
A Comparison of Solubility, Stability, and Bioavailability between Astilbin and Neoastilbin Isolated from Smilax glabra Rhizoma


Abstract
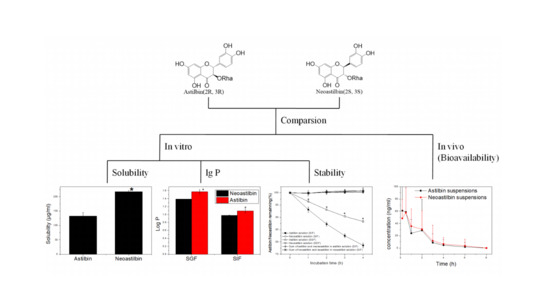
:
1. Introduction
2. Results and Discussion
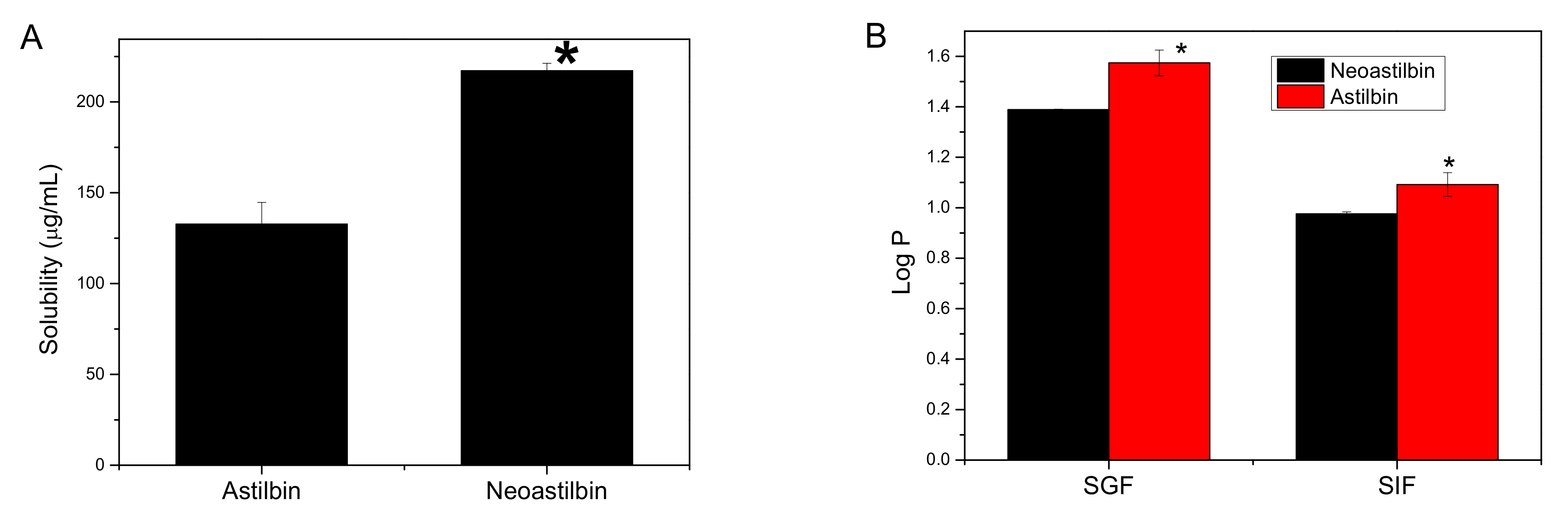
2.1. Solubility Comparison between Astilbin and Neoastilbin
2.2. Oil–Water Distribution Coefficient (Log P) Comparison between Astilbin and Neoastilbin
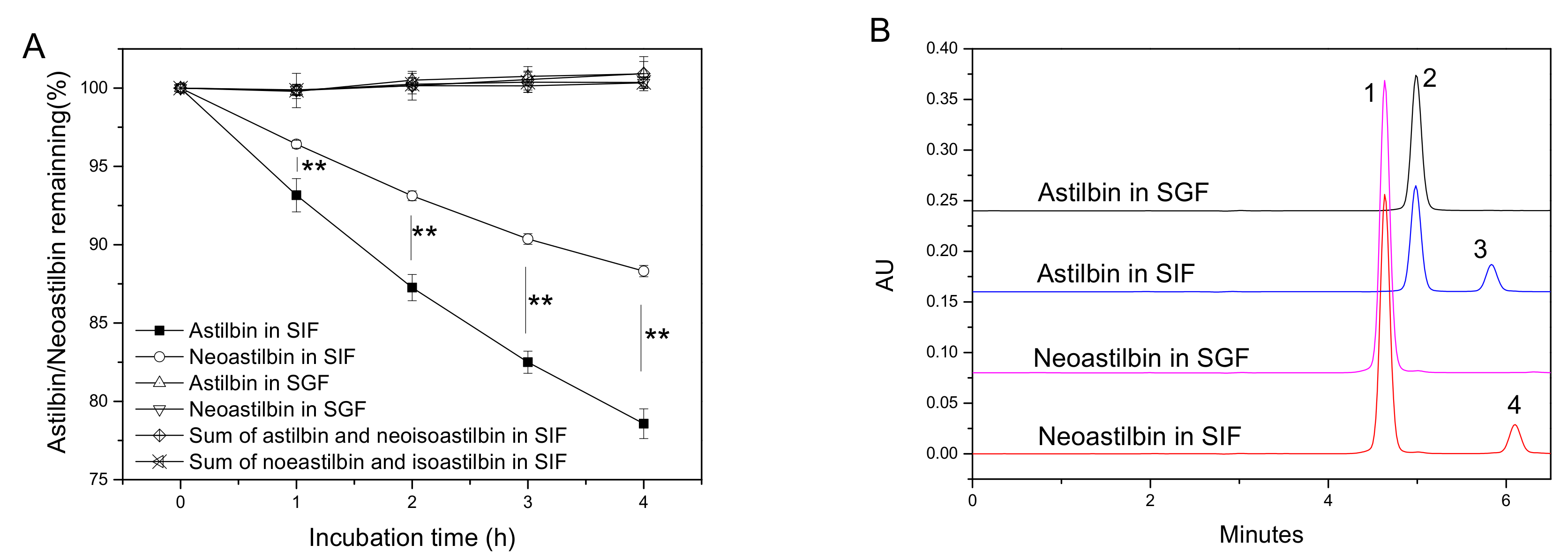
2.3. Stability Comparison between Astilbin and Neoastilbin in SGF and SIF
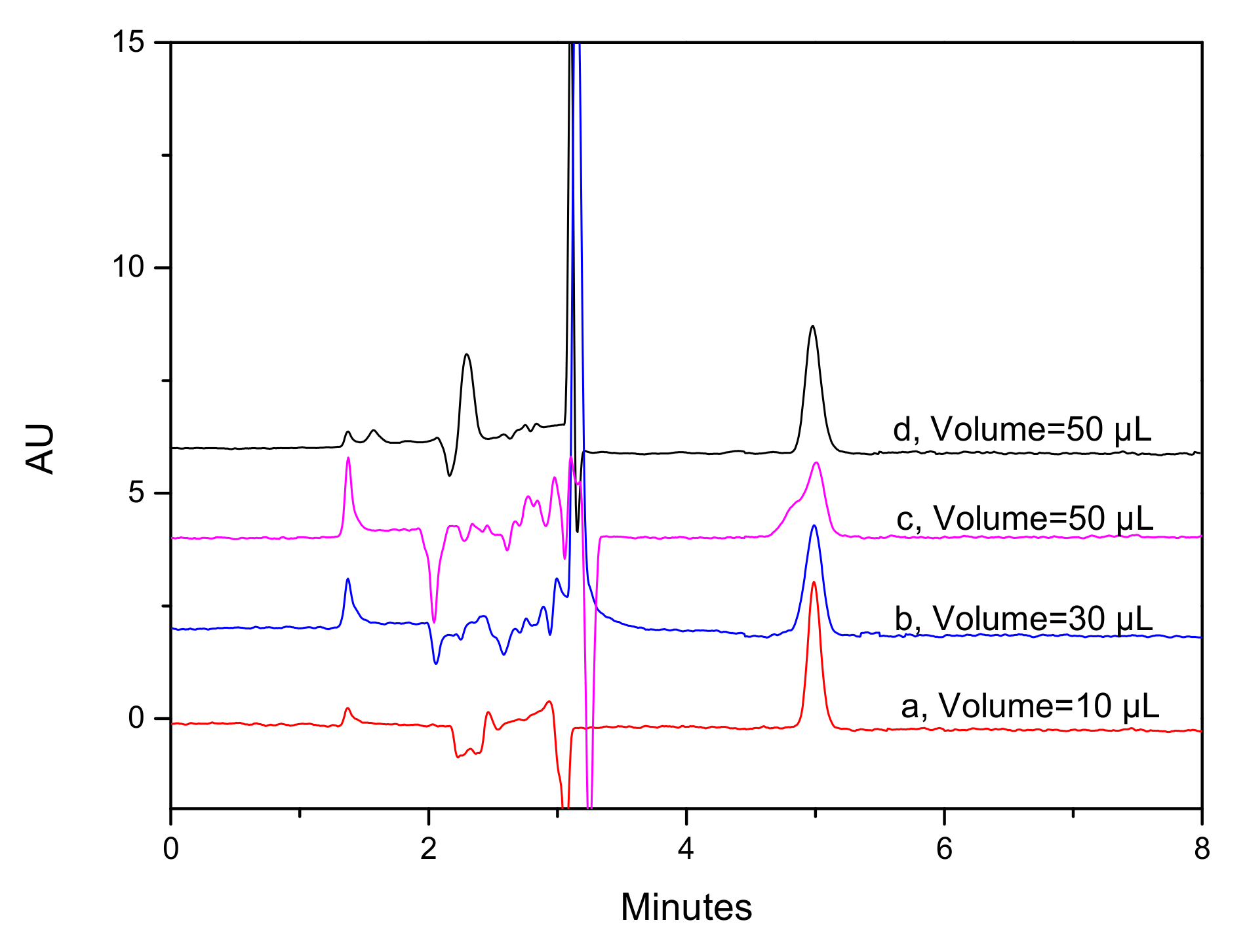
2.4. Plasma Sample Pretreatment Optimization for Pharmacokinetic Study
2.5. HPLC Method Development for Pharmacokinetic Study
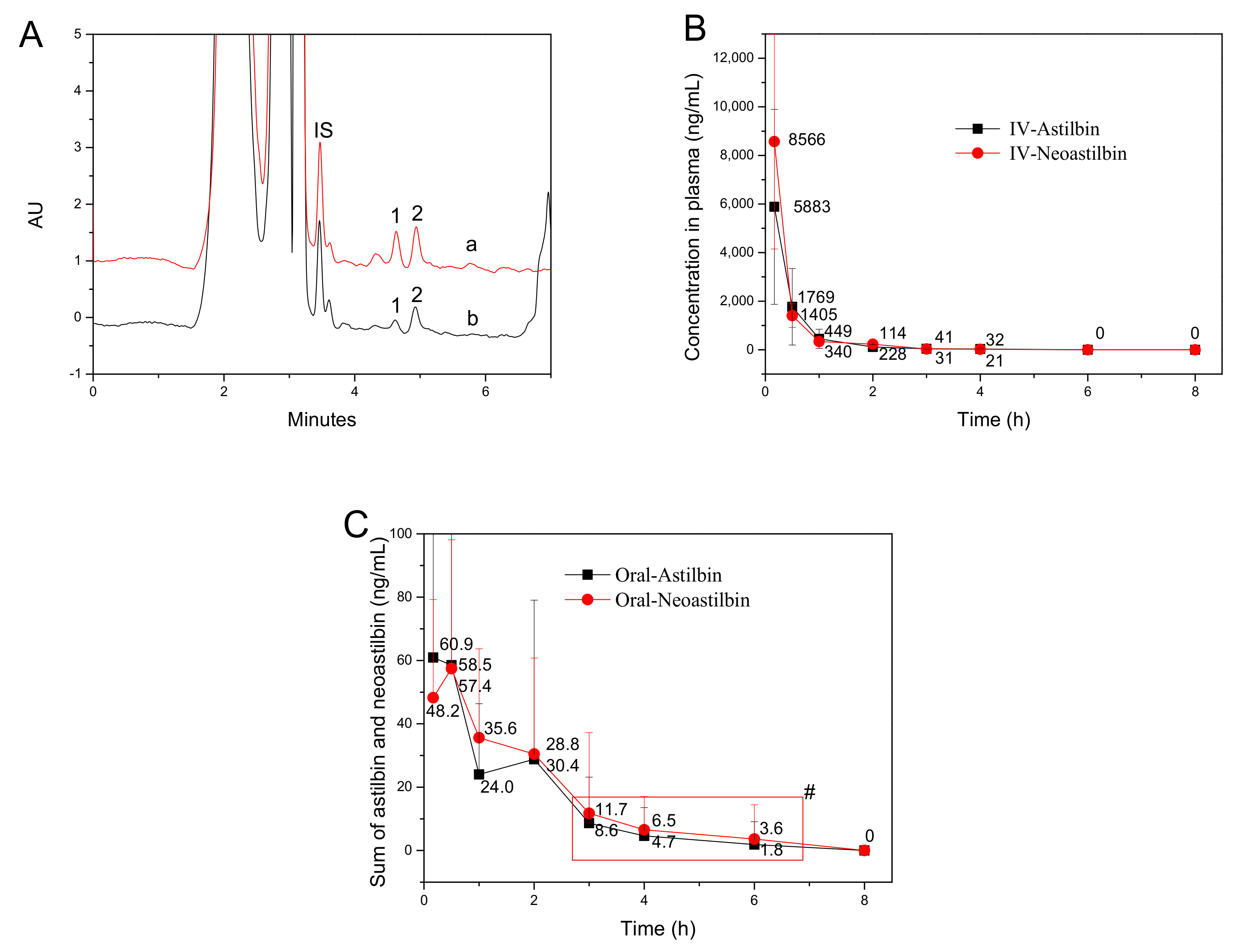
2.6. Bioavailability Comparison between Astilbin and Neoastilbin in Rat
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Solubility Test
3.3. Oil–Water Distribution Coefficient Determination
3.4. Stability Comparison of Astilbin and Neoastilbin in SGF (pH 1.2) and SIF (pH 6.8)
3.5. Bioavailability Comparison of Astilbin and Neoastilbin in Rats
3.6. Plasma Sample Pretreatment
3.6.1. Preparation of Spiked Plasma Sample
3.6.2. Plasma Sample Pretreatment Method Comparison
3.6.3. Astilbin/Neoastilbin Stability in Plasma Sample
3.6.4. Plasma Sample Pretreatment for Astilbin/Neoastilbin Stability and Pharmacokinetic Study
3.7. HPLC Method Development for Pharmacokinetic Study
3.7.1. HPLC Conditions
3.7.2. Calibration Curve
3.7.3. Precision and Accuracy
