
Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
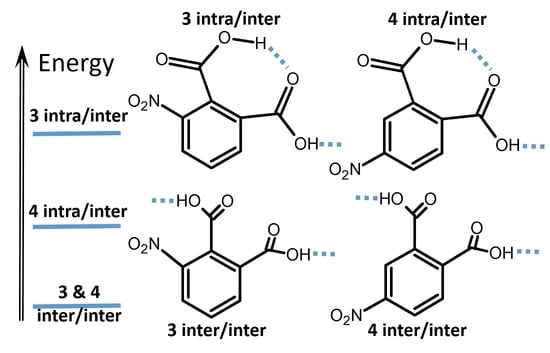
2.1. DFT Study of H-Bond, Nitro, and Carboxyl Groups’ Dynamics of Nitrophthalic Acids
2.2. Dynamics of Hydrogen Bonding within the Framework of Molecular Dynamics
2.3. NMR Studies of Nitrophthalic Acids
2.4. H-Bonding Vibrational Modes in Carboxyl Dimers
3. Materials and Methods
3.1. Compounds and Deuteration
3.2. Infrared and Raman Measurements
3.3. Incoherent Inelastic Neutron Scattering (IINS) Measurements
3.4. NMR Measurements
3.5. Car–Parrinello Molecular Dynamics’ Simulations
3.6. DFT Calculations
3.7. PED Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Di(hydroperoxy)cycloalkane Adducts of Triarylphosphine Oxides: A Comprehensive Study Including Solid-State Structures and Association in Solution. Inorg. Chem. 2020, 59, 13719–13732. [Google Scholar] [CrossRef]
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide adducts of triarylphosphine oxides. Dalton Trans. 2019, 48, 14312. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Limbach, H.-H.; Smirnov, S.N.; Tolstoy, P.M.; Denisov, G.S.; Golubev, N.S. H/D isotope effects on the low-temperature NMR parameters and hydrogen bond geometries of (FH)2F− and (FH)3F− dissolved in CDF3/CDF2Cl. Phys. Chem. Chem. Phys. 2002, 4, 5488–5497. [Google Scholar] [CrossRef]
- Mauder, D.; Akcakayiran, D.; Lesnichin, S.B.; Findenegg, G.H.; Shenderovich, I.G. Acidity of Sulfonic and Phosphonic Acid-Functionalized SBA-15 under Almost Water-Free Conditions. J. Phys. Chem. C 2009, 113, 19185–19192. [Google Scholar] [CrossRef]
- Melikova, S.M.; Voronin, A.P.; Panek, J.; Frolov, N.E.; Shishkina, A.V.; Rykounov, A.A.; Tretyakov, P.Y.; Vener, M.V. Interplay of pi-stacking and inter-stacking interactions in two-component crystals of neutral closed-shell aromatic compounds: Periodic DFT study. RSC Adv. 2020, 10, 27899–27910. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bonds with π-Electrons Acting as Lewis BasesTheoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. Triel Bonds, pi-Hole-pi-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules 2013, 20, 11297–11316. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Zhurov, V.V.; Stash, A.I.; Vener, M.V.; Pinkerton, A.A.; Tsirelson, V.G. Noncovalent Interactions in Crystalline Picolinic Acid N-Oxide: Insights from Experimental and Theoretical Charge Density Analysis. Cryst. Growth Des. 2013, 13, 816–828. [Google Scholar] [CrossRef]
- Palusiak, M.; Grabowski, S.J. Do intramolecular halogen bonds exist? Ab initio calculations and crystal structures’ evidences. Struct. Chem. 2008, 19, 5–11. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Rozhkova, Y.A.; Zukal, A.; Čejka, J.; Shenderovich, I.G. Mutable Lewis and Brønsted Acidity of Aluminated SBA-15 as Revealed by NMR of Adsorbed Pyridine-15N. Langmuir 2011, 27, 12115–12123. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. For Whom a Puddle Is the Sea? Adsorption of Organic Guests on Hydrated MCM-41 Silica. Langmuir 2020, 36, 11383–11392. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, B.; Emmler, T.; Gedat, E.; Shenderovich, I.; Findenegg, G.H.; Limbach, H.-H.; Buntkowsky, G. Hydrogen Bonding of Water Confined in Mesoporous Silica MCM-41 and SBA-15 Studied by 1H Solid-State NMR. Chem. Eur. J. 2004, 10, 5689–5696. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W. Specific ion effects in colloidal and biological systems. Curr. Opin. Colloid Interface Sci. 2010, 15, 34–39. [Google Scholar] [CrossRef]
- Shenderovich, I.G. The Partner Does Matter: The Structure of Heteroaggregates of Acridine Orange in Water. Molecules 2019, 24, 2816. [Google Scholar] [CrossRef] [Green Version]
- Heyne, B. Self-assembly of organic dyes in supramolecular aggregates. Photochem. Photobiol. Sci. 2016, 15, 1103–1114. [Google Scholar] [CrossRef]
