
Engineering Stem Cell Factor Ligands with Different c-Kit Agonistic Potencies

 , and
, and
Abstract
:1. Introduction
2. Results
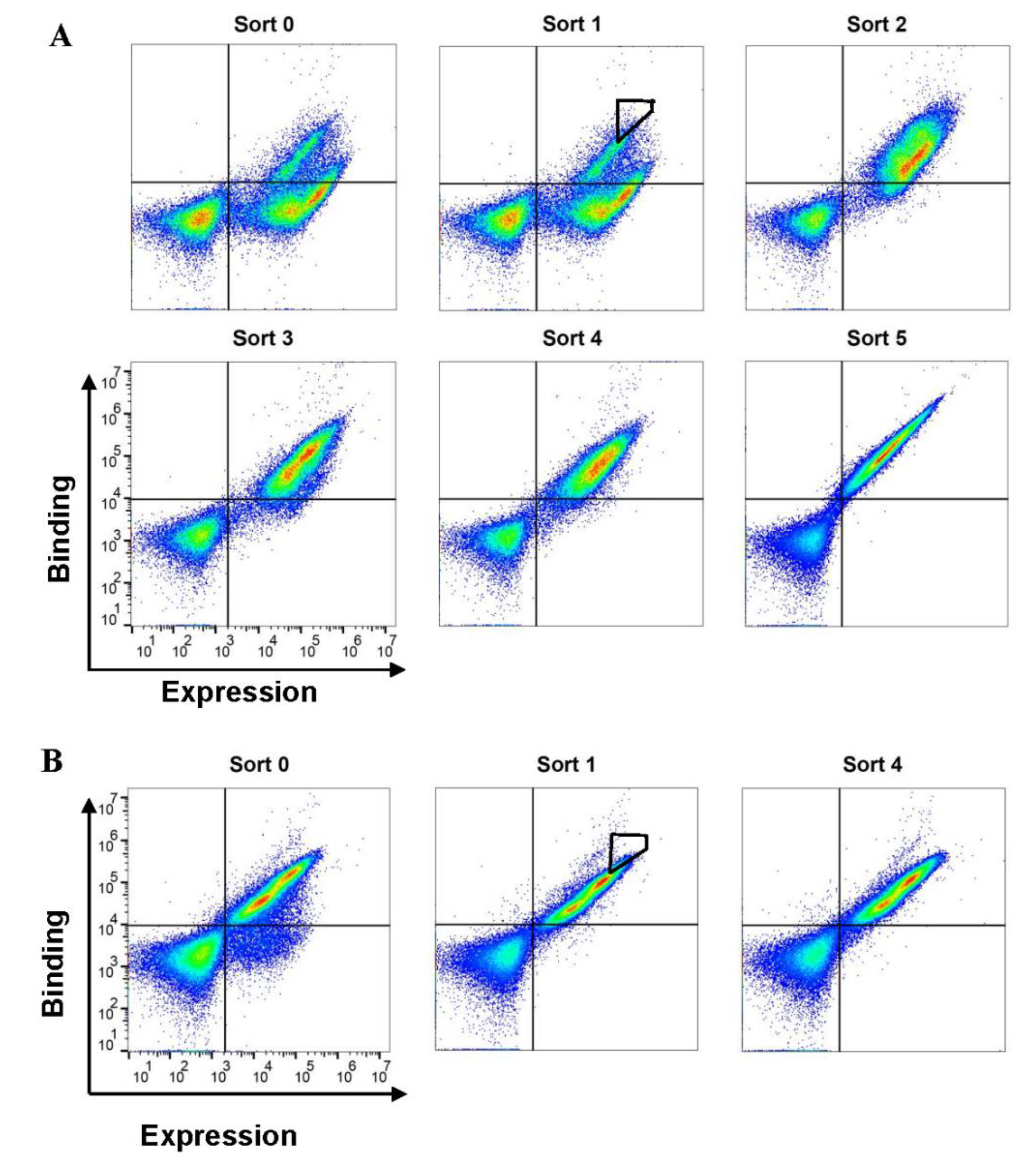
2.1. Sorting of SCFM Libraries for Variants with a High Affinity for c-Kit
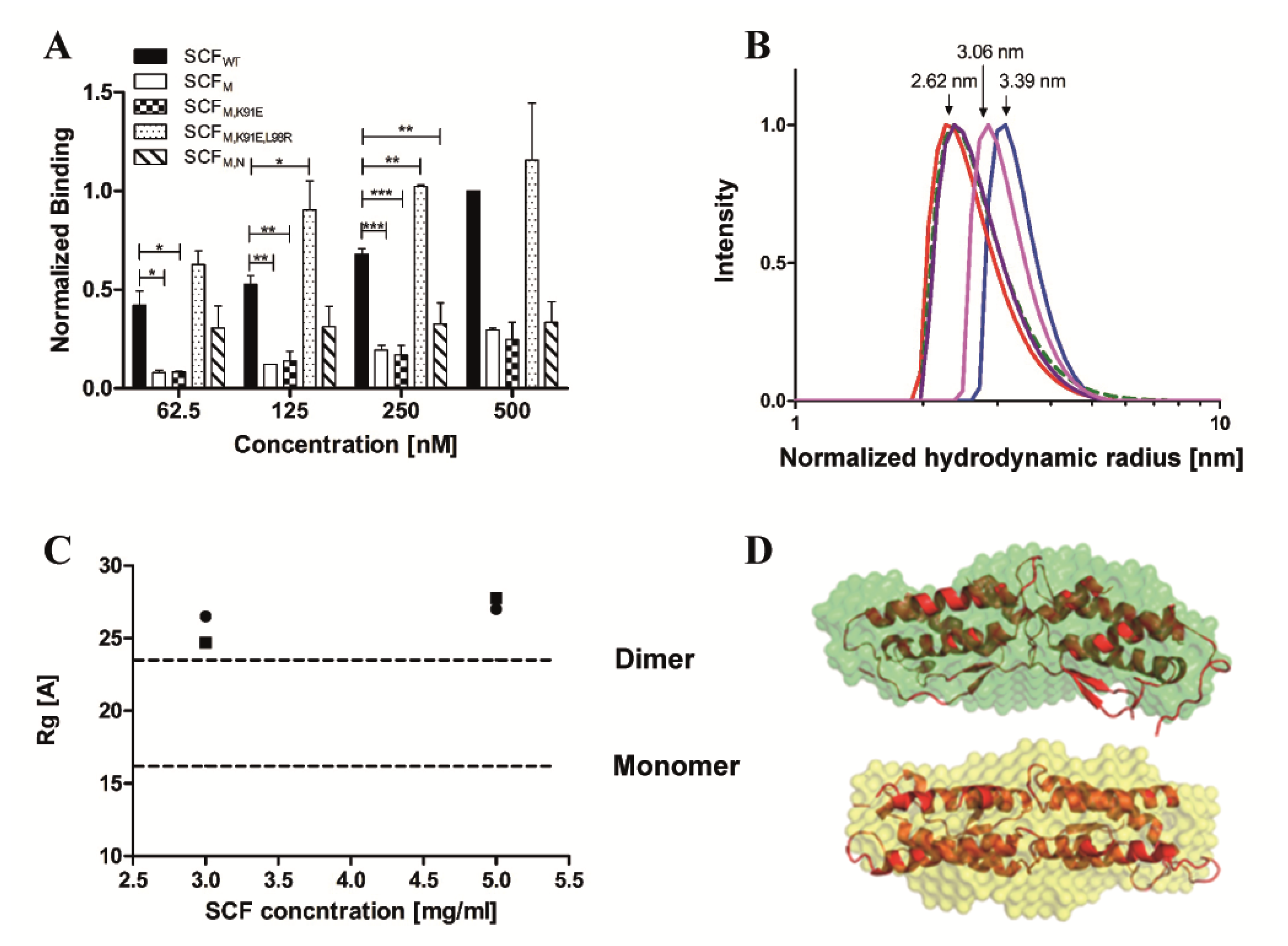
2.2. The Dimeric State of SCF Proteins is Concentration Dependent
2.3. SCFM,K91E and SCFM,K91E,L98R Exhibit High Affinity for c-Kit
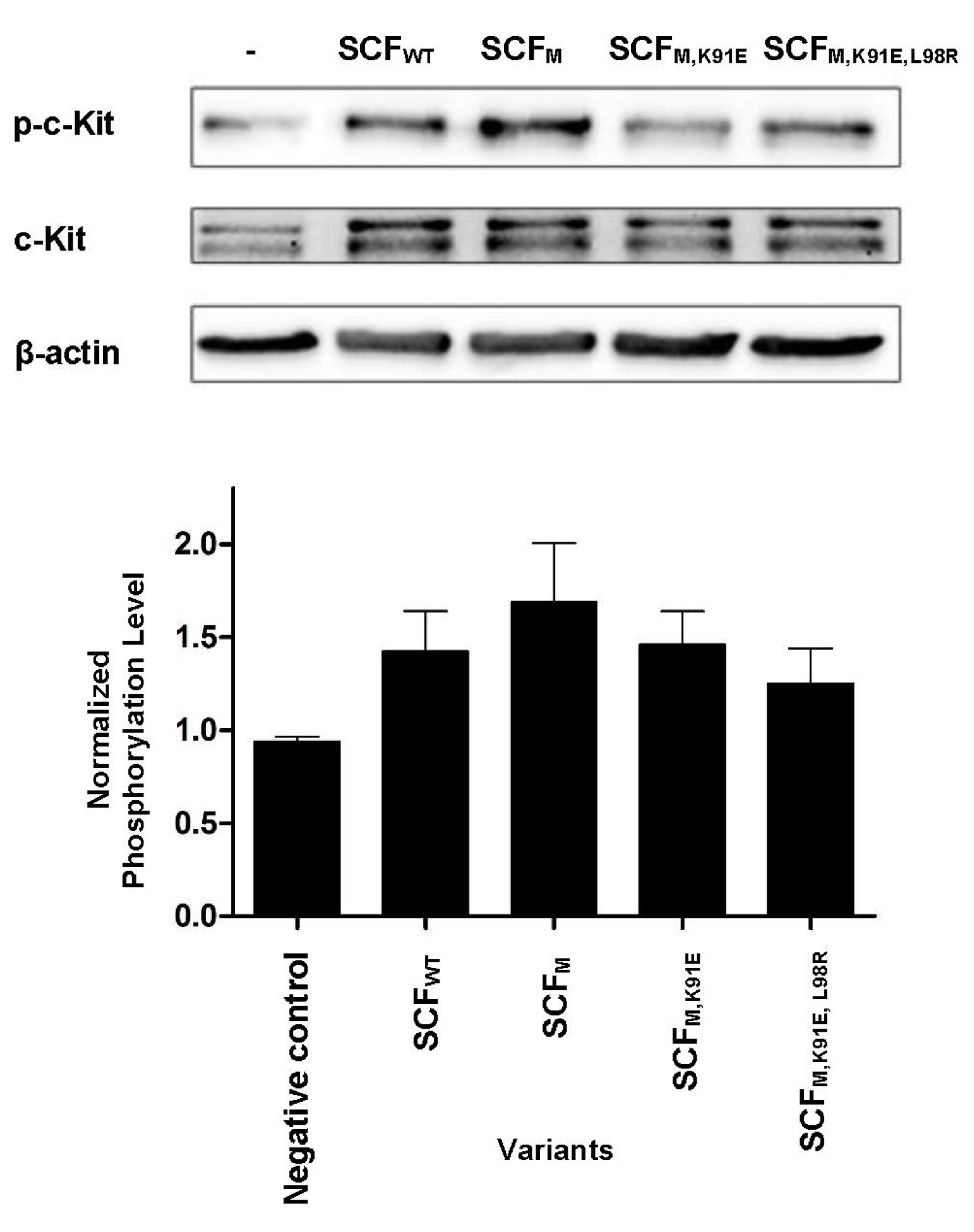
2.4. SCF-Dimerization is the Major Determinant for c-Kit Phosphorylation in Human Umbilical Vein Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Generating Random Mutagenesis SCFM Libraries in Yeast
4.2. Screening of SCF Libraries
4.3. Dimerization Assays Using Flow Cytometry
4.4. Dynamic Light Scattering
4.5. Small Angle X-ray Scattering
4.6. Surface Plasmon Resonance
4.7. Cell Binding Assays
4.8. c-Kit Phosphorylation
4.9. Matrigel Endothelial Tube Formation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baselga, J. Targeting tyrosine kinases in cancer: The second wave. Science 2006, 312, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Belli, S.; Esposito, D.; Servetto, A.; Pesapane, A.; Formisano, L.; Bianco, R. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers 2020, 12, 1489. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Park, M. Receptor tyrosine kinases: Role in cancer progression. Curr. Oncol. 2006, 13, 191–193. [Google Scholar] [PubMed]
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta Biomembr. 2017, 1859 Pt A, 1417–1429. [Google Scholar] [CrossRef]
- Changeux, J.P.; Christopoulos, A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, K.M.; Hu, C.; Lemmon, M.A. Insulin and epidermal growth factor receptor family members share parallel activation mechanisms. Protein Sci. 2020, 29, 1331–1344. [Google Scholar] [CrossRef]
- Kazi, J.U.; Ronnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef]