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, L.; Yin, Y.; Yi, H.; Xu, Q.; Chen, T. Simultaneous quantification of five major bioactive flavonoids in Rhizoma Smilacis glabrae by high-performance liquid chromatography. J. Pharm. Biomed. 2007, 43, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cheng, Z.; Shi, H.; Xin, W.; Wang, T.T.Y.; Yu, L. Isolation and characterization of two flavonoids, engeletin and astilbin, from the leaves of Engelhardtia roxburghiana leaf and their potential anti-inflammatory properties. J. Agric. Food Chem. 2011, 59, 4562–4569. [Google Scholar] [CrossRef] [PubMed]
- Landrault, N.; Larronde, F.; Delaunay, J.C.; Castagnino, C.; Vercauteren, J.; Merillon, J.M.; Gasc, F.; Cros, G.; Teissedre, P.L. Levels of stilbene oligomers and astilbin in french varietal wines and in grapes during noble rot development. J. Agric. Food Chem. 2002, 50, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Gaffield, W.; Waiss, A.C., Jr.; Tominaga, T. Structural relations and interconversions of isomeric astilbins. J. Org. Chem. 1975, 40, 1057–1061. [Google Scholar] [CrossRef]
- Du, Q.; Li, L.; Jerz, G. Purification of astilbin and isoastilbin in the extract of Smilax glabra rhizome by high-speed counter-current chromatography. J. Chromatogr. A. 2005, 1077, 98–101. [Google Scholar] [CrossRef]
- Wu, J.B.; Chen, Z.; Zhao, Y.M.; Wang, H.Z. One-step purification of astilbin from Smilax glabra by high-performance centrifugal partition chromatography. J. Liq. Chromatogr. Relat. Technol. 2013, 36, 1540–1548. [Google Scholar] [CrossRef]
- Haraguchi, H.; Ohmi, I.; Fukuda, A.; Tamura, Y.; Mizutani, K.; Tanaka, O.; Chou, W.H. Inhibition of aldose reductase and sorbitol accumulation by astilbin and taxifolin dihydroflavonols in Engelhardtia chrysolepis. Biosci. Biotechnol. Biochem. 1997, 61, 651–654. [Google Scholar] [CrossRef]