- Sharif, S.; Shenderovich, I.G.; González, L.; Denisov, G.S.; Silverman, D.N.; Limbach, H.-H. NMR and Ab initio Studies of Small Complexes Formed between Water and Pyridine Derivatives in Solid and Liquid Phase. J. Phys. Chem. A 2007, 111, 6084–6093. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Effect of Non-Covalent Interactions on the 31P Chemical Shift Tensor of Phosphine Oxides, Phosphinic, Phosphonic, and Phosphoric Acids and Their Complexes with Lead(II). J. Phys. Chem. C 2013, 117, 26689–26702. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P=O Moiety as an Ambidextrous Hydrogen Bond Acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Nagy, P.I. Competing Intramolecular vs. Intermolecular Hydrogen Bonds in Solution. Int. J. Mol. Sci. 2014, 15, 19562–19633. [Google Scholar] [CrossRef] [Green Version]
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-Dependent Mechanisms of Intramolecular Proton Transfer in Aprotic Solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379. [Google Scholar] [CrossRef]
- Benner, S.A. Unusual Hydrogen Bonding Patterns and the Role of the Backbone in Nucleic Acid Information Transfer. ACS Cent. Sci. 2016, 2, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, J.P. Short strong hydrogen bonds: Can they explain enzymic catalysis? Chem. Biol. 1996, 3, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Bruchmann, B.; Lehn, J.-M. DYNAMERS: Dynamic polymers as self-healing materials. Chem. Soc. Rev. 2015, 44, 3786–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyalikh, A.; Emmler, T.; Shenderovich, I.; Zeng, Y.; Findenegg, G.H.; Buntkowsky, G. 2H-Solid State NMR and DSC Study of Isobutyric Acid in Mesoporous Silica Materials. Phys. Chem. Chem. Phys. 2007, 9, 2249–2257. [Google Scholar] [CrossRef]
- Li, Z.-T.; Wu, L.-Z. Hydrogen Bonded Supramolecular Materials; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Chan-Huot, M.; Dos, A.; Zander, R.; Sharif, S.; Tolstoy, P.M.; Compton, S.; Fogle, E.; Toney, M.D.; Shenderovich, I.; Denisov, G.S.; et al. NMR Studies of Protonation and Hydrogen Bond States of Internal Aldimines of Pyridoxal 5′-Phosphate Acid−Base in Alanine Racemase, Aspartate Aminotransferase, and Poly-L-lysine. J. Am. Chem. Soc. 2013, 135, 18160–18175. [Google Scholar] [CrossRef]
- Hynes, J.T.; Klinman, J.P.; Limbach, H.-H.; Schowen, R.L. Hydrogen-Transfer Reactions; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Segura, J.L.; Mancheno, M.J.; Zamora, F. Covalent Organic Frameworks Based on Schiff-base Chemistry: Synthesis, Properties and Potential Applications. Chem. Soc. Rev. 2016, 45, 5635–5671. [Google Scholar] [CrossRef]
- Burrows, A.D. Crystal Engineering Using Multiple Hydrogen Bonds. Struct. Bonds 2004, 108, 55–96. [Google Scholar]
- Pietrzak, M.; Wehling, J.P.; Kong, S.; Tolstoy, P.M.; Shenderovich, I.G.; Lopez, C.; Claramunt, R.M.; Elguero, J.; Denisov, G.S.; Limbach, H.-H. Symmetrization of Cationic Hydrogen Bridges of Protonated Sponges Induced by Solvent and Counteranion Interactions as Revealed by NMR Spectroscopy. Chem. Eur. J. 2010, 16, 1679–1690. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowski, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [Green Version]
- Montis, R.; Hursthouse, M.B. Surprisingly complex supramolecular behaviour in the crystal structures of a family of mono-substituted salicylic acids. CrystEngComm 2012, 14, 5242–5254. [Google Scholar] [CrossRef]
- Golubev, N.S.; Smirnov, S.N.; Schah-Mohammedi, P.; Shenderovich, I.G.; Denisov, G.S.; Gindin, V.A.; Limbach, H.-H. Study of Acid-Base Interaction by Means of Low-Temperature NMR Spectra. Structure of Salicylic Acid. Russ. J. Gen. Chem. 1997, 67, 1082–1087. [Google Scholar]