- Paul, M.D.; Hristova, K. The transition model of RTK activation: A quantitative framework for understanding RTK signaling and RTK modulator activity. Cytokine Growth Factor Rev. 2019, 49, 23–31. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Boesen, T.P.; Soni, B.; Schwartz, T.W.; Halkier, T. Single-chain vascular endothelial growth factor variant with antagonistic activity. J. Biol. Chem. 2002, 277, 40335–40341. [Google Scholar] [CrossRef] [Green Version]
- Fuh, G.; Li, B.; Crowley, C.; Cunningham, B.; Wells, J.A. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J. Biol. Chem. 1998, 273, 11197–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.R.; Wu, G.M.; Mendiaz, E.A.; Syed, R.; Wypych, J.; Toso, R.; Mann, M.B.; Boone, T.C.; Narhi, L.O.; Lu, H.S.; et al. The majority of stem cell factor exists as monomer under physiological conditions. Implications for dimerization mediating biological activity. J. Biol. Chem. 1997, 272, 6406–6415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zur, Y.; Rosenfeld, L.; Keshelman, C.A.; Dalal, N.; Guterman-Ram, G.; Orenbuch, A.; Einav, Y.; Levaot, N.; Papo, N. A dual-specific macrophage colony-stimulating factor antagonist of c-FMS and alphavbeta3 integrin for osteoporosis therapy. PLoS Biol. 2018, 16, e2002979. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Wang, Y.L.; Pattengale, P.K.; Rettenmier, C.W. The role of individual cysteine residues in the processing, structure, and function of human macrophage colony-stimulating factor. Biochem. Biophys. Res. Commun. 1996, 228, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, L.; Shirian, J.; Zur, Y.; Levaot, N.; Shifman, J.M.; Papo, N. Combinatorial and Computational Approaches to Identify Interactions of Macrophage Colony-stimulating Factor (M-CSF) and Its Receptor c-FMS. J. Biol. Chem. 2015, 290, 26180–26193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, K.; Jeppesen, M.G.; Malmgaard-Clausen, M.; Krzywkowski, K.; Dulal, K.; Cheng, T.; Hjorto, G.M.; Larsen, O.; Burg, J.S.; Jarvis, M.A.; et al. Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 8427–8432. [Google Scholar] [CrossRef] [Green Version]