- Xu, Q.; Sun, Y. Novel immunosuppression stemming from the selective activity of Chinese herbal drugs. Progr. Chem. 2009, 21, 55–62. [Google Scholar]
- Kasai, B.; Hirono, S.; Chou, W.H. An additional sweet dihydroflavonol glycoside from leaves of Engelhardtia chrysolepi, a Chinese folk medicine, Huang-qi. Chem. Pharm. Bull. 1991, 39, 1871–1872. [Google Scholar] [CrossRef] [Green Version]
- He, J.W.; Bondy, G.S.; Zhou, T.; Caldwell, D.; Boland, G.J.; Scott, P.M. Toxicology of 3-epi-deoxynivalenol, a deoxynivalenol-transformation product by Devosia mutans 17-2-E-8. Food. Chem. Toxicol. 2015, 84, 250–259. [Google Scholar] [CrossRef]
- Kakumanu, V.K.; Arora, V.; Bansal, A.K. Investigation on physicochemical and biological differences of cefpodoxime proxetil enantiomers. Eur. J. Pharm. Biopharm. 2006, 64, 255–259. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, L.; Zhang, Q.F. Isomerization of astilbin and its application for preparation of the four stereoisomers from Rhizoma Smilacis glabra. J. Pharmaceut. Biomed. 2018, 155, 202–209. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Nie, H.C.; Shangguang, X.C.; Yin, Z.P.; Zheng, G.D.; Chen, J.G. Aqueous solubility and stability enhancement of astilbin through complexation with cyclodextrins. J. Agric. Food Chem. 2013, 61, 151–156. [Google Scholar] [CrossRef]
- National Commission of Chinese Pharmacopoeia. Pharmacopoeia of Peoples Republic of China, Vol. I; China Medical Science Press: Beijing, China, 2010; p. 17. [Google Scholar]
- Li, Z.; Jiang, H.; Xu, C.; Gu, L. A review: Using nanoparticles to enhance absorption and bioavailability of phenolic phytochemicals. Food Hydrocoll. 2015, 43, 153–164. [Google Scholar] [CrossRef]
