis of use in cancer treatment and prevention. Gen. Pharmacol. 1996, 5, 923. [Google Scholar]
- Kilbourn, B.T.; Mais, R.H.B.; Owston, P.G. Identification of isomers of a substituted triarylethylene: The crystal structure of 1-p-(2-dimethylaminoethoxyphenyl)-1,2-cis-diphenylbut-1-ene hydrobromide. Chem. Commun. 1968, 1, 291. [Google Scholar] [CrossRef]
- Harper, M.J.K.; Walpole, A.L. Contrasting Endocrine Activities of cis and trans Isomers in a Series of Substituted Triphenylethylenes. Nature 1966, 212, 87. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.J.K.; Walpole, A.L. A New Derivative Of Triphenylethylene: Effect On Implantation And Mode Of Action In Rats. Reproduction 1967, 13, 101–119. [Google Scholar] [CrossRef]
- Harper, M.J.K.; Walpole, A.L.; Heap, R.B.; Holzbauer, M.; Newport, H.M. Mode of Action of I.C.I. 46,474 in Preventing Implantation in Rats. J. Endocrinol. 1967, 37, 83–92. [Google Scholar] [CrossRef]
- Klopper, A.; Hall, M. New Synthetic Agent for the Induction of Ovulation: Preliminary Trials in Women. BMJ 1971, 1, 152–154. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.G.; Ellis, J.D. The Induction Of Ovulation by Tamoxifen. BJOG: Int. J. Obstet. Gynaecol. 1973, 80, 844–847. [Google Scholar] [CrossRef]
- Obrero, M. Estrogen Receptor-dependent and Estrogen Receptor-independent Pathways for Tamoxifen and 4-Hydroxytamoxifen-induced Programmed Cell Death. J. Boil. Chem. 2002, 277, 45695–45703. [Google Scholar] [CrossRef] [Green Version]
- Furr, B.; Jordan, V. The pharmacology and clinical uses of tamoxifen. Pharmacol. Ther. 1984, 25, 127–205. [Google Scholar] [CrossRef]
- Lippman, M.E.; Bolan, G. Oestrogen-responsive human breast cancer in long term tissue culture. Nature 1975, 256, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.R.; Kang, Y.; Greaves, B. Effects of tamoxifen on growth and apoptosis of oestrogen-dependent and -independent human breast cancer cells. Ann. Surg. Oncol. 1995, 2, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Maximov, P.Y.; McDaniel, R.E.; Jordan, V.C. Tamoxifen Goes Forward Alone. In Bipolar Depression: Molecular Neurobiology, Clinical Diagnosis, and Pharmacotherapy; Springer: Berlin, Germany, 2013; Volume 2013, pp. 31–46. [Google Scholar]
- Viedma-Rodriguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Esparza-Garrido, R.R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (Review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLozier, T.; Julien, J.-P.; Juret, P.; Veyret, C.; Couëtte, J.-E.; Graïc, Y.; Ollivier, J.-M.; De Ranieri, E. Adjuvant tamoxifen in postmenopausal breast cancer: Preliminary results of a randomized trial. Breast Cancer Res. Treat. 1986, 7, 105–109. [Google Scholar] [CrossRef]
- Tormey, U.C.; Jordan, V.C. Long-term tamoxifen adjuvant therapy in node-positive breast cancer: A metabolic and pilot clinical study. Breast Cancer Res. Treat. 1984, 4, 297–302. [Google Scholar] [CrossRef]
- Tormey, U.C.; Rasmussen, P.; Jordan, V.C. Long-term adjuvant tamoxifen study: Clinical update. Breast Cancer Res. Treat. 1987, 9, 157–158. [Google Scholar] [CrossRef]
- The Scottish Trial. Adjuvant tamoxifen in the management of operable breast cancer: Breast cancer trials committee Lancet. Lancet 1987, 2, 171–175. [Google Scholar]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Alencar, V.H.M.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.J.; Prescott, R.J.; Forrest, A.P.M. Scottish adjuvant tamoxifen trial: A randomized study updated to 15 years. J. Natl. Cancer Inst. 2001, 93, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.E.; Clark, E.; Jordan, V. Evidence for the metabolic activation of non-steroidal antioestrogens: A study of structure-activity relationships. Br. J. Pharmacol. 1980, 71, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.T.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for Prevention of Breast Cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef] [PubMed]
- Quirke, V.M. Tamoxifen from Failed Contraceptive Pill to Best-Selling Breast Cancer Medicine: A Case-Study in Pharmaceutical Innovation. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Maximov, P.Y.; McDaniel, R.E.; Jordan, V.C. Carcinogenesis and Tamoxifen. In Tamoxifen: Pioneering Medicine in Breast Cancer; Springer: Berlin, Germany, 2013; pp. 101–114. [Google Scholar]
