
Bcr-Abl Allosteric Inhibitors: Where We Are and Where We Are Going to

 , and
, and 
Abstract
:1. Introduction
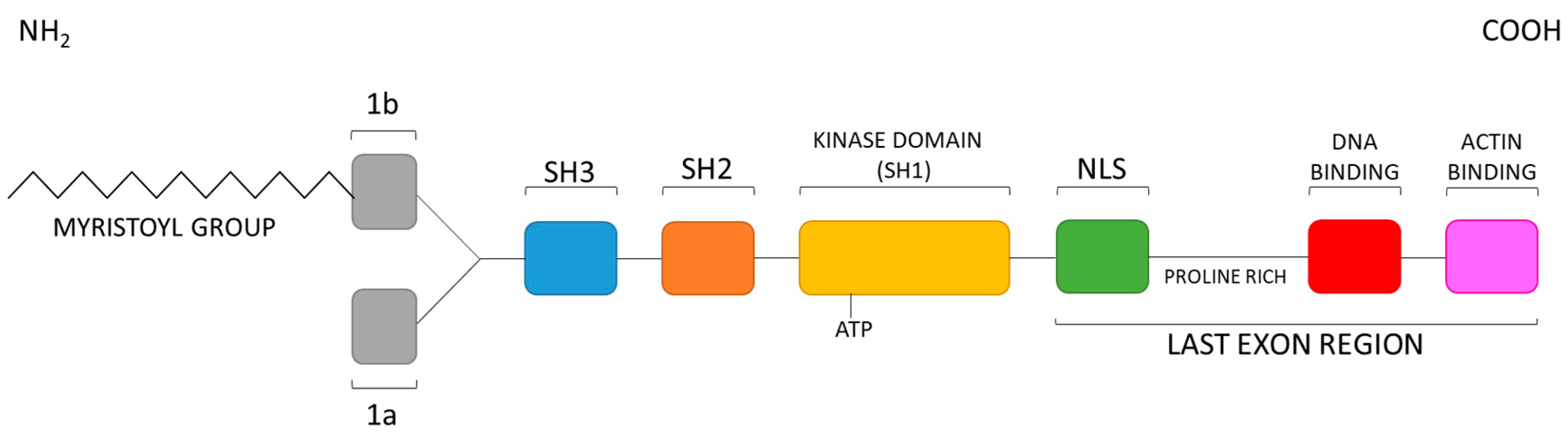
2. Structural Bases for the Auto-Inhibition of c-Abl Tyrosine Kinase
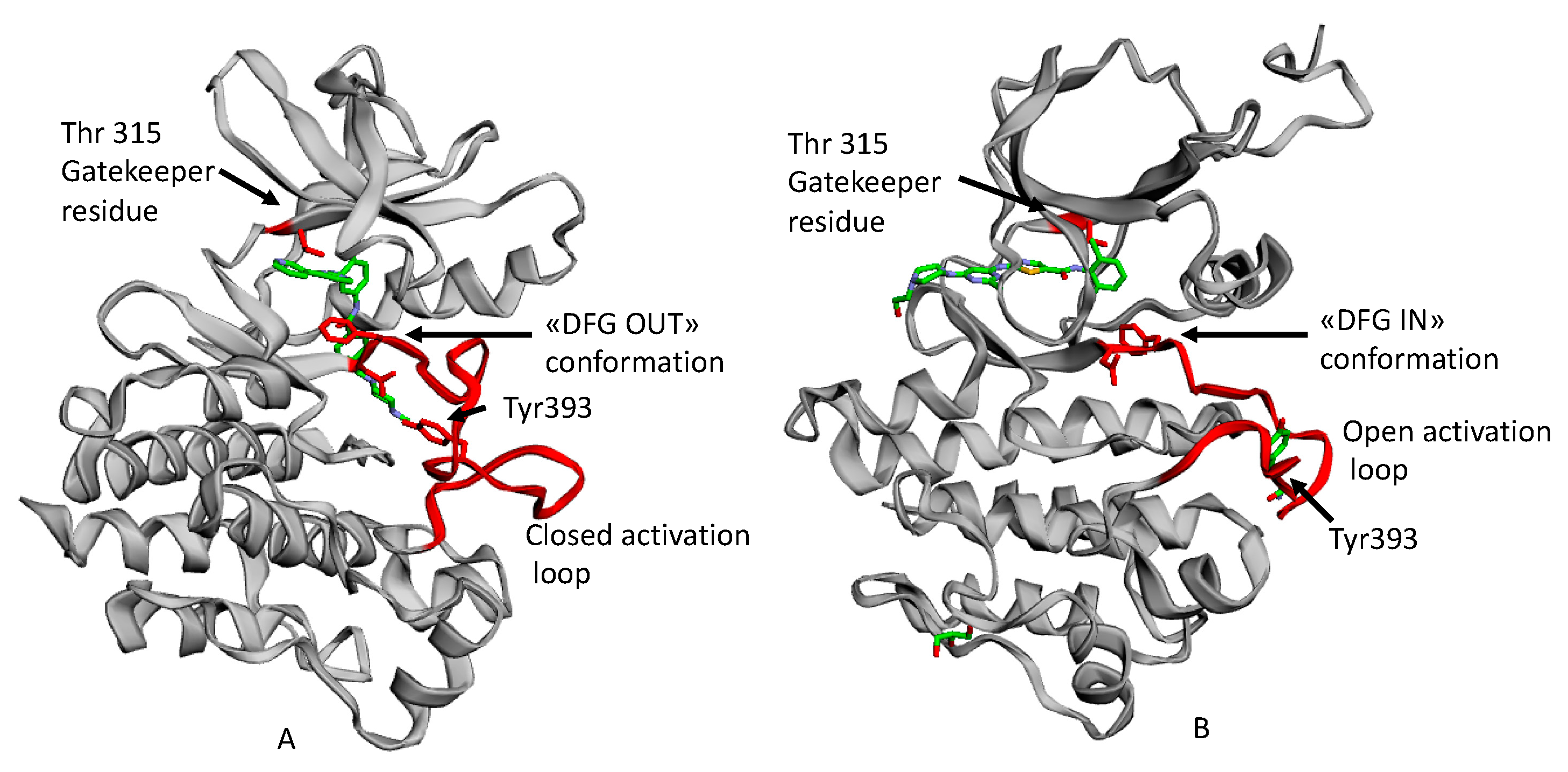
3. Bcr-Abl Tyrosine Kinase and Related Inhibitors
4. Bcr-Abl Orthosteric Inhibitors
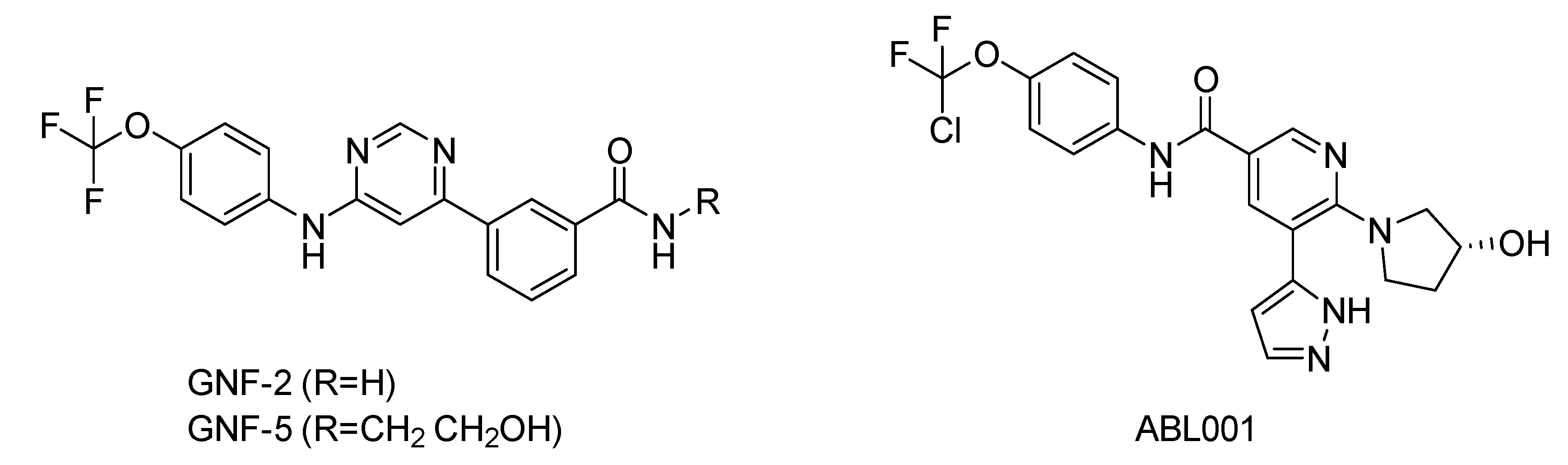
5. Bcr-Abl Allosteric Inhibitors
6. Non-Small Molecule Bcr-Abl Allosteric Inhibitors
7. In Silico Approaches for the Design of Allosteric Inhibitors