- Xue, J.; Jia, X.B.; Tan, X.B.; Jia, D.S.; Jiang, J.; Zhang, L.Y. Determination of apparent oil/water partition coefficient and absorption prediction of Triptolide. Chin. Pharm. J. 2009, 44, 1560–1563. [Google Scholar]
- Abraham, D.J. Burger’s Medicinal Chemistry and Drug Discovery; John Wiley & Sons Inc.: Auckland, New Zealand, 2003; pp. 259–293. [Google Scholar]
- Tian, X.J.; Yang, X.W.; Yang, X.D.; Wang, K. Studies of intestinal permeability of 36 flavonoids using Caco-2 cell monolayer model. Int. J. Pharm. 2009, 367, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.F.; Fu, Y.J.; Huang, Z.W.; Shangguang, X.C.; Guo, Y.X. Aqueous stability of astilbin: Effects of pH, temperature, and solvent. J. Agric. Food Chem. 2013, 61, 12085–12091. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Chen, R.; Sun, W.; Huang, C.; Lin, X.; Dong, Y.; Wen, C.; Wang, X. Determination and pharmacokinetics of engeletin in rat plasma by ultra-high performance liquid chromatography with tandem mass spectrometry. J. Chromatogr. B 2013, 1060, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, Q.; Chen, T. Quantitative determination of astilbin in rabbit plasma by liquid chromatography. J. Chromatogr. B 2004, 805, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D. Studies on the Transport of Taxifolin and Astilbin In Vitro and Pharmacokinetics in Rats. Ph.D. Thesis, Zhejiang University, Zhejiang, China, 2009. [Google Scholar]
- Yin, L.; Zhang, Y.-H.; Zhao, S.; Cheng, L.-M.; Shi, M.-Y.; Yang, Y.; Sun, Y.-T.; Liu, X.-D.; Fawcett, J.P. Rapid quantification of astilbin in rat plasma by liquid chromatography-tandem mass spectrometry and its application to pharmacokinetic study. Chem. Res. Chin. Univ. 2013, 29, 1078–1082. [Google Scholar] [CrossRef]