- Shlamkovich, T.; Aharon, L.; Barton, W.A.; Papo, N. Utilizing combinatorial engineering to develop Tie2 targeting antagonistic angiopoetin-2 ligands as candidates for anti-angiogenesis therapy. Oncotarget 2017, 8, 33571–33585. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Das, A.V.; James, J.; Bhattacharya, S.; Zhao, X. Neural stem cells in the mammalian eye: Types and regulation. Semin Cell Dev. Biol. 2004, 15, 53–62. [Google Scholar] [CrossRef]
- Das, A.V.; James, J.; Zhao, X.; Rahnenfuhrer, J.; Ahmad, I. Identification of c-Kit receptor as a regulator of adult neural stem cells in the mammalian eye: Interactions with Notch signaling. Dev. Biol. 2004, 273, 87–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broudy, V.C.; Smith, F.O.; Lin, N.; Zsebo, K.M.; Egrie, J.; Bernstein, I.D. Blasts from patients with acute myelogenous leukemia express functional receptors for stem cell factor. Blood 1992, 80, 60–67. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, G.; Steinert, D.M.; McAuliffe, J.C.; Trent, J.C. Update on the biology and therapy of gastrointestinal stromal tumors. Cancer Control 2005, 12, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Blanke, C.D.; Druker, B.J.; Corless, C.L. Inhibition of KIT tyrosine kinase activity: A novel molecular approach to the treatment of KIT-positive malignancies. J. Clin. Oncol. 2002, 20, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Hongyo, T.; Li, T.; Syaifudin, M.; Baskar, R.; Ikeda, H.; Kanakura, Y.; Aozasa, K.; Nomura, T. Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res. 2000, 60, 2345–2347. [Google Scholar] [PubMed]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 1991, 78, 2962–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.N.; Liang, L.; Ren, S.; Wu, M.; Shi, D.C.; Mo, W.J.; Chen, G. Cd117 Expression is Correlated with Poor Survival of Patients and Progression of Lung Carcinoma: A Meta-Analysis with a Panel of 2645 Patients. Pol. J. Pathol. 2019, 70, 63–78. [Google Scholar] [CrossRef]
