Cathepsin B-Cleavable Cyclopeptidic Chemotherapeutic Prodrugs


Abstract
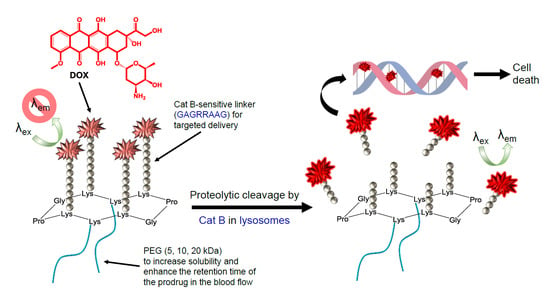
:
1. Introduction
2. Results and Discussion
2.1. Syntheses
2.2. Fluorescence Quenching
2.3. Cleavability with Cat B from Human Placenta
2.4. Cytotoxicity of cPCPs
2.5. Uptake and Intracellular Accumulation of DOX-Containing cPCPs
2.6. Differences between the Intracellular and Nuclear DOX Fluorescence Measurements
2.7. Cell Cycle Analysis
2.8. Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Synthesis of DBCO-GAGRRAAG
3.4. General Procedure for the Synthesis of the PEGylated Cyclopeptidic Conjugates
3.4.1. Coupling of the DBCO Anchor(s)
3.4.2. PEGylation
3.4.3. Deprotection and Coupling of the Azido Anchors
3.4.4. Coupling of the DBCO-GAGRRAAG-DOX Moieties
3.5. General Procedure for the Synthesis of the Pentasubstituted Cyclopeptidic Conjugate
3.5.1. Deprotection and Coupling of the Azido Anchors
3.5.2. Coupling of the DBCO-GAGRRAAG-DOX Moieties
3.6. Fluorescence Quenching
3.7. Evaluation of DOX Release by Cat B
3.8. In Vitro Cell Studies
3.8.1. Cell Culture
3.8.2. Cytotoxicity Assays
3.8.3. Confocal Fluorescence Microscopy of cPCP Uptake and Accumulation in Cells
3.8.4. Flow Cytometry Measurements of Intracellular and Nuclear Accumulation of DOX from cPCPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ringsdorf, H. Structure and Properties of Pharmacologically Active Polymers. J. Polym. Sci. Polym. Symp. 1975, 51, 135–153. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nature Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Garanger, E.; Boturyn, D.; Renaudet, O.; Defrancq, E.; Dumy, P. Chemoselectively addressable template: A valuable tool for the engineering of molecular conjugates. J. Org. Chem. 2006, 71, 2402–2410. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherer, G.; Servis, C.; Corradin, G.; Blum, U.; Rivier, J.; Mutter, M. Total chemical synthesis, characterization, and immunological properties of an MHC class I model using the TASP concept for protein de novo design. Protein Sci. 1992, 1, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Floegel, R.; Mutter, M. Molecular dynamics conformational search of six cyclic peptides used in the template assembled synthetic protein approach for protein de novo design. Biopolymers 1992, 32, 1283–1310. [Google Scholar] [CrossRef]
- Tuchscherer, G.; Grell, D.; Mathieu, M.; Mutter, M. Extending the concept of template-assembled synthetic proteins. J. Peptide Res. 1999, 54, 185–194. [Google Scholar] [CrossRef]
- Mutter, M.; Tuchscherer, G. Non-native architectures in protein design and mimicry. Cell. Mol. Life Sci. 1997, 53, 851–863. [Google Scholar] [CrossRef]
- Garanger, E.; Boturyn, D.; Coll, J.L.; Favrot, M.C.; Dumy, P. Multivalent RGD synthetic peptides as potent alphaVbeta3 integrin ligands. Org. Biomol. Chem. 2006, 4, 1958–1965. [Google Scholar] [CrossRef]
- Foillard, S.; Jin, Z.H.; Garanger, E.; Boturyn, D.; Favrot, M.C.; Coll, J.L.; Dumy, P. Synthesis and biological characterisation of targeted pro-apoptotic peptide. Chembiochem 2008, 9, 2326–2332. [Google Scholar] [CrossRef] [PubMed]
- Foillard, S.; Sancey, L.; Coll, J.L.; Boturyn, D.; Dumy, P. Targeted delivery of activatable fluorescent pro-apoptotic peptide into live cells. Org. Biomol. Chem. 2009, 7, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Boturyn, D.; Coll, J.L.; Garanger, E.; Favrot, M.C.; Dumy, P. Template assembled cyclopeptides as multimeric system for integrin targeting and endocytosis. J. Am. Chem. Soc. 2004, 126, 5730–5739. [Google Scholar] [CrossRef] [PubMed]
- Sancey, L.; Ardisson, V.; Riou, L.M.; Ahmadi, M.; Marti-Batlle, D.; Boturyn, D.; Dumy, P.; Fagret, D.; Ghezzi, C.; Vuillez, J.P. In vivo imaging of tumour angiogenesis in mice with the alpha(v)beta (3) integrin-targeted tracer 99mTc-RAFT-RGD. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Galibert, M.; Sancey, L.; Renaudet, O.; Coll, J.L.; Dumy, P.; Boturyn, D. Application of click-click chemistry to the synthesis of new multivalent RGD conjugates. Org. Biomol. Chem. 2010, 8, 5133–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galibert, M.; Jin, Z.H.; Furukawa, T.; Fukumura, T.; Saga, T.; Fujibayashi, Y.; Dumy, P.; Boturyn, D. RGD-cyclam conjugate: Synthesis and potential application for positron emission tomography. Bioorg. Med. Chem. Lett. 2010, 20, 5422–5425. [Google Scholar] [CrossRef] [PubMed]