- Machado, T.C.; Kuminek, G.; Cardoso, S.G.; Rodríguez-Hornedo, N. The role of pH and dose/solubility ratio on cocrystal dissolution, drug supersaturation and precipitation. Eur. J. Pharm. Sci. 2020, 152, 105422. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Chen, J.-M.; Lub, T.-B. Two polymorphs and one hydrate of a molecular salt involving phenazopyridine and salicylic acid. CrystEngComm 2013, 15, 7852–7855. [Google Scholar] [CrossRef]
- Bolla, G.; Sanphui, P.; Nangia, A. Solubility Advantage of Tenoxicam Phenolic Cocrystals Compared to Salts. Cryst. Growth Des. 2013, 13, 1988–2003. [Google Scholar] [CrossRef]
- Glidewell, C.; Low, J.N.; Skakle, J.M.S.; Wardell, J.L. 3-Nitrophthalic acid: C(4) and R22(8) motifs of O-H⋯O hydrogen bonds generate sheets which are linked by C-H⋯O hydrogen bonds. Acta Cryst. 2003, C59, o144–o146. [Google Scholar]
- Smith, G.; Wermuth, U.D.; Young, D.J.; White, J.M. The 1:1 proton-transfer compounds of 4-(phenyldiazenyl)aniline (aniline yellow) with 3-nitrophthalic, 4-nitrophthalic and 5-nitroisophthalic acids. Acta Cryst. 2008, C64, o123–o127. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D. Proton-transfer compounds of isonipecotamide with the aromatic dicarboxylic acids 4-nitrophthalic, 4,5-dichlorophthalic, 5-nitroisophthalic and terephthalic acid. Acta Cryst. 2011, 67, o259–o264. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Wang, D.; Du, S.; Linhe, Q.; Fu, M.; Wu, S. Crystal and Molecular Structure of Two Proton Transfer Compounds from Quinolin-8-ol, 4-nitro-phthalic Acid, and 1,5-Naphthalenedisulfonic Acid. J. Chem. Crystallogr. 2014, 44, 435–441. [Google Scholar] [CrossRef]
- Saunders, L.K.; Nowell, H.; Hatcher, L.E.; Shepherd, H.J.; Teat, S.J.; Allan, D.R.; Raithby, P.R.; Wilson, C.C. Exploring short strong hydrogen bonds engineered in organic acid molecular crystals for temperature dependent proton migration behaviour using single crystal synchrotron X-ray diffraction (SCSXRD). CrystEngComm 2019, 21, 5249–5260. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P.; Halperin, O.; Pros, E.; Schwarzkopf, V. Additionsprodukte von Zinnhalogeniden an Carbonylverbindungen, I. Beitrag zur Theorie der Halochromieerscheinungen. J. Liebegs Ann. Chem. 1910, 376, 285–310. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Friedmann, B.; Goldberg, Z.; Pros, E.; Schwarzkopf, V. Beitrag zur Theorie der Halochromieerscheinungen II. Unter experimenteller Mitarbeit. J. Liebegs Ann. Chem. 1911, 383, 92–155. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P. Zur Kenntnis der sauren Salze der Carbonsäuren. Ber. Deutsch. Chem. Ges. 1914, 47, 1580–1596. [Google Scholar] [CrossRef] [Green Version]
- Marechal, Y.; Witkowski, A. Infrared Spectra of H-Bonded Systems. J. Chem. Phys. 1968, 48, 3637. [Google Scholar] [CrossRef]
- Marechal, Y.; Durig, J. Vibration Spectra and Structure; Elsevier: Amsterdam/Holland, The Netherlands, 1997. [Google Scholar]
- Wójcik, M.J. Infrared Spectra of Hydrogen-Bonded Salicylic Acid and Its Derivatives. Salicylic Acid and Acetylsalicylic Acid. Chem. Phys. Lett. 1981, 83, 503–507. [Google Scholar] [CrossRef]
- Issaoui, N.; Rekik, N.; Oujia, B.; Wójcik, M.J. Theoretical Infrared Line Shapes of H-Bonds within the Strong Anharmonic Coupling Theory. Fermi Resonances Effects. Int. J. Quant. Chem. 2010, 110, 2583–2602. [Google Scholar] [CrossRef]
- Wójcik, M.J.; Szczeponek, K.; Boczar, M. Theoretical Study of Multidimensional Proton Tunnelling in Benzoic Acid Dimer. Int. J. Mol. Sci. 2003, 4, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Kearley, G.J.; Fillaux, F.; Baron, M.-H.; Bennington, S.; Tomkinson, J. A New Look at Proton Transfer Dynamics along the Hydrogen Bonds in Amides and Peptides. Science 1994, 264, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Stepanian, S.G.; Reva, I.D.; Radchenko, E.D.; Sheina, G.G. Infrared spectra of benzoic acid monomers and dimers in argon matrix. Vib. Spectrosc. 1996, 11, 123–133. [Google Scholar] [CrossRef]