- Makhlouf, A.M.; Chitikova, Z.; Pusztaszeri, M.; Berczy, M.; Delucinge-Vivier, C.; Triponez, F.; Meyer, P.; Philippe, J.; Dibner, C. Identification of CHEK1, SLC26A4, c-KIT, TPO and TG as new biomarkers for human follicular thyroid carcinoma. Oncotarget 2016, 7, 45776–45788. [Google Scholar] [CrossRef] [Green Version]
- Mattiolo, P.; Hong, S.M.; Paolino, G.; Rusev, B.C.; Marchegiani, G.; Salvia, R.; Andrianello, S.; Capelli, P.; Piccoli, P.; Parolini, C.; et al. CD117 Is a Specific Marker of Intraductal Papillary Mucinous Neoplasms (IPMN) of the Pancreas, Oncocytic Subtype. Int. J. Mol. Sci. 2020, 21, 5794. [Google Scholar] [CrossRef] [PubMed]
- Opatowsky, Y.; Lax, I.; Tome, F.; Bleichert, F.; Unger, V.M.; Schlessinger, J. Structure, domain organization, and different conformational states of stem cell factor-induced intact KIT dimers. Proc. Natl. Acad. Sci. USA 2014, 111, 1772–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, R.; Joachimiak, A.; Schlessinger, J.; Kong, X.P. Crystal structure of human stem cell factor: Implication for stem cell factor receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 2000, 97, 7732–7737. [Google Scholar] [CrossRef] [Green Version]
- Blume-Jensen, P.; Wernstedt, C.; Heldin, C.H.; Ronnstrand, L. Identification of the major phosphorylation sites for protein kinase C in kit/stem cell factor receptor in vitro and in intact cells. J. Biol. Chem. 1995, 270, 14192–14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, S.; Yarden, Y.; Givol, D. Dimerization and activation of the kit receptor by monovalent and bivalent binding of the stem cell factor. J. Biol. Chem. 1992, 267, 15970–15977. [Google Scholar]
- Philo, J.S.; Wen, J.; Wypych, J.; Schwartz, M.G.; Mendiaz, E.A.; Langley, K.E. Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, Kit. J. Biol. Chem. 1996, 271, 6895–6902. [Google Scholar] [CrossRef] [Green Version]
