
The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions



Abstract
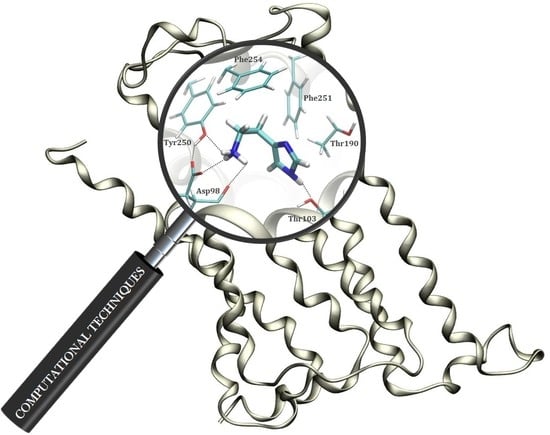
:
1. Introduction
2. Results and Discussion
2.1. Docking Simulation
2.2. Molecular Dynamics Simulation of Histamine in Aqueous Solution
2.3. Molecular Dynamics Simulation within the H2 Receptor
2.4. Quantum-Chemical Calculations
3. Computational Details
3.1. Docking Analysis
3.2. Molecular Dynamics Simulation
3.3. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walter, M.; Stark, H. Histamine receptor subtypes: A century of rational drug design. Front. Biosci. 2012, 4, 461–488. [Google Scholar] [CrossRef]
- Cong, X.; Topin, J.; Golebiowski, J. Class A GPCRs: Structure, function, modeling and structure-based ligand design. Curr. Pharm. Des. 2017, 23, 4390–4409. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Langmead, C.J.; Mason, J.S.; Marshall, F.H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011, 54, 4283–4311. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, A.; Solis, G.P.; Hersch, M.; Koval, A.; Kryuchkov, M.; Bergmann, S.; Katanaev, V.L. High capacity in G protein-coupled receptor signaling. Nat. Commun. 2018, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Ubbelohde, A.R.; Gallagher, K.J. Acid-base effects in hydrogen bonds in crystals. Acta Cryst. 1955, 8, 71–83. [Google Scholar] [CrossRef]
- De Souza, J.M.; Freire, P.T.C.; Bordallo, H.N.; Argyriou, D.N. Structural isotopic effects in the smallest chiral amino acid: Observation of a structural phase transition in fully deuterated alanine. J. Phys. Chem. B 2007, 111, 5034–5039. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, X.; Yu, C.-H.; Yao, Y.-F.; Zhang, W. Geometric isotope effect of deuteration in a hydrogen-bonded host-guest crystal. Nat. Commun. 2018, 9, 481. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Wang, Z.; McElmurry, B.A.; Willaert, F.F.; Lucchese, R.R.; Bevan, J.W.; Suenram, R.D.; Lovas, F.J. A ground state morphed intermolecular potential for the hydrogen bonded and van der Waals isomers in OC:HI and a prediction of an anomalous deuterium isotope effect. J. Chem. Phys. 2010, 133, 184305. [Google Scholar] [CrossRef]
- Mishra, A.K.; Murli, C.; Sharma, S.M. High pressure Raman spectroscopic study of deuterated γ-glycine. J. Phys. Chem. B 2008, 112, 15867–15874. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.O.; Freire, P.T.C.; Bordallo, H.N.; Lima, J.A., Jr.; Melo, F.E.A.; Mendes Filho, J.; Argyriou, D.N.; Lima, R.J.C. High-pressure Raman spectra of deuterated L-alanine crystal. J. Raman Spectrosc. 2009, 40, 958–963. [Google Scholar] [CrossRef]
- Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Benedict, H.; Schah-Mohammedi, P.; Limbach, H.-H. Hydrogen/deuterium isotope effects on the NMR chemical shifts and geometries of intermolecular low-barrier hydrogen-bonded complexes. J. Am. Chem. Soc. 1996, 118, 4094–4101. [Google Scholar] [CrossRef]
- Yarnell, A.T. Heavy-hydrogen drugs turn heads, again. Chem. Eng. News 2009, 87, 36–39. [Google Scholar] [CrossRef]
- Halford, B. Deuterium switcheroo breathes life into old drugs. Chem. Eng. News 2016, 94, 32–36. [Google Scholar]
- Belleau, B.; Burba, J.; Pindell, M.; Reiffenstein, J. Effect of Deuterium Substitution in Sympathomimetic Amines on Adrenergic Responses. Science 1961, 133, 102–104. [Google Scholar] [CrossRef]
- Falconnet, J.B.; Brazier, J.L.; Desage, M. Synthesis of seven deuteromethyl-caffeine analogues; observation of deuterium isotope effects on CMR analysis. J. Label. Compd. Radiopharm. 1986, 23, 267–276. [Google Scholar] [CrossRef]
- Brazier, J.L.; Ribon, B.; Falconnet, J.B.; Cherrah, Y.; Benchekroun, Y. Etude et utilisation des effets isotopiques en pharmacologie. Therapie 1987, 42, 445–450. [Google Scholar]
- Cherrah, Y.; Falconnet, J.B.; Desage, M.; Brazier, J.L.; Zini, R.; Tillement, J.P. Study of deuterium isotope effects on protein binding by gas chromatography/mass spectrometry. Caffeine and deuterated isotopomers. Biomed. Environ. Mass Spectrom. 1987, 14, 653–657. [Google Scholar] [CrossRef]