- Fillaux, F.; Limage, M.H.; Romain, F. Quantum proton transfer and interconversion in the benzoic acid crystal: Vibrational spectra, mechanism and theory. Chem. Phys. 2002, 276, 181–210. [Google Scholar] [CrossRef]
- Shipman, S.T.; Douglass, P.C.; Yoo, H.S.; Hinkle, C.E.; Mierzejewski, E.L.; Pate, B.H. Vibrational dynamics of carboxylic acid dimers in gas and dilute solution. Phys. Chem. Chem. Phys. 2007, 9, 4572–4586. [Google Scholar] [CrossRef]
- Worley, J.D. A Family of Hydrogen Bonds in the Model System Salicylic Acid-Toluene-Water. J. Chem. Educ. 1993, 70, 417–420. [Google Scholar] [CrossRef]
- Zhu, L.; Al-Kaysi, R.O.; Dillon, R.J.; Tham, F.S.; Bardeen, C.J. Crystal Structures and Photophysical Properties of 9-Anthracene Carboxylic Acid Derivatives for Photomechanical Applications. Cryst. Growth Des. 2011, 11, 4975–4983. [Google Scholar] [CrossRef]
- Neumann, M.A.; Craciun, S.; Corval, A.; Johnson, M.R.; Horsewil, A.J.; Benderskii, V.A.; Trommsdorff, H.P. Proton Dynamics and the Tautomerization Potential in Benzoic Acid Crystals. Ber. Busenges. Phys. Chem 1998, 102, 325–334. [Google Scholar] [CrossRef]
- Marushkevich, K.; Khriachtchev, L.; Rasanen, M.; Melavuori, M.; Lundell, J. Dimers of the Higher-Energy Conformer of Formic Acid: Experimental Observation. J. Phys. Chem. A 2012, 116, 2101–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marushkevich, K.; Khriachtchev, L.; Lundell, J.; Rasanen, M. cis-trans Formic Acid Dimer: Experimental Observation and Improved Stability against Proton Tunneling. J. Am. Chem. Soc. 2006, 128, 12060–12061. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, M.G.; Bushmarinova, I.S.; Lyssenko, K.A. Z-effect reversal in carboxylic acid associates. Chem. Commun. 2016, 52, 6593–6596. [Google Scholar] [CrossRef]
- Radosavljevic Evans, I.; Howard, J.A.K.; Evans, J.S.O.; Postlethwaite, S.R.; Johnson, M.R. Polymorphism and hydrogen bonding in cinchomeronic acid: A variable temperature experimental and computational study. CrystEngComm 2008, 10, 1404–1409. [Google Scholar] [CrossRef]
- Thomas, L.H.; Craig, G.A.; Gutmann, M.J.; Parkin, A.; Shanklande, K.; Wilson, C.C. Conformational polymorphism of the molecular complex of 3-fluorobenzoic acid with 4-acetylpyridine. CrystEngComm 2011, 13, 3349–3354. [Google Scholar] [CrossRef]
- Kubitschke, J.; Lange, H.; Strutz, H. Carboxylic Acids, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Akcakayiran, D.; Mauder, D.; Hess, C.; Sievers, T.K.; Kurth, D.G.; Shenderovich, I.; Limbach, H.-H.; Findenegg, G.H. Carboxylic Acid-Doped SBA-15 Silica as a Host for Metallo-supramolecular Coordination Polymers. J. Phys. Chem. B 2008, 112, 14637–14647. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. First-Principle Molecular Dynamics Study of Selected Schiff and Mannich Bases: Application of Two-Dimensional Potential of Mean Force to Systems with Strong Intramolecular Hydrogen Bonds. J. Chem. Theory Comput. 2008, 4, 375–384. [Google Scholar] [CrossRef]
- Sobczyk, L.; Chudoba, D.M.; Tolstoy, T.M.; Filarowski, A. Some Brief Notes on Theoretical and Experimental Investigations of Intramolecular Hydrogen Bonding. Molecules 2016, 21, 1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. Simplified Calculation Approaches Designed to Reproduce the Geometry of Hydrogen Bonds in Molecular Complexes in Aprotic Solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef] [Green Version]
- Tolstoy, P.M.; Schah-Mohammedi, P.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. Characterization of Fluxional Hydrogen-Bonded Complexes of Acetic Acid and Acetate by NMR: Geometries and Isotope and Solvent Effects. J. Am. Chem. Soc. 2004, 126, 5621–5634. [Google Scholar] [CrossRef]
- Leiserowitz, L. Molecular Packing Modes. Carboxylic Acids. Acta Cryst. B 1976, 32, 775–802. [Google Scholar] [CrossRef] [Green Version]