- Potgens, A.J.; Lubsen, N.H.; van Altena, M.C.; Vermeulen, R.; Bakker, A.; Schoenmakers, J.G.; Ruiter, D.J.; de Waal, R.M. Covalent dimerization of vascular permeability factor/vascular endothelial growth factor is essential for its biological activity. Evidence from Cys to Ser mutations. J. Biol. Chem. 1994, 269, 32879–32885. [Google Scholar]
- Colby, D.W.; Kellogg, B.A.; Graff, C.P.; Yeung, Y.A.; Swers, J.S.; Wittrup, K.D. Engineering antibody affinity by yeast surface display. Methods Enzymol. 2004, 388, 348–358. [Google Scholar]
- Liu, Y.C.; Kawagishi, M.; Kameda, R.; Ohashi, H. Characterization of a fusion protein composed of the extracellular domain of c-kit and the Fc region of human IgG expressed in a baculovirus system. Biochem. Biophys. Res. Commun. 1993, 197, 1094–1102. [Google Scholar] [CrossRef]
- Fratto, M.E.; Imperatori, M.; Vincenzi, B.; Tomao, F.; Santini, D.; Tonini, G. New perspectives: Role of Sunitinib in Breast Cancer. Clin. Ter. 2011, 162, 251–257. [Google Scholar]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G.; et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanopoulou, E.; Judson, I. The safety profile of imatinib in CML and GIST: Long-term considerations. Arch. Toxicol. 2012, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weigel, M.T.; Dahmke, L.; Schem, C.; Bauerschlag, D.O.; Weber, K.; Niehoff, P.; Bauer, M.; Strauss, A.; Jonat, W.; Maass, N.; et al. In vitro effects of imatinib mesylate on radiosensitivity and chemosensitivity of breast cancer cells. BMC Cancer 2010, 10, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmes, L.J.; Pallavicini, M.G.; Fleming, L.M.; Gibbs, J.; Wang, D.; Li, K.L.; Partridge, S.C.; Henry, R.G.; Shalinsky, D.R.; Hu-Lowe, D.; et al. AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging. Magn. Reason. Imaging 2007, 25, 319–327. [Google Scholar] [CrossRef]