- Grigalevicius, S.; Chierici, S.; Renaudet, O.; Lo-Man, R.; Deriaud, E.; Leclerc, C.; Dumy, P. Chemoselective assembly and immunological evaluation of multiepitopic glycoconjugates bearing clustered Tn antigen as synthetic anticancer vaccines. Bioconjugate Chem. 2005, 16, 1149–1159. [Google Scholar] [CrossRef]
- Boturyn, D.; Defrancq, E.; Dolphin, G.T.; Garcia, J.; Labbe, P.; Renaudet, O.; Dumy, P. RAFT Nano-constructs: Surfing to biological applications. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2008, 14, 224–240. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharm. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Zunino, F.; Di Marco, A.; Zaccara, A.; Gambetta, R.A. The interaction of daunorubicin and doxorubicin with DNA and chromatin. Biochim. Biophys. Acta 1980, 607, 206–214. [Google Scholar] [CrossRef]
- Foglesong, P.D.; Reckord, C.; Swink, S. Doxorubicin inhibits human DNA topoisomerase I. Cancer Chemother Pharm. 1992, 30, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Bigioni, M.; Cipollone, A.; Rossi, C.; Goso, C.; Maggi, C.A.; Capranico, G.; Animati, F. Anthracyclines: Selected new developments. Curr. Med. Chem. Anticancer Agents 2001, 1, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Cairo, G.; Monti, E. Role of iron in anthracycline cardiotoxicity: New tunes for an old song? FASEB J. 1999, 13, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Benchekroun, M.N.; Sinha, B.K.; Robert, J. Doxorubicin-induced oxygen free radical formation in sensitive and doxorubicin-resistant variants of rat glioblastoma cell lines. FEBS Lett. 1993, 326, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Karukstis, K.K.; Thompson, E.H.; Whiles, J.A.; Rosenfeld, R.J. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys. Chem. 1998, 73, 249–263. [Google Scholar] [CrossRef]
- Perego, P.; Corna, E.; De Cesare, M.; Gatti, L.; Polizzi, D.; Pratesi, G.; Supino, R.; Zunino, F. Role of apoptosis and apoptosis-related genes in cellular response and antitumor efficacy of anthracyclines. Curr. Med. Chem. 2001, 8, 31–37. [Google Scholar] [CrossRef]
- Raj, S.; Franco, V.I.; Lipshultz, S.E. Anthracycline-induced cardiotoxicity: A review of pathophysiology, diagnosis, and treatment. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 315. [Google Scholar] [CrossRef]
- Zhong, Y.J.; Shao, L.H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Berdowska, I. Cysteine proteases as disease markers. Clin. Chim. Acta 2004, 342, 41–69. [Google Scholar] [CrossRef]
- Jedeszko, C.; Sloane, B.F. Cysteine cathepsins in human cancer. Biol. Chem. 2004, 38, 1017–1027. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef]
- Mohan, P.; Rapoport, N. Doxorubicin as a molecular nanotheranostic agent: Effect of doxorubicin encapsulation in micelles or nanoemulsions on the ultrasound-mediated intracellular delivery and nuclear trafficking. Mol. Pharm. 2010, 7, 1959–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouilloux, J.; Yushchenko, O.; Dereka, B.; Boso, G.; Babic, A.; Zbinden, H.; Vauthey, E.; Lange, N. Correction: Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: Part II—Co-loading of pheophorbide A and black hole quencher. Photochem. Photobiol. Sci. 2019, 18, 2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouilloux, J.; Yushchenko, O.; Dereka, B.; Boso, G.; Zbinden, H.; Vauthey, E.; Babic, A.; Lange, N. Correction: Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: Part I—Self-quenched prodrugs. Photochem. Photobiol. Sci. 2019, 18, 2814. [Google Scholar] [CrossRef] [Green Version]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; et al. PEG-doxorubicin conjugates: Influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activity. Bioconjug. Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Ruzza, P.; Quintieri, L.; Osler, A.; Calderan, A.; Biondi, B.; Floreani, M.; Guiotto, A.; Borin, G. Fluorescent, internally quenched, peptides for exploring the pH-dependent substrate specificity of cathepsin B. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2006, 12, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hulkower, K.I.; Butler, C.C.; Linebaugh, B.E.; Klaus, J.L.; Keppler, D.; Giranda, V.L.; Sloane, B.F. Fluorescent microplate assay for cancer cell-associated cathepsin B. Eur. J. Biochem. 2000, 267, 4165–4170. [Google Scholar] [CrossRef] [PubMed]