- Bissett, D.; Davis, J.; George, W. Gynaecological monitoring during tamoxifen therapy. Lancet 1994, 344, 1244. [Google Scholar] [CrossRef]
- Magriples, U.; Naftolin, F.; Schwartz, P.E.; Carcangiu, M.L. High-grade endometrial carcinoma in tamoxifen-treated breast cancer patients. J. Clin. Oncol. 1993, 11, 485–490. [Google Scholar] [CrossRef] [PubMed]
- White, I.N. The tamoxifen dilemma. Carcinogenesis 1999, 20, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Jena, S.K.; Sangamwar, A.T. Polymeric micelles: A promising tool for tamoxifen delivery in cancer? Ther. Deliv. 2017, 8, 109–111. [Google Scholar] [CrossRef] [Green Version]
- Day, C.M.; Parikh, A.; Song, Y.; Garg, S. Nanotechnology and Nature’s Miracle Compound: Curcumin. In NanoNutraceuticals; Taylor & Francis: Milton Park, UK, 2018; pp. 213–254. [Google Scholar]
- Xuan, Q.-J.; Wang, J.-X.; Nanding, A.; Wang, Z.-P.; Liu, H.; Lian, X.; Zhang, Q.-Y. Tumor-Associated Macrophages are Correlated with Tamoxifen Resistance in the Postmenopausal Breast Cancer Patients. Pathol. Oncol. Res. 2014, 20, 619–624. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. P T Peer-Rev. J. Formul. Manag. 2017, 42, 742–755. [Google Scholar]
- Dai, Y.; Xu, C.; Sun, X.; Chen, X. Nanoparticle design strategies for enhanced anticancer therapy by exploiting the tumour microenvironment. Chem. Soc. Rev. 2017, 46, 3830–3852. [Google Scholar] [CrossRef]
- Sharma, A.; Jain, N.; Sareen, R. Nanocarriers for Diagnosis and Targeting of Breast Cancer. BioMed Res. Int. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layek, B.; Mukherjee, B. Tamoxifen Citrate Encapsulated Sustained Release Liposomes: Preparation and Evaluation of Physicochemical Properties. Sci. Pharm. 2010, 78, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-L.; Chen, C.-H.; Wu, H.-Y.; Tsai, N.-M.; Jian, T.-Y.; Chang, Y.-C.; Lin, C.-H.; Wu, C.-H.; Hsu, F.-T.; Leung, T.K.; et al. Inhibition of breast cancer with transdermal tamoxifen-encapsulated lipoplex. J. Nanobiotechnol. 2016, 14, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosco, D.; Paolino, D.; Cilurzo, F.; Casale, F.; Fresta, M. Gemcitabine and tamoxifen-loaded liposomes as multidrug carriers for the treatment of breast cancer diseases. Int. J. Pharm. 2012, 422, 229–237. [Google Scholar] [CrossRef]
- Deepak, M.; Basavaraj, N.; Manvi, F. Formulation and characterization of tamoxifen loaded stealth liposomes for breast cancer. Int. J. Drug Dev. Res. 2013, 5, 271. [Google Scholar]
- Bhatia, A.; Bhushan, S.; Singh, B.; Katare, O.P. Studies on tamoxifen encapsulated in lipid vesicles: Effect on the growth of human breast cancer MCF-7 cells. J. Liposome Res. 2009, 19, 169–172. [Google Scholar] [CrossRef]
- Guo, J.; Lü, W.-L. Effects of stealth liposomal daunorubicin plus tamoxifen on the breast cancer and cancer stem cells. J. Pharm. Pharm. Sci. 2010, 13, 136–151. [Google Scholar] [CrossRef] [Green Version]
- Jose, A.; Ninave, K.M.; Karnam, S.; Venuganti, V.V.K. Temperature-sensitive liposomes for co-delivery of tamoxifen and imatinib for synergistic breast cancer treatment. J. Liposome Res. 2018, 29, 153–162. [Google Scholar] [CrossRef]
- Rana, S.; Bhattacharjee, J.; Barick, K.C.; Verma, G.; Hassan, P.A.; Yakhmi, J.V. Interfacial engineering of nanoparticles for cancer therapeutics. Nanostruct. Cancer Ther. 2017, 2017, 177–209. [Google Scholar]
- Kim, S.; Shi, Y.; Kim, J.Y.; Park, K.; Cheng, J.-X. Overcoming the barriers in micellar drug delivery: Loading efficiency, in vivostability, and micelle–cell interaction. Expert Opin. Drug Deliv. 2009, 7, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Soga, O.; Van Nostrum, C.F.; Fens, M.; Rijcken, C.J.; Schiffelers, R.M.; Storm, G.; Hennink, W.E. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J. Control. Release 2005, 103, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lukyanov, A.N.; Singhal, A.; Torchilin, V.P. Diacyllipid-Polymer Micelles as Nanocarriers for Poorly Soluble Anticancer Drugs. Nano Lett. 2002, 2, 979–982. [Google Scholar] [CrossRef]
- Lukyanov, A.N.; Gao, Z.; Torchilin, V.P. Micelles from polyethyleneglycol/phosphatidylethanolamine conjugates for tumour drug delivery. J. Control. Release. 2003, 91, 97–102. [Google Scholar] [CrossRef]