- Middlemiss, D.S.; Facchini, M.; Morrison, C.A.; Wilson, C.C. Small energy differences in molecular crystals: A first principles study of tautomerism and dynamics in benzoic acid derivatives. CrystEngComm 2007, 9, 777–785. [Google Scholar] [CrossRef]
- Borissova, A.O.; Lyssenko, K.A.; Gurinov, A.A.; Shenderovich, I.G. Energy Analysis of Competing Non-Covalent Interaction in 1:1 and 1:2 Adducts of Collidine with Benzoic Acids by Means of X-Ray Diffraction. Z. Phys. Chem. 2013, 227, 775–790. [Google Scholar] [CrossRef]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. [Google Scholar] [CrossRef]
- Schuster, P. Energy surfaces for hydrogen bonded systems. In The Hydrogen Bond; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland: Amsterdam/Holland, The Netherlands, 1976; Volume II. [Google Scholar]
- Emel’yanenko, V.N.; Stange, P.; Feder-Kubis, J.; Verevkin, S.P.; Ludwig, R. Dissecting intermolecular interactions in the condensed phase of ibuprofen and related compounds: The specific role and quantification of hydrogen bonding and dispersion forces. Phys. Chem. Chem. Phys. 2020, 22, 4896–4904. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. Anti-Electrostatic Hydrogen Bonds. Angew. Chem. Int. Ed. 2014, 53, 11214–11217. [Google Scholar] [CrossRef]
- Olovsson, I.; Ptasiewicz-Bak, H.; Gustafsson, T.; Majerz, I. Asymmetric hydrogen bonds in centrosymmetric environment: Neutron study of very short hydrogen bonds in potassium hydrogen dichloromaleate. Acta Cryst. 2001, B57, 311–316. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. Towards an uniced hydrogen-bond theory. J. Mol. Struct. 2000, 552, 1–15. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The Nature of the Hydrated Excess Proton in Water. Nature 1999, 397, 601–604. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Marx, D.; Klein, M.L.; Parrinello, M. On the quantum nature of the shared proton in hydrogen bonds. Science 1997, 275, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Chandra, A.; Tuckerman, M.E. Aqueous basic solutions: Hydroxide solvation, structural diffusion, and comparison to the hydrated proton. Chem. Rev. 2010, 110, 2174–2216. [Google Scholar] [CrossRef]
- Dopieralski, P.P.; Perrin, C.L.; Latajka, Z. On the Intramolecular Hydrogen Bond in Solution: Car–Parrinello and Path Integral Molecular Dynamics Perspective. J. Chem. Theory Comput. 2011, 7, 3505–3513. [Google Scholar] [CrossRef]
- Jezierska-Mazzarello, A.; Vuilleumier, R.; Panek, J.J.; Ciccotti, G. Molecular Property Investigations of an ortho-Hydroxy Schiff Base Type Compound with the First-Principle Molecular Dynamics Approach. J. Phys. Chem. B 2010, 114, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Panek, J.J.; Jezierska-Mazzarello, A.B.; Lipkowski, P.; Martyniak, A.; Filarowski, A. Comparison of resonance assisted and charge assisted effects in strengthening of hydrogen bonds in dipyrrins. J. Chem. Inf. Model. 2014, 54, 86–95. [Google Scholar] [CrossRef]
- Brela, M.; Stare, J.; Pirc, G.; Sollner-Dolenc, M.; Boczar, M.; Wójcik, M.J.; Mavri, J. Car–Parrinello Simulation of the Vibrational Spectrum of a Medium Strong Hydrogen Bond by Two-Dimensional Quantization of the Nuclear Motion: Application to 2-Hydroxy-5-nitrobenzamide. J. Phys. Chem. B 2012, 116, 4510–4518. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Viloca, M.; Gonzalez-Lafont, A.; Lluch, J.M. Asymmetry of the Hydrogen Bond of Hydrogen Phthalate Anion in Solution. A QM/MM Study. J. Am. Chem. Soc. 1999, 121, 9198–9207. [Google Scholar] [CrossRef]