- Li, M.; Dyett, B.; Zhang, X. Automated Femtoliter Droplet-Based Determination of Oil–Water Partition Coefficient. Anal. Chem. 2019, 91, 10371–10375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannava, M.K.; Suresh, K.; Kumar Bommaka, M.; Bhavani Konga, D.; Nangia, A. Curcumin-artemisinin coamorphous solid: Xenograft model preclinical study. Pharmaceutics 2018, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, M.G.; Stehly, G.R.; Hayton, W.L. Pharmacokinetic modeling in aquatic animals I. Models and concepts. Aquat. Toxicol. 1990, 17, 187–212. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds of astilbin and neoastilbin are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Stability | Relative Recovery (%) 1 | |
|---|---|---|---|
| Astilbin | Neoastilbin | ||
| Short-time (Ambient Atmosphere) | 2 h | 106.16 ± 2.76 | 92.38 ± 1.04 * |
| 4 h | 98.02 ± 3.48 | 89.38 ± 1.73 * | |
| 6 h | 101.10 ± 5.71 | 89.23 ± 4.48 * | |
| 8 h | 95.60 ± 5.28 | 96.30 ± 6.03 | |
| Long-time (−80 °C) | 7 days | 86.34 ± 2.17 | 87.78 ± 8.01 |
| 14 days | 91.52 ± 2.48 | 92.40 ± 4.46 | |
| 21 days | 89.04 ± 8.91 | 89.57 ± 9.25 | |
| 28 days | 86.65 ± 3.35 | 86.87 ± 6.60 | |
| Freeze/Thaw | Three freeze/thaw | 97.15 ± 7.45 | 93.29 ± 10.32 |
| Compound | Migration Time (RSD,%) | Peak Area Ratio (RSD,%) | Accuracy (%) | Regression Equation | Linear Range (ng/mL) | Linearity (R) | LLOD (ng/mL) | LLOQ (ng/mL) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | Intra-Day | Inter-Day | ||||||