- Yadav, H.; Kumar, P.; Sharma, V.; Sharma, G.; Raza, K.; Katare, O.P. Enhanced efficacy and a better pharmacokinetic profile of tamoxifen employing polymeric micelles. RSC Adv. 2016, 6, 53351–53357. [Google Scholar] [CrossRef]
- Jena, S.K.; Sangamwar, A. Polymeric micelles of amphiphilic graft copolymer of α-tocopherol succinate-g-carboxymethyl chitosan for tamoxifen delivery: Synthesis, characterization and in vivo pharmacokinetic study. Carbohydr. Polym. 2016, 151, 1162–1174. [Google Scholar] [CrossRef]
- Thotakura, N.; Dadarwal, M.; Kumar, R.; Singh, B.; Sharma, G.; Kumar, P.; Katare, O.P.; Raza, K. Chitosan-palmitic acid based polymeric micelles as promising carrier for circumventing pharmacokinetic and drug delivery concerns of tamoxifen. Int. J. Boil. Macromol. 2017, 102, 1220–1225. [Google Scholar] [CrossRef]
- Thakur, C.K.; Thotakura, N.; Kumar, R.; Kumar, P.; Singh, B.; Chitkara, D.; Raza, K. Chitosan-modified PLGA polymeric nanocarriers with better delivery potential for tamoxifen. Int. J. Boil. Macromol. 2016, 93, 381–389. [Google Scholar] [CrossRef]
- Day, C.M.; Barclay, T.G.; Song, Y.; Garg, S. Swelling-controlled Drug Delivery Systems. In Biomaterials Science Series; The Royal Society of Chemistry: London, UK, 2018; Volume 2018, pp. 232–264. [Google Scholar]
- Agudelo, D.; Sanyakamdhorn, S.; Nafisi, S.; Tajmir-Riahi, H.A. Transporting Antitumor Drug Tamoxifen and Its Metabolites, 4-Hydroxytamoxifen and Endoxifen by Chitosan Nanoparticles. PLoS ONE 2013, 8, e60250. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; Maji, R.; Dey, N.S.; Satapathy, B.S.; Mondal, S. Preparation and characterization of Tamoxifen citrate loaded nanoparticles for breast cancer therapy. Int. J. Nanomed. 2014, 9, 3107–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumara, N.; Bharadwaj, M.; Madhusudhan, B. Tamoxifen citrate-loaded poly(d,l) lactic acid nanoparticles: Evaluation for their anticancer activity in vitro and in vivo. J. Biomater. Appl. 2016, 31, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Abbasalipourkabir, R.; Salehzadeh, A.; Abdullah, R. Tamoxifen-loaded solid lipid nanoparticles-induced apoptosis in breast cancer cell lines. J. Exp. Nanosci. 2015, 11, 1–14. [Google Scholar] [CrossRef]
- Eskiler, G.G.; Cecener, G.; Dikmen, G.; Egeli, U.; Tunca, B. Solid lipid nanoparticles: Reversal of tamoxifen resistance in breast cancer. Eur. J. Pharm. Sci. 2018, 120, 73–88. [Google Scholar] [CrossRef]
- Jain, S.; Heeralal, B.; Swami, R.; Swarnakar, N.K.; Jain, S. Improved Oral Bioavailability, Therapeutic Efficacy, and Reduced Toxicity of Tamoxifen-Loaded Liquid Crystalline Nanoparticles. AAPS Pharm. Sci. Tech. 2017, 19, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, V.; Balakumar, S.; Amutha, S. Biopolymer mediated nanoparticles synthesized from Adenia hondala for enhanced tamoxifen drug delivery in breast cancer cell line. ANSN 2017, 8, 35011. [Google Scholar]
- Nosrati, H.; Rashidi, N.; Danafar, H.; Manjili, H.K. Anticancer Activity of Tamoxifen Loaded Tyrosine Decorated Biocompatible Fe3O4 Magnetic Nanoparticles Against Breast Cancer Cell Lines. J. Inorg. Organomet. Polym. Mater. 2017, 28, 1178–1186. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-binding Specificity of Human Serum Albumin. J. Mol. Boil. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Ahmed Ouameur, A.; Diamantoglou, S.; Sedaghat-Herati, M.R.; Nafisi, S.; Carpentier, R.; Tajmir-Riahi, H.A. An overview of drug binding to human serum albumin: Protein folding and unfolding. Cell Biochem. Biophys. 2006, 45, 203–213. [Google Scholar] [CrossRef]
- Chanphai, P.; Vesper, A.; Bekale, L.; Berube, G.; Tajmir-Riahi, H. Transporting testosterone and its dimers by serum proteins. J. Photochem. Photobiol. B Boil. 2015, 153, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, P.; Kanakis, C.D.; Tarantilis, P.; Pollissiou, M.G.; Tajmir-Riahi, H.A. Resveratrol, Genistein, and Curcumin Bind Bovine Serum Albumin. J. Phys. Chem. B 2010, 114, 3348–3354. [Google Scholar] [CrossRef]
- Charbonneau, D.M.; Tajmir-Riahi, H.-A. Study on the Interaction of Cationic Lipids with Bovine Serum Albumin. J. Phys. Chem. B 2010, 114, 1148–1155. [Google Scholar] [CrossRef]