- Malugin, A.; Kopeckova, P.; Kopecek, J. Liberation of doxorubicin from HPMA copolymer conjugate is essential for the induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J. Control. Release 2007, 124, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rihova, B.; Strohalm, J.; Hovorka, O.; Subr, V.; Etrych, T.; Chytil, P.; Pola, R.; Plocova, D.; Boucek, J.; Ulbrich, K. Doxorubicin release is not a prerequisite for the in vitro cytotoxicity of HPMA-based pharmaceuticals: In vitro effect of extra drug-free GlyPheLeuGly sequences. J. Control. Release 2008, 127, 110–120. [Google Scholar] [CrossRef]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T.; et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 83–94. [Google Scholar]
- Khandare, J.; Minko, T. Polymer-drug conjugates: Progress in polymeric prodrugs. Prog. Polym. Sci. 2006, 31, 359–397. [Google Scholar] [CrossRef]
- Hovorka, O.; Subr, V.; Vetvicka, D.; Kovar, L.; Strohalm, J.; Strohalm, M.; Benda, A.; Hof, M.; Ulbrich, K.; Rihova, B. Spectral analysis of doxorubicin accumulation and the indirect quantification of its DNA intercalation. Eur. J. Pharm. Biopharm. 2010, 76, 514–524. [Google Scholar] [CrossRef]
- Kiyomiya, K.; Matsuo, S.; Kurebe, M. Mechanism of specific nuclear transport of adriamycin: The mode of nuclear translocation of adriamycin-proteasome complex. Cancer Res. 2001, 61, 2467–2471. [Google Scholar]
- Pavesi, A.; Adriani, G.; Tay, A.; Warkiani, M.E.; Yeap, W.H.; Wong, S.C.; Kamm, R.D. Engineering a 3D microfluidic culture platform for tumor-treating field application. Sci. Rep. 2016, 6, 26584. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Adler, J.; Parmryd, I. Quantifying colocalization by correlation: The Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A 2010, 77, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Vindelov, L.L. Flow Microfluorometric Analysis of Nuclear-DNA in Cells from Solid Tumors and Cell-Suspensions—New Method for Rapid Isolation and Staining of Nuclei. Virchows Arch. B 1977, 24, 227–242. [Google Scholar]
Sample Availability: Not available. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugate | Starting RAFT | N° of DOX Moieties | N° of PEG Side Chains | Mw of Each PEG Side Chain (kDa) | Mw (kDa) |
|---|---|---|---|---|---|
| cPCP4/52 (8) | cP24 | 4 | 2 | 5 | 17.4 |
| cPCP4/102 (9) | cP24 | 4 | 2 | 10 | 28.4 |
| cPCP2/20 (13) | cP12 | 2 | 1 | 20 | 24.2 |
| cPCP5 (15) | cP14 | 5 | - | - | 9.1 |
| Product Name | % G1 | % S | % G2 |
|---|---|---|---|
| CTR | 51.5 ± 3.7 | 25.6 ± 3.0 | 21.0 ± 0.4 |
| DOX | 30.4 ± 2.3 | 36.7 ± 11.9 | 31.1 ± 14.8 |
| cPCP4/52 | 48.5 ± 3.6 | 30.9 ± 1.1 | 18.1 ± 2.2 |
| cPCP4/102 | 51.2 ± 1.1 | 26.3 ± 0.9 | 20.2 ± 0.3 |
| cPCP2/20 | 49.5 ± 1.0 | 27.2 ± 1.0 | 21.1 ± 0.9 |
| cPCP5 | 45.2 ± 0.5 | 30.3 ± 5.4 | 22.1 ± 5.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herceg, V.; Bouilloux, J.; Janikowska, K.; Allémann, E.; Lange, N. Cathepsin B-Cleavable Cyclopeptidic Chemotherapeutic Prodrugs. Molecules 2020, 25, 4285. https://doi.org/10.3390/molecules25184285
Herceg V, Bouilloux J, Janikowska K, Allémann E, Lange N. Cathepsin B-Cleavable Cyclopeptidic Chemotherapeutic Prodrugs. Molecules. 2020; 25(18):4285. https://doi.org/10.3390/molecules25184285
Chicago/Turabian StyleHerceg, Viktorija, Jordan Bouilloux, Karolina Janikowska, Eric Allémann, and Norbert Lange. 2020. "Cathepsin B-Cleavable Cyclopeptidic Chemotherapeutic Prodrugs" Molecules 25, no. 18: 4285. https://doi.org/10.3390/molecules25184285