- Lev, S.; Yarden, Y.; Givol, D. A recombinant ectodomain of the receptor for the stem cell factor (SCF) retains ligand-induced receptor dimerization and antagonizes SCF-stimulated cellular responses. J. Biol. Chem. 1992, 267, 10866–10873. [Google Scholar]
- Fleischman, R.A. From white spots to stem cells: The role of the Kit receptor in mammalian development. Trends Genet. 1993, 9, 285–290. [Google Scholar] [CrossRef]
- Babaei, M.A.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.C.M.; Chhabra, A.; Starkl, P.; Schnorr, P.J.; Wilmes, S.; Moraga, I.; Kwon, H.S.; Gaudenzio, N.; Sibilano, R.; Wehrman, T.S.; et al. Decoupling the Functional Pleiotropy of Stem Cell Factor by Tuning c-Kit Signaling. Cell 2017, 168, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.P.; Ku, W.Y.; Wu, M.H.; Tsai, Y.L.; Chen, C.Y.; Kuo, C.A.; Huang, L.L. Full-length recombinant human SCF1-165 is more thermostable than the truncated SCF1-141 form. PLoS ONE 2014, 9, e103251. [Google Scholar] [CrossRef] [Green Version]
- Abrams, T.; Connor, A.; Fanton, C.; Cohen, S.B.; Huber, T.; Miller, K.; Hong, E.E.; Niu, X.H.; Kline, J.; Ison-Dugenny, M.; et al. Preclinical Antitumor Activity of a Novel Anti-c-KIT Antibody-Drug Conjugate against Mutant and Wild-type c-KIT-Positive Solid Tumors. Clin. Cancer Res. 2018, 24, 4297–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobba, A.; Cavatorta, P.; Attimonelli, M.; Riccio, P.; Masotti, L.; Quagliariello, E. Estimation of protein secondary structure from circular dichroism spectra: A critical examination of the CONTIN program. Protein Seq. Data Anal. 1990, 3, 7–10. [Google Scholar] [PubMed]
- Aliyu, A.; Kariim, I.; Abdulkareem, S.A. Effects of aspect ratio of multi-walled carbon nanotubes on coal washery waste water treatment. J. Environ. Manag. 2017, 202 Pt 1, 84–93. [Google Scholar] [CrossRef]