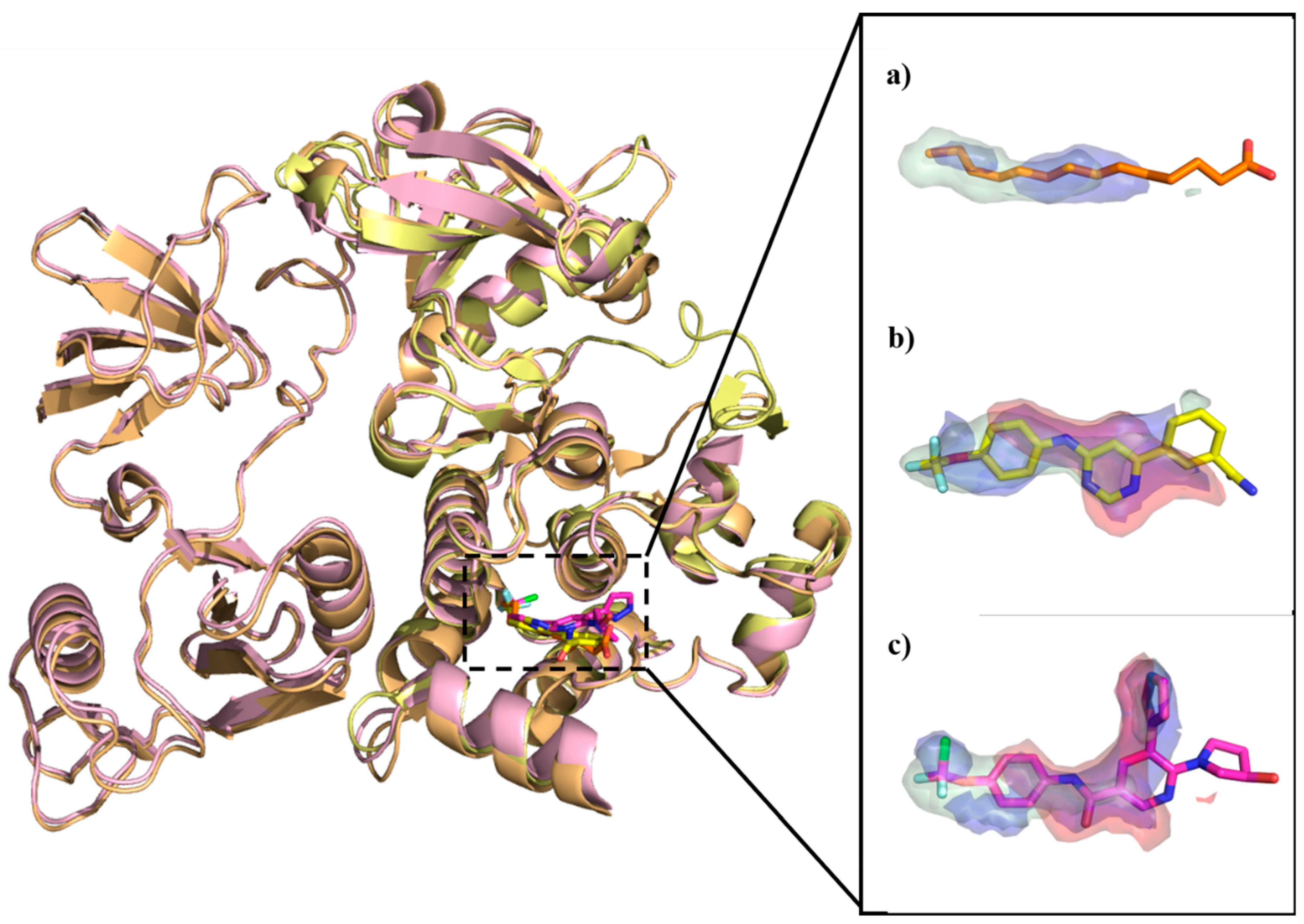
7.1. Case Study I: Molecular Interaction Fields Analysis
7.2. Case Study II: de novo Drug Design Based on Artifcial Intelligence
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zanforlin, E.; Zagotto, G.; Ribaudo, G. A Chemical Approach to Overcome Tyrosine Kinase Inhibitors Resistance: Learning from Chronic Myeloid Leukemia. Curr. Med. Chem. 2019, 26, 6033–6052. [Google Scholar]
- Tsai, C.-J.; Nussinov, R. A Unified View of “How Allostery Works. ” PLOS Comput. Biol. 2014, 10, e1003394. [Google Scholar]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar]
- Changeux, J.-P.; Christopoulos, A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. [Google Scholar]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar]
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-Aided Drug Design in Epigenetics. Front. Chem. 2018, 6, 57. [Google Scholar]
- Guo, J.; Zhou, H.-X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar]
- National Library of Medicine. Available online: https://pubmed.ncbi.nlm.nih.gov/23582321/ (accessed on 6 August 2020).