- Cherrah, Y.; Zini, R.; Falconnet, J.B.; Desage, M.; Tillement, J.P.; Brazier, J.L. Study of deutero-isotopomer-induced inhibition of caffeine and phenobarbitone binding to human serum albumin. Biochem. Pharmacol. 1988, 37, 1311–1315. [Google Scholar] [CrossRef]
- Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 2017, 35, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Bowers, S.G.; Truong, A.P.; Probst, G. The Role and Significance of Unconventional Hydrogen Bonds in Small Molecule Recognition by Biological Receptors of Pharmaceutical Relevance. Curr. Pharm. Des. 2007, 13, 3476–3493. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Mavri, J. Microsolvation of the histamine monocation in aqueous solution: The effect on structure, hydrogen bonding ability and vibrational spectrum. New J. Chem. 2012, 36, 954–962. [Google Scholar] [CrossRef]
- Stare, J.; Mavri, J.; Grdadolnik, J.; Zidar, J.; Maksić, Z.B.; Vianello, R. Hydrogen bond dynamics of histamine monocation in aqueous solution: Car–Parrinello molecular dynamics and vibrational spectroscopy study. J. Phys. Chem. B 2011, 115, 5999–6010. [Google Scholar] [CrossRef] [PubMed]
- Kržan, M.; Vianello, R.; Maršavelski, A.; Repič, M.; Zakšek, M.; Kotnik, K.; Fijan, E.; Mavri, J. The quantum nature of drug-receptor interactions: Deuteration changes binding affinities for histamine receptor ligands. PLoS ONE 2016, 11, e0154002. [Google Scholar] [CrossRef] [Green Version]
- Kržan, M.; Keuschler, J.; Mavri, J.; Vianello, R. Relevance of Hydrogen Bonds for the Histamine H2 Receptor-Ligand Interactions: A Lesson from Deuteration. Biomolecules 2020, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.C. The power issue: Determination of KB or Ki from IC50—A closer look at the Cheng–Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 2001, 46, 61–71. [Google Scholar] [CrossRef]
- Singh, V.; Gohil, N.; Ramírez-García, R. New insight into the control of peptic ulcer by targeting the histamine H2 receptor. J. Cell Biochem. 2018, 119, 2003–2011. [Google Scholar] [CrossRef]
- Strasser, A.; Wittmann, H.J. Molecular Modelling Approaches for the Analysis of Histamine Receptors and Their Interaction with Ligands. In Histamine and Histamine Receptors in Health and Disease. Handbook of Experimental Pharmacology; Hattori, Y., Seifert, R., Eds.; Springer: Cham, Switzerland, 2017; Volume 241, pp. 31–61. [Google Scholar]
- Sun, X.; Li, Y.; Li, W.; Xu, Z.; Tang, Y. Computational investigation of interactions between human H2 receptor and its agonists. J. Mol. Graph. Model. 2011, 29, 693–701. [Google Scholar] [CrossRef]
- Schreeb, A.; Łażewska, D.; Dove, S.; Buschauer, A.; Kieć-Kononiwcz, K.; Stark, H. Histamine H4 receptor ligands. In Histamine H4 Receptor: A Novel Drug Target for Immunoregulation and Inflammation; Stark, H., Ed.; Versita: London, UK, 2013; pp. 21–61. [Google Scholar]
- Collado, J.A.; Tuñón, I.; Silla, E.; Ramírez, F.J. Vibrational Dynamics of Histamine Monocation in Solution: An Experimental (FT-IR, FT-Raman) and Theoretical (SCRF-DFT) Study. J. Phys. Chem. A 2000, 104, 2120–2131. [Google Scholar] [CrossRef]
- Collado, J.A.; Ramírez, F.J. Vibrational spectra and assignments of histamine dication in the solid state and in solution. J. Raman Spectrosc. 2000, 31, 925–931. [Google Scholar] [CrossRef]
- Drozdzewski, P.; Kordon, E. Isotope effects in the far-infrared spectra of histamine complexes with palladium(II). Vib. Spectrosc. 2000, 24, 243–248. [Google Scholar] [CrossRef]
- Xerri, B.; Flament, J.-P.; Petitjean, H.; Berthomieu, C.; Berthomieu, D. Vibrational modeling of copper-histamine complexes: Metal-ligand IR modes investigation. J. Phys. Chem. B 2009, 113, 15119–15127. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Birdsall, N.J. Cloning and structure-function of the H2 histamine receptor. Trends Pharmacol. Sci. 1991, 12, 9–10. [Google Scholar] [CrossRef]
- Gantz, I.; DelValle, J.; Wang, L.D.; Tashiro, T.; Munzert, G.; Guo, Y.J.; Konda, Y.; Yamada, T. Molecular basis for the interaction of histamine with the histamine H2 receptor. J. Biol. Chem. 1992, 267, 20840–20843. [Google Scholar]