- N’Soukpoé-Kossi, C.; Sedaghat-Herati, R.; Ragi, C.; Hotchandani, S.; Tajmir-Riahi, H. Retinol and retinoic acid bind human serum albumin: Stability and structural features. Int. J. Boil. Macromol. 2007, 40, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Mandeville, J.-S.; Froehlich, E.; Tajmir-Riahi, H. Study of curcumin and genistein interactions with human serum albumin. J. Pharm. Biomed. Anal. 2009, 49, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Akdoğan, Y.; Reichenwallner, J.; Hinderberger, D. Evidence for Water-Tuned Structural Differences in Proteins: An Approach Emphasizing Variations in Local Hydrophilicity. PLoS ONE 2012, 7, e45681. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.; Benito-Miguel, M.; Iglesias, I.; Teijón, J.M.; Blanco, M.D. Tamoxifen-loaded thiolated alginate-albumin nanoparticles as antitumoral drug delivery systems. J. Biomed. Mater. Res. Part A 2012, 100, 1467–1476. [Google Scholar] [CrossRef]
- Bourassa, P.; Dubeau, S.; Maharvi, G.M.; Fauq, A.H.; Thomas, T.; Tajmir-Riahi, H. Locating the binding sites of anticancer tamoxifen and its metabolites 4-hydroxytamoxifen and endoxifen on bovine serum albumin. Eur. J. Med. Chem. 2011, 46, 4344–4353. [Google Scholar] [CrossRef]
- Safavi, M.S.; Shojaosadati, S.A.; Dorkoosh, F.A.; Jo, H.J.; Kwon, Y.; Lee, K.C.; Yang, H.G.; Park, E.J.; Na, D.H. The synthesis of tamoxifen-loaded albumin nanoparticles by homogenizers: Optimization and in vitro characterization. J. Drug Deliv. Sci. Technol. 2017, 41, 20–30. [Google Scholar] [CrossRef]
- Akim, A.M.; Tung, E.E.; Chong, P.P.; Hamzah, M.Y.; Dahlan, K.Z.M. Nanoparticle-Encapsulated Tamoxifen Inducing Cytotoxic Effect on Mcf-7 Breast Cancer Cell Lines. In 4th Latin American Congress on Biomedical Engineering 2007, Bioengineering Solutions for Latin America Health; Springer: Berlin, Germany, 2013; pp. 226–228. [Google Scholar]
- Othayoth, R.; Karthik, V.; Kumar, K.S. Development and characterization of chitosan-pluronic nanoparticles for tamoxifen delivery and cytotoxicity to MCF-7 cells. In Proceedings of the International Conference on Advanced Nanomaterials & Emerging Engineering Technologies, Chennai, India, 24–26 July 2013; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2013; pp. 394–401. [Google Scholar]
- Vivek, R.; Babu, V.N.; Thangam, R.; Subramanian, K.; Kannan, S. pH-responsive drug delivery of chitosan nanoparticles as Tamoxifen carriers for effective anti-tumor activity in breast cancer cells. Colloids Surfaces B Biointerfaces 2013, 111, 117–123. [Google Scholar] [CrossRef]
- Roberta, C.; Bisazza, A.; Bussano, R.; Trotta, M.; Civra, A.; Lembo, D.; Ranucci, E.; Ferruti, P. Poly(amidoamine)-Cholesterol Conjugate Nanoparticles Obtained by Electrospraying as Novel Tamoxifen Delivery System. J. Drug Deliv. 2011, 2011, 587604. [Google Scholar]
- Chauhan, D.; Srivastava, R. Synthesis and characterization of gold encapsulated and tamoxifen loaded PLGA nanoparticles for breast cancer theranostics. In Proceedings of the 2015 9th IEEE International Conference on Nano/Molecular Medicine & Engineering (NANOMED), Honolulu, HI, USA, 15–18 November 2015; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2015; pp. 143–146. [Google Scholar]
- Sarmah, J.K.; Mahanta, R.; Bhattacharjee, S.K.; Mahanta, R.; Biswas, A. Controlled release of tamoxifen citrate encapsulated in cross-linked guar gum nanoparticles. Int. J. Boil. Macromol. 2011, 49, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, D.B.; Chawla, J.S.; Amiji, M.M. Biodegradable Polymeric Nanoparticles for Tumor-Selective Tamoxifen Delivery: In Vitro and in Vivo Studies. In Proceedings of the MRS Proceedings; Cambridge University Press: Cambridge, UK, 2004; Volume 845. [Google Scholar]
- Jain, A.K.; Thanki, K.; Jain, S. Co-encapsulation of Tsame for 100amoxifen and Quercetin in Polymeric Nanoparticles: Implications on Oral Bioavailability, Antitumor Efficacy, and Drug-Induced Toxicity. Mol. Pharm. 2013, 10, 3459–3474. [Google Scholar] [CrossRef]
- Varthya, M.; Pawar, H.; Singh, C.; Dora, C.P.; Jena, S.K.; Suresh, S.; Mansingh, V.; Harish, P.; Charan, S.; Parkash, D.C.; et al. Development of Novel Polymer-Lipid Hybrid Nanoparticles of Tamoxifen: In Vitro and In Vivo Evaluation. J. Nanosci. Nanotechnol. 2016, 16, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ostad, S.N.; Dehnad, S.; Nazari, Z.E.; Fini, S.T.; Mokhtari, N.; Shakibaie, M.; Shahverdi, A.R. Cytotoxic Activities of Silver Nanoparticles and Silver Ions in Parent and Tamoxifen-Resistant T47D Human Breast Cancer Cells and Their Combination Effects with Tamoxifen against Resistant Cells. Avicenna J. Med Biotechnol. 2010, 2, 187–196. [Google Scholar] [PubMed]