- Pirc, G.; Stare, J.; Mavri, J. Car–Parrinello simulation of hydrogen bond dynamics in sodium hydrogen bissulfate. J. Chem. Phys. 2010, 132, 224506. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Burtsev, A.P.; Denisov, G.S.; Golubev, N.S.; Limbach, H.-H. Influence of the Temperature-Dependent Dielectric Constant on the H/D Isotope Effects on the NMR Chemical Shifts and the Hydrogen Bond Geometry of Collidine-HF Complex in CDF3/CDClF2 Solution. Magn. Reson. Chem. 2001, 39, S91–S99. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Giba, I.S.; Kostin, M.A.; Denisov, G.; Shenderovich, I.G.; Tolstoy, P. Cyclic Trimers of Phosphinic Acids in Polar Aprotic Solvent: Symmetry, Chirality and H/D Isotope Effects on NMR Chemical Shifts. Phys. Chem. Chem. Phys. 2018, 20, 4901–4910. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Lesnichin, S.B.; Limbach, H.-H.; Shenderovich, I.G. How Short is the Strongest Hydrogen Bond in the Proton-Bound Homodimers of Pyridine Derivatives? J. Phys. Chem. A 2014, 118, 10804–10812. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Ustimchuk, D.O.; Filarowski, A.; Tolstoy, P.M. H/D Isotope Effects on 1H NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules 2020, 25, 1907. [Google Scholar] [CrossRef] [Green Version]
- Tolstoy, P.M.; Smirnov, S.N.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. NMR studies of solid state, solvent and H/D isotope effects on hydrogen bond geometries of 1:1 complexes of collidine with carboxylic acids. J. Mol. Struct. 2004, 700, 19–27. [Google Scholar] [CrossRef]
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G.; Limbach, H.-H. Geometrical Features of Hydrogen Bonded Complexes Involving Sterically Hindered Pyridines. J. Phys. Chem. A 2006, 110, 10872–10879. [Google Scholar] [CrossRef]
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D Isotope Effects on NMR Chemical Shifts of [FHF]- Ion Based on Calculations of Nuclear Magnetic Shielding Tensor Surface. Z. Phys. Chem. 2003, 217, 1549–1563. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Maximum Value of the Chemical Shift in the 1H NMR Spectrum of a Hydrogen-Bonded Complex. Russ. J. Gen. Chem. 2006, 76, 501–506. [Google Scholar] [CrossRef]
- Maréchal, Y. The Hydrogen Bond and the Water Molecule: The Physics and Chemistry of Water, Aqueous and Bio Media; Elsevier: Amsterdam, The Netherlands; Oxford, UK, 2007. [Google Scholar]
- Nelson, D.L.; Cox, M.M. Principles of Biochemistry, 4th ed.; W.H. Freeman: New York, NY, USA, 2005. [Google Scholar]
- Flakus, H.T.; Hachuła, B.; Hołaj-Krzak, J.T.; Al-Agel, F.A.; Rekik, N. “Long-distance” H/D isotopic self-organization phenomena in scope of the infrared spectra of hydrogen-bonded terephthalic and phthalic acid crystals. Spectrochim. Acta A 2017, 173, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Flakus, H.T.; Chełmecki, M. Infrared spectra of the hydrogen bond in benzoic acid crystals: Temperature and polarization effects. Spectrochim. Acta A 2002, 58, 179–196. [Google Scholar] [CrossRef]
- Ghalla, H.; Rekik, N.; Michta, A.; Oujia, B.; Flakus, H.T. Theoretical modeling of infrared spectra of the hydrogen and deuterium bond in aspirin crystal. Spectrochim. Acta A 2010, 173, 37–47. [Google Scholar] [CrossRef]
- Flakus, H.T.; Hachuła, B. The source of similarity of the IR spectra of acetic acid in the liquid and solid-state phases. Vib. Spectrosc. 2011, 56, 170–176. [Google Scholar] [CrossRef]
- Flakus, H.T.; Rekik, N.; Jarczyk, A. Polarized IR Spectra of the Hydrogen Bond in 2-Thiopheneacetic Acid and 2-Thiopheneacrylic Acid Crystals: H/D Isotopic and Temperature Effects. J. Phys. Chem. A 2012, 116, 2117–2130. [Google Scholar] [CrossRef]
- Flakus, H.T.; Hachuła, B.; Hołaj-Krzak, J.T. Long-distance inter-hydrogen bond coupling effects in the polarized IR spectra of succinic acid crystals. Spectrochim. Acta A 2015, 142, 126–134. [Google Scholar] [CrossRef]
- Flakus, H.T.; Jarczyk-Jędryka, A. Temperature and H/D Isotopic Effects in the IR Spectra of the Hydrogen Bond in Solid-State 2-Furanacetic Acid and 2-Furanacrylic Acid. J. Atom. Mol. Opt. Phys. 2012, 2012, 125471. [Google Scholar] [CrossRef] [Green Version]