- Ramesh Kumara, G.; Gokul Raj, S.; Saxena, A.; Karnal, A.K.; Raghavalu, T.; Mohan, R. Deuteration effects on structural, thermal, linear and nonlinear properties of L-threonine single crystals. Mater. Chem. Phys. 2008, 108, 359–363. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, T.; Wei, J. Homology Modeling and Antagonist Binding Site Study of the Human Histamine H2 Receptor. Med. Chem. 2012, 8, 1084–1092. [Google Scholar]
- Strasser, A. Molecular modeling and QSAR-based design of histamine receptor ligands. Expert Opin. Drug Discov. 2009, 4, 1061–1075. [Google Scholar] [CrossRef]
- Catalan, J.; Abboud, J.L.M.; Elguero, J. Basicity and Acidity of Azoles. Adv. Heterocycl. Chem. 1897, 41, 187–274. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N∙log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational Insight into the Mechanism of the Irreversible Inhibition of Monoamine Oxidase Enzymes by the Antiparkinsonian Propargylamine Inhibitors Rasagiline and Selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines—Harmine and Cinnamic Acid Hybrids as Novel Antiplasmodial Hits. Eur. J. Med. Chem. 2020, 187, 111927. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein-Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras-Raf and Ras-RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Georgieva, P.; Yu, J.G.; Himo, F. Mechanism of mycolic acid cyclopropane synthase: A theoretical study. Biochemistry 2011, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 36, 7057–7065. [Google Scholar] [CrossRef]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef]
- Himo, F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modeling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Brás, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wires Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Quesne, M.G.; Silveri, F.; de Leeuw, N.H.; Catlow, C.R.A. Advances in sustainable catalysis: A computational perspective. Front. Chem. 2019, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Gupta, M. Deuteration as a tool for optimization of metabolic stability and toxicity of drugs. Glob. J. Pharm. Sci. 2017, 1, 555566. [Google Scholar]
- Tung, R.D. Deuterium medicinal chemistry comes of age. Future Med. Chem. 2016, 8, 491–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Don, C.G.; Riniker, S. Scents and sense: In silico perspectives on olfactory receptors. J. Comput. Chem. 2014, 35, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histamine | |||
|---|---|---|---|
| ΔGBIND = −14.2 kcal mol−1 | |||
| Favorable Contributions | Unfavorable Contributions | ||
| Asp98 | −5.45 | Leu193 | +0.02 |
| Val99 | −1.78 | Gly157 | +0.02 |
| Phe254 | −1.60 | Lys166 | +0.02 |
| Thr103 | −1.29 | Lys83 | +0.02 |
| Phe251 | −0.97 | Val273 | +0.02 |
| Thr190 | −0.84 | Asn108 | +0.02 |
| Tyr250 | −0.83 | Lys88 | +0.02 |
| Gly187 | −0.64 | Tyr192 | +0.03 |
| Asp186 | −0.64 | Arg260 | +0.03 |
| Met100 | −0.20 | Ala253 | +0.05 |
| Glu270 | −0.16 | Ser153 | +0.05 |
| Cys102 | −0.14 | Arg161 | +0.06 |
| Glu267 | −0.11 | Lys173 | +0.11 |
| Val178 | −0.10 | Gln177 | +0.11 |
| Tyr94 | −0.08 | Gly183 | +0.13 |
| Val189 | −0.08 | Arg257 | +0.15 |
| Val185 | −0.07 | Leu97 | +0.19 |
| Val255 | −0.06 | Lys175 | +0.29 |
| Ligand | In H2O | In D2O | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEHYDR | ΔEINTER | ΔEBIND | ΔEHYDR | ΔEINTER | ΔEBIND | ΔΔEBIND,calc | ΔΔEBIND,exp | |
| Histamine | −71.63 | −82.31 | −10.69 | −71.20 | −82.73 | −11.54 | −0.85 | −0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hok, L.; Mavri, J.; Vianello, R. The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules 2020, 25, 6017. https://doi.org/10.3390/molecules25246017
Hok L, Mavri J, Vianello R. The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules. 2020; 25(24):6017. https://doi.org/10.3390/molecules25246017
Chicago/Turabian StyleHok, Lucija, Janez Mavri, and Robert Vianello. 2020. "The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions" Molecules 25, no. 24: 6017. https://doi.org/10.3390/molecules25246017