- Berezovsky, I.N.; Guarnera, E.; Zheng, Z.; Eisenhaber, B.; Eisenhaber, F. Protein function machinery: From basic structural units to modulation of activity. Curr. Opin. Struct. Biol. 2017, 42, 67–74. [Google Scholar]
- Goodey, N.M.; Benkovic, S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008, 4, 474–482. [Google Scholar]
- Palmieri, L.; Rastelli, G. αC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar]
- Nussinov, R.; Tsai, C.-J. Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol. Sci. 2014, 35, 256–264. [Google Scholar]
- Udi, Y.; Fragai, M.; Grossman, M.; Mitternacht, S.; Arad-Yellin, R.; Calderone, V.; Melikian, M.; Toccafondi, M.; Berezovsky, I.N.; Luchinat, C.; et al. Unraveling Hidden Regulatory Sites in Structurally Homologous Metalloproteases. J. Mol. Biol. 2013, 425, 2330–2346. [Google Scholar]
- Szczylik, C.; Skorski, T.; Nicolaides, N.C.; Manzella, L.; Malaguarnera, L.; Venturelli, D.; Gewirtz, A.M.; Calabretta, B. Selective inhibition of leukemia cell proliferation by BCR-ABL antisense oligodeoxynucleotides. Science 1991, 253, 562–565. [Google Scholar]
- Wang, J.Y.J. Nuclear protein tyrosine kinases. Trends Biochem. Sci. 1994, 19, 373–376. [Google Scholar]
- Van Etten, R.A.; Jackson, P.; Baltimore, D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell 1989, 58, 669–678. [Google Scholar]
- Abelson, H.T.; Rabstein, L.S. Lymphosarcoma: Virus-induced Thymic-independent Disease in Mice. Cancer Res. 1970, 30, 2213–2222. [Google Scholar]
- Renshaw, M.W.; Capozza, M.A.; Wang, J.Y. Differential expression of type-specific c-abl mRNAs in mouse tissues and cell lines. Mol. Cell. Biol. 1988, 8, 4547–4551. [Google Scholar]
- Nagar, B.; Hantschel, O.; Young, M.A.; Scheffzek, K.; Veach, D.; Bornmann, W.; Clarkson, B.; Superti-Furga, G.; Kuriyan, J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003, 112, 859–871. [Google Scholar]
- Hantschel, O.; Nagar, B.; Guettler, S.; Kretzschmar, J.; Dorey, K.; Kuriyan, J.; Superti-Furga, G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 2003, 112, 845–857. [Google Scholar]
- Nagar, B.; Hantschel, O.; Seeliger, M.; Davies, J.M.; Weis, W.I.; Superti-Furga, G.; Kuriyan, J. Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 2006, 21, 787–798. [Google Scholar]
- Skora, L.; Mestan, J.; Fabbro, D.; Jahnke, W.; Grzesiek, S. NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. USA 2013, 110, E4437–E4445. [Google Scholar]
- Sonti, R.; Hertel-Hering, I.; Lamontanara, A.J.; Hantschel, O.; Grzesiek, S. ATP Site Ligands Determine the Assembly State of the Abelson Kinase Regulatory Core via the Activation Loop Conformation. J. Am. Chem. Soc. 2018, 140, 1863–1869. [Google Scholar]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 385, 595–602. [Google Scholar]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. eLife 2015, 4, e06074. [Google Scholar]
- Reynolds, C.R.; Islam, S.A.; Sternberg, M.J.E. EzMol: A Web Server Wizard for the Rapid Visualization and Image Production of Protein and Nucleic Acid Structures. J. Mol. Biol. 2018, 430, 2244–2248. [Google Scholar]
- Hantschel, O.; Superti-Furga, G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 33–44. [Google Scholar]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar]