- Flakus, H.T. Vibronic model for H/D isotopic self-organization effects in centrosymmetric dimers of hydrogen bonds. J. Mol. Struct. 2003, 646, 15–23. [Google Scholar] [CrossRef]
- Rodziewicz, P.; Doltsinis, N.L. Formic Acid Dimerization: Evidence for Species Diversity from First Principles Simulations. J. Phys. Chem. A 2009, 113, 6266–6274. [Google Scholar] [CrossRef]
- Majerz, I. Proton Transfer Influence on Geometry and Electron Density in Benzoic Acid–Pyridine Complexes. Helv. Chim. Acta 2016, 99, 286–295. [Google Scholar] [CrossRef]
- Bournay, J.; Marechal, Y. Anamalous isotope effect in the H bonds of acetic acid dimers. J. Chem. Phys. 1973, 59, 5077–5087. [Google Scholar] [CrossRef]
- Vener, M.V.; Kuhn, O.; Bowman, J.M. Vibrational spectrum of the formic acid in the OH stretch region. A model 3D study. Chem. Phys. Lett. 2001, 349, 562–570. [Google Scholar] [CrossRef]
- Kwocz, A.; Panek, J.J.; Jezierska, A.; Hetmańczyk, Ł.; Pawlukojć, A.; Kochel, A.; Lipkowski, P.; Filarowski, A. A molecular roundabout: Triple cycle-arranged hydrogen bonds in light of experiment and theory. New J. Chem. 2018, 42, 19467–19477. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A. Intergrated intensity of νs(OH) absorption bands in bent hydrogen bonds in ortho-dialkylaminomethyl phenols. Vib. Spectrosc. 1996, 12, 15–24. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Lipkowski, P.; Pawlukojć, A. Inelastic neutron scattering and vibrational spectra of 2-(N-methyl-α-iminoethyl)-phenol and 2-(N-methyliminoethyl)-phenol: Experimental and theoretical approach. J. Mol. Struct. 2008, 880, 97–108. [Google Scholar] [CrossRef]
- Zeegers-Huyskens, T. Influence of the nature of the hydrogen bond on the isotopic ratio νAH/νAD. J. Mol. Struct. 1992, 217, 239–252. [Google Scholar] [CrossRef]
- Pająk, J.; Maes, G.; De Borggraeve, W.M.; Boens, N.; Filarowski, A. Matrix-isolation FT-IR and theoretical investigation of the vibrational properties of the sterically hindered ortho-hydroxy acylaromatic Schiff bases. J. Mol. Struct. 2007, 844–845, 83–93. [Google Scholar] [CrossRef]
- Grzegorzek, J.; Filarowski, A.; Mielke, Z. The photoinduced isomerization and its implication in the photo-dynamical processes in two simple Schiff bases isolated in solid argon. Phys. Chem. Chem. Phys. 2011, 13, 16596–16605. [Google Scholar] [CrossRef]
- Majerz, I.; Pawlukojć, A.; Sobczyk, L.; Dziembowska, T.; Grech, E.; Szady-Chełmieniecka, A. The infrared, Raman and inelastic neutron scattering studies on 5-nitro-N-salicylideneethylamine. J. Mol. Struct. 2000, 552, 243–247. [Google Scholar] [CrossRef]
- Grzegorzek, J.; Mielke, Z.; Filarowski, A. C=N–N=C conformational isomers of 2′-hydroxyacetophenone azine: FTIR matrix isolation and DFT study. J. Mol. Struct. 2010, 976, 371–376. [Google Scholar] [CrossRef]
- Pająk, J.; Maes, G.; De Borggraeve, W.M.; Boens, N.; Filarowski, A. Matrix-isolation FT-IR and theoretical investigation of the competitive intramolecular hydrogen bonding in 5-methyl-3-nitro-2-hydroxyacetophenone. J. Mol. Struct. 2008, 880, 86–96. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons, with Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005. [Google Scholar]
- Marques, M.P.M.; Batista de Carvalho, L.A.E.; Valero, R.; Machado, N.F.L.; Parker, S.F. An inelastic neutron scattering study of dietary phenolic acids. Phys. Chem. Chem. Phys. 2014, 16, 7491–7500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine-Vive, F.; Johnson, M.R.; Kearley, G.J.; Cowan, J.A.; Howard, J.A.K.; Parker, S.F. Phonon driven proton transfer in crystals with short strong hydrogen bonds. J. Chem. Phys. 2006, 124, 234503. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.R.; Trommsdorff, H.P. Vibrational spectra of crystalline formic and acetic acid isotopologues by inelastic neutron scattering and numerical simulations. Chem. Phys. 2009, 355, 118–122. [Google Scholar] [CrossRef]