- Majd, M.H.; Asgari, D.; Barar, J.; Valizadeh, H.; Kafil, V.; Abadpour, A.; Moumivand, E.; Mojarrad, S.J.; Rashidi, M.-R.; Coukos, G.; et al. Tamoxifen loaded folic acid armed PEGylated magnetic nanoparticles for targeted imaging and therapy of cancer. Colloids Surfaces B Biointerfaces 2013, 106, 117–125. [Google Scholar] [CrossRef]
- Fang, C.-L.; Al-Suwayeh, S.A.; Fang, J.-Y. Nanostructured Lipid Carriers (NLCs) for Drug Delivery and Targeting. Recent Patents Nanotechnol. 2013, 7, 41–55. [Google Scholar] [CrossRef]
- Beh, C.Y.; Abdullah, R.; Selvarajah, G.T.; Yazan, L.S.; Omar, A.R.; Foong, J.N.; How, C.W.; Foo, J.B. Enhanced anti-mammary gland cancer activities of tamoxifen-loaded erythropoietin-coated drug delivery system. PLoS ONE 2019, 14, e0219285. [Google Scholar] [CrossRef] [Green Version]
- How, C.W.; Rasedee, A.; Manickam, S.; Rosli, R. Tamoxifen-loaded nanostructured lipid carrier as a drug delivery system: Characterization, stability assessment and cytotoxicity. Colloids Surfaces B Biointerfaces 2013, 112, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Talat, M.; Upadhyay, P.; Omray, P.; Koteshwara, K.B.; Srivastava, O.N. Preparation and Characterization of Nanosuspension of Tamoxifen Citrate for Intra-Venous Administration in Drug Resistant Breast Cancer Cells. Adv. Sci. Lett. 2014, 20, 1483–1489. [Google Scholar] [CrossRef]
- Tagne, J.-B.; Kakumanu, S.; Ortiz, D.; Shea, T.; Nicolosi, R.J. A Nanoemulsion Formulation of Tamoxifen Increases Its Efficacy in a Breast Cancer Cell Line. Mol. Pharm. 2008, 5, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Buhleier, E.; Wehner, W.; Vögtle, F. “Cascade”- and “Nonskid-Chain-like” Syntheses of Molecular Cavity Topologies. Synthesis 1978, 1978, 155–158. [Google Scholar] [CrossRef]
- Matai, I.; Gopinath, P. Hydrophobic myristic acid modified PAMAM dendrimers augment the delivery of tamoxifen to breast cancer cells. RSC Adv. 2016, 6, 24808–24819. [Google Scholar] [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Rittner, K.; Benavente, A.; Bompard-Sorlet, A.; Heitz, F.; Divita, G.; Brasseur, R.; Jacobs, E. Fr New Basic Membrane-Destabilizing Peptides for Plasmid-Based Gene Delivery in Vitro and in Vivo. Mol. Ther. 2002, 5, 104–114. [Google Scholar] [CrossRef]
- Mecke, A.; Majoros, I.J.; Patri, A.K.; Baker, J.R.; Holl, M.B.; Orr, B.G. Lipid Bilayer Disruption by Polycationic Polymers: The Roles of Size and Chemical Functional Group. Langmuir 2005, 21, 10348–10354. [Google Scholar] [CrossRef]
- Chen, H.-T.; Neerman, M.F.; Parrish, A.; Simanek, E.E. Cytotoxicity, Hemolysis, and Acute in Vivo Toxicity of Dendrimers Based on Melamine, Candidate Vehicles for Drug Delivery. J. Am. Chem. Soc. 2004, 126, 10044–10048. [Google Scholar] [CrossRef]
- Domański, D.; Klajnert-Maculewicz, B.; Bryszewska, M. Influence of PAMAM dendrimers on human red blood cells. Bioelectrochemistry 2004, 63, 189–191. [Google Scholar] [CrossRef]
- Jain, K.; Kesharwani, P.; Gupta, U.; Jain, N.K. Dendrimer toxicity: Let’s meet the challenge. Int. J. Pharm. 2010, 394, 122–142. [Google Scholar] [CrossRef]
- Majd, M.H.; Akbarzadeh, A.; Sargazi, A. Evaluation of host–guest system to enhance the tamoxifen efficiency. Artif. Cells Nanomed. Biotechnol. 2016, 45, 1–7. [Google Scholar]
- Oskoueian, A.; Matori, K.A.; Bayat, S.; Oskoueian, E.; Ostovan, F.; Toozandehjani, M. Fabrication, Characterization, and Functionalization of Single-Walled Carbon Nanotube Conjugated with Tamoxifen and Its Anticancer Potential against Human Breast Cancer Cells. J. Nanomater. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Badea, I.; Kaur, R. Nanodiamonds as novel nanomaterials for biomedical applications: Drug delivery and imaging systems. Int. J. Nanomed. 2013, 8, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, J.; Li, W.; Zhang, Y.; Yang, X.; Chen, N.; Sun, Y.; Zhao, Y.; Fan, C.; Huang, Q. The Biocompatibility of Nanodiamonds and Their Application in Drug Delivery Systems. Theranostics 2012, 2, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Mostofizadeh, A.; Li, Y.; Song, B.; Huang, Y. Synthesis, Properties, and Applications of Low-Dimensional Carbon-Related Nanomaterials. J. Nanomater. 2011, 2011, 1–21. [Google Scholar] [CrossRef]