- Sherbenou, D.W.; Hantschel, O.; Kaupe, I.; Willis, S.; Bumm, T.; Turaga, L.P.; Lange, T.; Dao, K.-H.; Press, R.D.; Druker, B.J.; et al. BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib. Blood 2010, 116, 3278–3285. [Google Scholar]
- Lamontanara, A.J.; Georgeon, S.; Tria, G.; Svergun, D.I.; Hantschel, O. The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat. Commun. 2014, 5, 5470. [Google Scholar]
- Lee, B.J.; Shah, N.P. Identification of TKI-Sensitive Point Mutations That Activate c-ABL Kinase Activity and Transformation Potential and Confer in Vitro Resistance to the Allosteric ABL Inhibitor GNF-5. Blood 2015, 126, 17. [Google Scholar]
- Lu, S.; Qiu, Y.; Ni, D.; He, X.; Pu, J.; Zhang, J. Emergence of allosteric drug-resistance mutations: New challenges for allosteric drug discovery. Drug Discov. Today 2020, 25, 177–184. [Google Scholar]
- Sawyers, C.L. Chronic myeloid leukemia. N. Engl. J. Med. 1999, 340, 1330–1340. [Google Scholar]
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar]
- Panjarian, S.; Iacob, R.E.; Chen, S.; Engen, J.R.; Smithgall, T.E. Structure and Dynamic Regulation of Abl Kinases. J. Biol. Chem. 2013, 288, 5443–5450. [Google Scholar]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar]
- Hantschel, O.; Grebien, F.; Superti-Furga, G. The Growing Arsenal of ATP-Competitive and Allosteric Inhibitors of BCR–ABL. Cancer Res. 2012. [Google Scholar]
- Kung, J.E.; Jura, N. Prospects for pharmacological targeting of pseudokinases. Nat. Rev. Drug Discov. 2019, 18, 501–526. [Google Scholar]
- Lu, X.; Smaill, J.B.; Ding, K. New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew. Chem. Int. Ed Engl. 2019. [Google Scholar]
- Liu, Y.; Shah, K.; Yang, F.; Witucki, L.; Shokat, K.M. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg. Med. Chem. 1998, 6, 1219–1226. [Google Scholar]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.Ø.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [Google Scholar]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.F.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar]
- Reddy, E.P.; Aggarwal, A.K. The ins and outs of bcr-abl inhibition. Genes Cancer 2012, 3, 447–454. [Google Scholar]
- Carofiglio, F.; Lopalco, A.; Lopedota, A.; Cutrignelli, A.; Nicolotti, O.; Denora, N.; Stefanachi, A.; Leonetti, F. Bcr-Abl Tyrosine Kinase Inhibitors in the Treatment of Pediatric CML. Int. J. Mol. Sci. 2020, 21, 4469. [Google Scholar]
- Hochhaus, A.; O’Brien, S.G.; Guilhot, F.; Druker, B.J.; Branford, S.; Foroni, L.; Goldman, J.M.; Müller, M.C.; Radich, J.P.; Rudoltz, M.; et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23, 1054–1061. [Google Scholar]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O’Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; et al. Nilotinib in Imatinib-Resistant CML and Philadelphia Chromosome–Positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.-S.; Xu, Q.; et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar]
- O’Hare, T.; Eide, C.A.; Tyner, J.W.; Corbin, A.S.; Wong, M.J.; Buchanan, S.; Holme, K.; Jessen, K.A.; Tang, C.; Lewis, H.A.; et al. SGX393 inhibits the CML mutant Bcr-AblT315I and preempts in vitro resistance when combined with nilotinib or dasatinib. Proc. Natl. Acad. Sci. USA 2008, 105, 5507–5512. [Google Scholar]
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar]
- Guarnera, E.; Tan, Z.W.; Zheng, Z.; Berezovsky, I.N. AlloSigMA: Allosteric signaling and mutation analysis server. Bioinforma. Oxf. Engl. 2017, 33, 3996–3998. [Google Scholar]