- Akabayov, B.; Akabayov, S.R.; Lee, S.J.; Tabor, S.; Kulczyk, A.W.; Richardson, C.C. Conformational dynamics of bacteriophage T7 DNA polymerase and its processivity factor, Escherichia coli thioredoxin. Proc. Natl. Acad. Sci. USA 2010, 107, 15033–15038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svergun, D.I.; Petoukhov, M.V.; Koch, M.H. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Joubert, A.M.; Byrd, A.S.; LiCata, V.J. Global conformations, hydrodynamics, and X-ray scattering properties of Taq and Escherichia coli DNA polymerases in solution. J. Biol. Chem. 2003, 278, 25341–25347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramyadevi, J.; Jeyasubramanian, K.; Marikani, A.; Rajakumar, G.; Rahuman, A.A.; Santhoshkumar, T.; Kirthi, A.V.; Jayaseelan, C.; Marimuthu, S. Copper nanoparticles synthesized by polyol process used to control hematophagous parasites. Parasitol. Res. 2011, 109, 1403–1415. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | KD (M) |
|---|---|
| SCFWT | 4.36 × 10−9 ± 1.46 × 10−9 |
| SCFWT glycosylated | 5.82 × 10−9 ± 2.93 × 10−9 |
| SCFM | 146 × 10−9 ± 18 × 10−9 |
| SCFM,K91E | 88.9 × 10−9 ± 19.9 × 10−9 |
| SCFM,K91E,L98R | 40.7 × 10−9 ± 0.7 × 10−9 |
| SCFM,S64P,F126S,V131A,E134G,V139I | 74.4 × 10−9 ± 5.1 × 10−9 |
Sample Availability: Samples of the compounds SCFWT, SCFM, SCFM,K91E, SCFM,K91E,L98R, and SCFM,S64P,F126S,V131A,E134G,V139I are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tilayov, T.; Hingaly, T.; Greenshpan, Y.; Cohen, S.; Akabayov, B.; Gazit, R.; Papo, N. Engineering Stem Cell Factor Ligands with Different c-Kit Agonistic Potencies. Molecules 2020, 25, 4850. https://doi.org/10.3390/molecules25204850
Tilayov T, Hingaly T, Greenshpan Y, Cohen S, Akabayov B, Gazit R, Papo N. Engineering Stem Cell Factor Ligands with Different c-Kit Agonistic Potencies. Molecules. 2020; 25(20):4850. https://doi.org/10.3390/molecules25204850
Chicago/Turabian StyleTilayov, Tal, Tal Hingaly, Yariv Greenshpan, Shira Cohen, Barak Akabayov, Roi Gazit, and Niv Papo. 2020. "Engineering Stem Cell Factor Ligands with Different c-Kit Agonistic Potencies" Molecules 25, no. 20: 4850. https://doi.org/10.3390/molecules25204850