- Xiao, J.; Duan, X.; Yin, Q.; Zhang, Z.; Yu, H.; Yu, H. Nanodiamonds-mediated doxorubicin nuclear delivery to inhibit lung metastasis of breast cancer. Biomater. 2013, 34, 9648–9656. [Google Scholar] [CrossRef]
- Ribelles, N.; Santonja, A.; Pajares, B.; Llácer, C.; Alba, E. The seed and soil hypothesis revisited: Current state of knowledge of inherited genes on prognosis in breast cancer. Cancer Treat. Rev. 2014, 40, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Bourassa, P.; Thomas, T.; Bariyanga, J.; Tajmir-Riahi, H. Breast anticancer drug tamoxifen and its metabolites bind tRNA at multiple sites. Int. J. Boil. Macromol. 2015, 72, 692–698. [Google Scholar] [CrossRef]
- Landeros-Martínez, L.-L.; Flores-Holguín, N.; Orrantia-Borunda, E.; Flores-Holguin, N. Construction of a Nanodiamond–Tamoxifen Complex as a Breast Cancer Drug Delivery Vehicle. J. Nanomater. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Torne, S.; Darandale, S.; Vavia, P.R.; Trotta, F.; Roberta, C. Cyclodextrin-based nanosponges: Effective nanocarrier for Tamoxifen delivery. Pharm. Dev. Technol. 2012, 18, 619–625. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.; El-Massik, M.; Abdallah, O.Y. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef]
- Ballerini, A.; Filgueira, C.; Nicolov, E.; Jain, P.; Bruno, G.; Hood, R.L.; Scaglione, F.; Grattoni, A. Sustained Delivery of Tamoxifen from a Nanofluidic Delivery Platform. Drug Deliv. Lett. 2017, 6, 127–133. [Google Scholar] [CrossRef]
- Jain, A.S.; Goel, P.N.; Shah, S.; Dhawan, V.V.; Nikam, Y.; Gude, R.P.; Nagarsenker, M.S. Tamoxifen guided liposomes for targeting encapsulated anticancer agent to estrogen receptor positive breast cancer cells: In vitro and in vivo evaluation. Biomed. Pharmacother. 2014, 68, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.; Mwakwari, S.C.; Sodji, Q.H.; Oyelere, A.; El-Sayed, M.A. Tamoxifen−Poly(ethylene glycol)−Thiol Gold Nanoparticle Conjugates: Enhanced Potency and Selective Delivery for Breast Cancer Treatment. Bioconjugate Chem. 2009, 20, 2247–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, T.G.; Day, C.M.; Petrovsky, N.; Garg, S. Review of polysaccharide particle-based functional drug delivery. Carbohydr. Polym. 2019, 221, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.A.; Thomas, E.; McLaughlin, R.A.; Flematti, G.; Fuller, R. A new selective fluorescent probe based on tamoxifen. Bioorganic Med. Chem. Lett. 2016, 26, 4879–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Song, M.-R.; Yuan, G.-K.; Ye, H.-N.; Tian, Y.; Huang, M.; Xue, J.-P.; Zhang, Z.; Liu, J. A Molecular Combination of Zinc(II) Phthalocyanine and Tamoxifen Derivative for Dual Targeting Photodynamic Therapy and Hormone Therapy. J. Med. Chem. 2017, 60, 6693–6703. [Google Scholar] [CrossRef]
- Rickert, E.L.; Oriana, S.; Hartman-Frey, C.; Long, X.; Webb, T.T.; Nephew, K.P.; Weatherman, R.V. Synthesis and Characterization of Fluorescent 4-Hydroxytamoxifen Conjugates with Unique Antiestrogenic Properties. Bioconjugate Chem. 2010, 21, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Caster, J.M.; Patel, A.N.; Zhang, T.; Wang, A.Z. Investigational nanomedicines in 2016: A review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 9, e1416. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Matthews, J.; Chang, P.H.; Niroui, F.; Dorkin, J.R.; Severgnini, M.; Anderson, D.G. In Vitro–In VivoTranslation of Lipid Nanoparticles for Hepatocellular siRNA Delivery. ACS Nano 2012, 6, 6922–6929. [Google Scholar] [CrossRef] [Green Version]
- Bharti, C.; Gulati, N.; Nagaich, U.; Pal, A.K. Mesoporous silica nanoparticles in target drug delivery system: A review. Int. J. Pharm. Investig. 2015, 5, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic Potential of Materials at the Nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Shvedova, A.A.; Kagan, V.E.; Fadeel, B. Close Encounters of the Small Kind: Adverse Effects of Man-Made Materials Interfacing with the Nano-Cosmos of Biological Systems. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 63–88. [Google Scholar] [CrossRef]