- Beran, G.J.O.; Chronister, E.L.; Daemen, L.L.; Moehlig, A.R.; Mueller, L.J.; Oomens, J.; Rice, A.; Santiago-Dieppa, D.R.; Tham, F.S.; Theel, K.; et al. Vibrations of a chelated proton in a protonated tertiary diamine. Phys. Chem. Chem. Phys. 2011, 13, 20380–20392. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and Spectral Properties of the Protonated Homodimer of Pyridine in the Liquid and Solid States. A Combined NMR, X-ray Diffraction and Inelastic Neutron Scattering Study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef]
- Miyazawa, T.; Pitzer, K.S. Internal Rotation and Infrared Spectra of Formic Acid Monomer and Normal Coordinate Treatment of Out-of-Plane Vibrations of Monomer, Dimer, and Polymer. J. Chem. Phys. 1959, 30, 1076–1086. [Google Scholar] [CrossRef]
- Novak, A. Hydrogen bonding in solids correlation of spectroscopic and crystallographic data. Struct. Bonding 1974, 18, 177–216. [Google Scholar]
- Howard, J.; Tomkinson, J.; Eckert, J.; Goldstone, J.A.; Taylor, A.D. Inelastic neutron scattering studies of some intramolecular hydrogen bonded complexes: A new correlation of γ(OHO) vs. R (OO). J. Chem. Phys. 1983, 78, 3150–3155. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Series on Neutron Techniques and Applications, Vibrational Spectroscopy with Neutrons; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2005. [Google Scholar]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Hockney, R.W. The potential calculation and some applications. Methods Comput. Phys. 1970, 9, 136–211. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- CPMD, Version 3.17.1; Copyright IBM Corp. (1990–2004) Copyright MPI für Festkörperforschung Stuttgart (1997–2001). Available online: http://www.cpmd.org/ (accessed on 14 October 2003).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gnuplot, Version 4.2; An Interactive Plotting Program; Thomas Williams and Colin Kelley. 2007. Available online: https://sourceforge.net/projects/gnuplot/ (accessed on 14 October 2007).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Soc. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Pulay, P.; Fogarasi, G.; Pang, F.; Boggs, J.E. Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc. 1979, 101, 2550–2560. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Van Alsenoy, C. Gar2ped; University of Antwerp: Antwerpen, Belgium, 1995. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Method | Bridge 1 | Bridge 2 | ||||
|---|---|---|---|---|---|---|---|
| d(OH) | d(OO) | d(OH) | d(OO) | OHO[°] | COH…O[°] | ||
| 3, monomer | CPMD | 0.993 ± 0.022 | 2.728 ± 0.151 | - | - | - | - |
| 3, dimer | - | 1.027 ± 0.032 | 2.634 ± 0.095 | 1.028 ± 0.034 | 2.633 ± 0.091 | - | - |
| 4, monomer | - | 1.005 ± 0.022 | 2.587 ± 0.088 | - | - | - | - |
| 4, dimer | - | 1.028 ± 0.036 | 2.650 ± 0.122 | 1.028 ± 0.037 | 2.653 ± 0.115 | - | - |
| 3, monomer | DFT | 0.978 | 2.670 | - | - | 150.2 | 64.4 |
| 3, dimer | - | 0.999 | 2.679 | 1.001 | 2.660 | 178.9 | 0.2 |
| 4, monomer | - | 0.985 | 2.583 | - | - | 160.0 | 50.1 |
| 4, dimer | - | 1.000 | 2.669 | 0.999 | 2.679 | 178.6 | 0.7 |
| 3, dimer | X-ray [38] | 0.84 | 2.698 | 0.84 | 2.698 | 155.5 | - |
| 3, oligomer | - | 0.84 | 2.681 | - | - | - | - |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates. Molecules 2020, 25, 4720. https://doi.org/10.3390/molecules25204720
Jóźwiak K, Jezierska A, Panek JJ, Goremychkin EA, Tolstoy PM, Shenderovich IG, Filarowski A. Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates. Molecules. 2020; 25(20):4720. https://doi.org/10.3390/molecules25204720
Chicago/Turabian StyleJóźwiak, Kinga, Aneta Jezierska, Jarosław J. Panek, Eugene A. Goremychkin, Peter M. Tolstoy, Ilya G. Shenderovich, and Aleksander Filarowski. 2020. "Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates" Molecules 25, no. 20: 4720. https://doi.org/10.3390/molecules25204720