| Astilbin | 0.34 | 0.21 | 1.49 | 3.71 | 94.56 | 92.75 | Y = 2.324X | 17.3–1105 | 0.9915 | 4.8 | 16.3 |
| Neoastilbin | 0.35 | 0.23 | 7.78 | 4.23 | 96.15 | 91.27 | Y = 2.189X | 31.6–1010 | 0.9975 | 5.3 | 17.5 |
| IV-Ast | IV-Neoast | PO-Ast | PO-Neoast | |
|---|---|---|---|---|
| Cmax (ng/L) | 5883.4 ± 2081.0 | 8566.7 ± 3091.8 | 60.9 ± 48.1 | 57.5 ± 25.3 |
| AUC(0-t) (ng/L·h) | 3487.1 ± 1813.2 | 4370.9 ± 1417.1 | 103.8 ± 56.8 | 118.3 ± 73.5 |
| AUC(0-∞) (ng/L·h) | 3493.4 ± 1793.2 | 4480.3 ± 1420.0 | 107.5 ± 50.4 | 122.1 ± 88.2 |
| Tmax (h) | 0.17 ± 0 | 0.17 ± 0 | 0.17 ± 0.5 | 0.5 ± 0.4 |
| t1/2 (h) | 0.5 ± 0.5 | 1.2 ± 0.1 | 1.36 ± 0.7 | 1.2 ± 0.7 |
| CL (L·h−1·kg−1) | 0.5 ± 0.4 | 0.5 ± 0.1 | 186.0 ± 22.8 | 163.7 ± 17.9 |
| MRT(0-t) | 0.4 ± 0.2 | 0.4 ± 0.1 | 1.5 ± 0.2 | 1.67 ± 0.3 |
| MRT(0-∞) | 0.4 ± 0.3 | 0.5 ± 0.1 | 1.7 ± 1.4 | 2.0 ± 0.7 |
| Fr(%) | 100 | 100 | 0.30 | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, D.; Ruan, Y.-T.; Yin, Z.-P.; Zhang, Q.-F. A Comparison of Solubility, Stability, and Bioavailability between Astilbin and Neoastilbin Isolated from Smilax glabra Rhizoma. Molecules 2020, 25, 4728. https://doi.org/10.3390/molecules25204728
Zheng D, Ruan Y-T, Yin Z-P, Zhang Q-F. A Comparison of Solubility, Stability, and Bioavailability between Astilbin and Neoastilbin Isolated from Smilax glabra Rhizoma. Molecules. 2020; 25(20):4728. https://doi.org/10.3390/molecules25204728
Chicago/Turabian StyleZheng, Dan, Yi-Ting Ruan, Zhong-Ping Yin, and Qing-Feng Zhang. 2020. "A Comparison of Solubility, Stability, and Bioavailability between Astilbin and Neoastilbin Isolated from Smilax glabra Rhizoma" Molecules 25, no. 20: 4728. https://doi.org/10.3390/molecules25204728