- Orrenius, S.; Nicotera, P.; Zhivotovsky, B. Cell Death Mechanisms and Their Implications in Toxicology. Toxicol. Sci. 2010, 119, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Cauda, V.; Argyo, C.; Bein, T. Impact of different PEGylation patterns on the long-term bio-stability of colloidal mesoporous silica nanoparticles. J. Mater. Chem. 2010, 20, 8693. [Google Scholar] [CrossRef]
- Yagüe, C.; Moros, M.; Grazú, V.; Arruebo, M.; Santamaría, J. Synthesis and stealthing study of bare and PEGylated silica micro- and nanoparticles as potential drug-delivery vectors. Chem. Eng. J. 2008, 137, 45–53. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Z.; Gao, F.; Yu, H.; Shi, J. In vivo Biodistribution and Urinary Excretion of Mesoporous Silica Nanoparticles: Effects of Particle Size and PEGylation. Small 2010, 7, 271–280. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef]
- Yang, Q.; Jones, S.; Parker, C.L.; Zamboni, W.C.; Bear, J.E.; Lai, S.K. Evading Immune Cell Uptake and Clearance Requires PEG Grafting at Densities Substantially Exceeding the Minimum for Brush Conformation. Mol. Pharm. 2014, 11, 1250–1258. [Google Scholar] [CrossRef]
- Lipsky, P.E.; Calabrese, L.; Kavanaugh, A.; Sundy, J.; Wright, D.; Wolfson, M.; Becker, M.A. Pegloticase immunogenicity: The relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res. Ther. 2014, 16, R60. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef]
- Ganson, N.J.; Kelly, S.J.; Scarlett, E.; Sundy, J.; Hershfield, M.S. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. Ther. 2005, 8, R12. [Google Scholar] [CrossRef] [Green Version]
- Longo, N.; Harding, C.O.; Burton, B.K.; Grange, R.K.; Vockley, J.; Wasserstein, M.; Rice, G.M.; Dorenbaum, A.; Neuenburg, J.K.; Musson, D.G.; et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: An open-label, multicentre, phase 1 dose-escalation trial. Lancet 2014, 384, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Hershfield, M.S.; Ganson, N.J.; Kelly, S.J.; Scarlett, E.L.; Jaggers, D.A.; Sundy, J.S. Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res. Ther. 2014, 16, R63. [Google Scholar] [CrossRef] [Green Version]
- Garay, R.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef]
- Ganson, N.J.; Povsic, T.J.; Sullenger, B.A.; Alexander, J.H.; Zelenkofske, S.L.; Sailstad, J.M.; Rusconi, C.P.; Hershfield, M.S. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J. Allergy Clin. Immunol. 2015, 137, 1610–1613.e7. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.R.; Webb, M.N.; Cadotte, T.H.; Gavette, M.N. Use of Targeted Liposome-based Chemotherapeutics to Treat Breast Cancer. Breast Cancer Basic Clin. Res. 2015, 9, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P. CHAPTER 1. Fundamentals of Stimuli-responsive Drug and Gene Delivery Systems. Biomater. Sci. Ser. 2018, 2018, 1–32. [Google Scholar]
- Raza, A.; Rasheed, T.; Nabeel, F.; Hayat, U.; Bilal, M.; Iqbal, H.M. Endogenous and Exogenous Stimuli-Responsive Drug Delivery Systems for Programmed Site-Specific Release. Molecules 2019, 24, 1117. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Chen, Q.; Chen, J.; Dong, Z.; Zhang, R.; Liu, J.; Liu, Z. Tumor-pH-Responsive Dissociable Albumin-Tamoxifen Nanocomplexes Enabling Efficient Tumor Penetration and Hypoxia Relief for Enhanced Cancer Photodynamic Therapy. Small 2018, 14, e1803262. [Google Scholar] [CrossRef]
- Meng, D.; Lei, H.; Zheng, X.; Han, Y.; Sun, R.; Zhao, D.; Liu, R. A temperature-sensitive phase-change hydrogel of tamoxifen achieves the long-acting antitumor activation on breast cancer cells. Onco. Targets Ther. 2019, 12, 3919–3931. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition a | Physiochemical Properties b | Advantages/Remarks | Ref |
|---|---|---|---|
| TAM, SPC, CH, span 20 | MD = 203.5 ± 19.5 nm PI = 0.442 ± 0.03 |
| [59] |
| TAM, PEG-PEI | PS < 270 nm ZP = 40 mV |
| [60] |
| TAM, CH and lipid (DMPC and/or DSPC) | PS = 482 + 0.013 nm to 887 ± 0.336 nm Smooth surface Multilamellar, disc shaped ZP = −37.2 to −41.2 mV |
| [62] |
| TAM, Saturated SPC, phospholipid GmbH | N/A |
| [63] |
| TAM, GEM, DPPC, CH, DMPG and DSPE-MPEG 2000c | PS = 150–200 nm Surface charge = 50 mV and −30 mV |
| [61] |
| TAM, DAU, EPC, PEG2000-DSPE, CH, SRB | EE = 95% (DAU) and 90% (TAM) PS = 100 nm |
| [64] |
| TAM, Imatinib, DPPC, MPPC | EE > 70% 80% drug release with temperature-responsive behaviour (within 30 min above transition temperature 34.9 °C). |
| [65] |
| Composition a | Physiochemical Properties b | Advantages/Remarks | Ref |
|---|---|---|---|
| TAM, PEG5000-PE | PS < 200 nm DL: 1:5 ratio drug/polymer w/w (20 wt % of TAM) |
| [69,70] |
| TAM, PLGA–PEG | Spherical PS = 76.4 ± 2.1 nm ZP = −4.89 mV (neutral) EE = 60.86 ± 3.21% |
| [71] |
| TAM, CS, TS | Spherical PS < 200 nm |
| [72] |
| TAM, Palmitic acid, CS | PS = 83.71 ± 0.15 nm ZP = +37 ± 0.14 mV EE = 93.76 ± 0.40% DL = 10.24 ± 0.21% |
| [73] |
| TAM, CS, PLGA | PS = 81.48 nm PDI = 0.209 ZP = +19.27 ± 4.34 mV |
| [74] |
| Composition a | Physiochemical Properties b | Advantages/Remarks | Ref |
|---|---|---|---|
| Examples of Polymeric NPs | |||
| TAM, LMW CMC, TS | LD = 8.08 ± 0.98% |
| [76] |
| TAM, PLGA | Smooth surface PS = 250–380 nm Maximal DL = 27.16 ± 2.08% |
| [77] |
| TAM, Poly(d,l-Lactide) | NPs prepared by emulsification solvent diffusion method PS = 271.4 nm ZP = 34 mV EE = 76.4% |
| [78] |
| TAM, NIPAAM, VP, PEG-DA | NPs prepared by gamma irradiation polymerization Core-shell structure Spherical or elliptical shaped Smooth surface Size distribution = 49.89 nm (SD ±1.82) |
| [96] |
| TAM, CS, Pluronic | Spherical shaped with positive charge MD = 150–300 nm EE = 8mg/mL TAM |
| [97] |
| TAM, CS | MD = 100–150 nm DL = 28% |
| [98] |
| TAM, PAA, CH | NPs prepared by electrospray technique PS <500 nm Spherical shaped DL = 40% |
| [99] |
| TAM, PLGA, AuNPs | EE = 30% |
| [100] |
| TAM, Guar gum (GG) | TAM, GG was crosslinked with glutaraldehyde NPs prepared by oil in water (o/w) emulsion polymer cross-linking method DL = 15% PS = 200–300 nm |
| [101] |
| TAM, PCL (MW ∼ 15, 000), Pluronic® F-68, F-108, PEO, PPO | NPs prepared by solvent displacement Spherical shaped ZP ∼25 mV PS = 200–300 nm |
| [102] |
| TAM, QT, PLGA | PS = 185.3 ± 1.20 nm PDI = 0.184 ± 0.004 EE = 67.16 ± 1.24% for TAM, 68.60 ± 1.58% for QT |
| [103] |
| Example of Polymer-lipid Hybrid NPs | |||
| TAM, Tween 80, CS, Lecithin | NPs prepared by modified solvent emulsification-evaporation method PS = 169.66 ± 4.84 nm |
| [104] |
| Examples of protein base-based NPs | |||
| TAM, SA, thiolated alginate (alginate-cysteine conjugate) | MD = 446, 430 and 498 nm ZP = −37 ± 9, −26 ± 7, and −7.2 ± 0.3 mV |
| [93] |
| TAM, 4-OHT, Endoxifen, HSA, BSA | EE = 45–52% for each drug-protein conjugate Free binding energy of hydrogen bonding was 11.79 to −11.25 Kcal/mol (drug-HSA) and −13.79 to −12.72 Kcal/mol (drug-BSA conjugates) |
| [94] |
| TAM, albumin | NPs prepared by HPH and HSH DL = 14.2 ± 1.9% (HPH); DL = 11.6 ± 2.3% (HSH) |
| [95] |
| Examples of SLNs | |||
| TAM, Hydrogenated palm oil, Hydrogenated soybean lecithin | NPs prepared by HPH PS = 251.65 ± 33.02 nm ZP = +10.16 ± 0.22 |
| [79] |
| TAM, Stearic acid 5%, Tween 80 2.5% | NPs prepared by HH Circular shaped PS = 2283 ± 1.88 nm PDI = 0.298 and Tam-SLNs |
| [80] |
| Example of Liquid Crystalline NPs | |||
| TAM, Glyceryl monooleate, Phytantriol, Oleic acid | Hexagonal PS = 154.93–235.76 nm PDI = 0.18–0.24 nm EE = 79–81% |
| [81] |
| Examples of Precious Metal NPs | |||
| TAM, Adenia hondala tuber extract, CS, AgNPs | MD = 60–140 nm |
| [82] |
| TAM, Ag+, AgNPs | PS = 1–28 nm |
| [105] |
| Examples of Magnetic NPs | |||
| TAM, Tyrosin, Fe3O4 | ZP = − 12.8 mV PS = 22.19 ± 3.58 nm DL = 11.34 ± 0.09% EE = 51.21 ± 0.41% |
| [83] |
| TAM, Fe3O4, APS-PEG-BrAc | PS = 40 nm DL = 49.1% |
| [106] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, C.M.; Hickey, S.M.; Song, Y.; Plush, S.E.; Garg, S. Novel Tamoxifen Nanoformulations for Improving Breast Cancer Treatment: Old Wine in New Bottles. Molecules 2020, 25, 1182. https://doi.org/10.3390/molecules25051182
Day CM, Hickey SM, Song Y, Plush SE, Garg S. Novel Tamoxifen Nanoformulations for Improving Breast Cancer Treatment: Old Wine in New Bottles. Molecules. 2020; 25(5):1182. https://doi.org/10.3390/molecules25051182
Chicago/Turabian StyleDay, Candace M., Shane M. Hickey, Yunmei Song, Sally E. Plush, and Sanjay Garg. 2020. "Novel Tamoxifen Nanoformulations for Improving Breast Cancer Treatment: Old Wine in New Bottles" Molecules 25, no. 5: 1182. https://doi.org/10.3390/molecules25051182