- Tan, Z.W.; Tee, W.-V.; Guarnera, E.; Booth, L.; Berezovsky, I.N. AlloMAPS: Allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res. 2019, 47, D265–D270. [Google Scholar]
- Tee, W.-V.; Guarnera, E.; Berezovsky, I.N. On the Allosteric Effect of nsSNPs and the Emerging Importance of Allosteric Polymorphism. J. Mol. Biol. 2019, 431, 3933–3942. [Google Scholar]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar]
- Nussinov, R.; Tsai, C.-J. The design of covalent allosteric drugs. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 249–267. [Google Scholar]
- Zhang, J.; Adrián, F.J.; Jahnke, W.; Cowan-Jacob, S.W.; Li, A.G.; Iacob, R.E.; Sim, T.; Powers, J.; Dierks, C.; Sun, F.; et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. [Google Scholar]
- Adrián, F.J.; Ding, Q.; Sim, T.; Velentza, A.; Sloan, C.; Liu, Y.; Zhang, G.; Hur, W.; Ding, S.; Manley, P.; et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2006, 2, 95–102. [Google Scholar]
- Choi, Y.; Seeliger, M.A.; Panjarian, S.B.; Kim, H.; Deng, X.; Sim, T.; Couch, B.; Koleske, A.J.; Smithgall, T.E.; Gray, N.S. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J. Biol. Chem. 2009, 284, 29005–29014. [Google Scholar]
- Fabbro, D.; Manley, P.W.; Jahnke, W.; Liebetanz, J.; Szyttenholm, A.; Fendrich, G.; Strauss, A.; Zhang, J.; Gray, N.S.; Adrian, F.; et al. Inhibitors of the Abl kinase directed at either the ATP- or myristate-binding site. Biochim. Biophys. Acta BBA - Proteins Proteomics 2010, 1804, 454–462. [Google Scholar]
- Flight, M.H. A winning combination against BCR–ABL. Nat. Rev. Drug Discov. 2010, 9, 194. [Google Scholar]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar]
- Qiang, W.; Antelope, O.; Zabriskie, M.S.; Pomicter, A.D.; Vellore, N.A.; Szankasi, P.; Rea, D.; Cayuela, J.M.; Kelley, T.W.; Deininger, M.W.; et al. Mechanisms of resistance to the BCR-ABL1 allosteric inhibitor asciminib. Leukemia 2017, 31, 2844–2847. [Google Scholar]
- Eide, C.A.; Zabriskie, M.S.; Savage Stevens, S.L.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D.; et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy Against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431–443.e5. [Google Scholar]
- Filippakopoulos, P.; Kofler, M.; Hantschel, O.; Gish, G.D.; Grebien, F.; Salah, E.; Neudecker, P.; Kay, L.E.; Turk, B.E.; Superti-Furga, G.; et al. Structural Coupling of SH2-Kinase Domains Links Fes and Abl Substrate Recognition and Kinase Activation. Cell 2008, 134, 793–803. [Google Scholar]
- Grebien, F.; Hantschel, O.; Wojcik, J.; Kaupe, I.; Kovacic, B.; Wyrzucki, A.M.; Gish, G.D.; Cerny-Reiterer, S.; Koide, A.; Beug, H.; et al. Targeting the SH2-Kinase Interface in Bcr-Abl Inhibits Leukemogenesis. Cell 2011, 147, 306–319. [Google Scholar]
- Wojcik, J.; Hantschel, O.; Grebien, F.; Kaupe, I.; Bennett, K.L.; Barkinge, J.; Jones, R.B.; Koide, A.; Superti-Furga, G.; Koide, S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010, 17, 519–527. [Google Scholar]
- Nicolotti, O.; Gillet, V.J.; Fleming, P.J.; Green, D.V.S. Multiobjective optimization in quantitative structure-activity relationships: Deriving accurate and interpretable QSARs. J. Med. Chem. 2002, 45, 5069–5080. [Google Scholar]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar]
- Nicolotti, O.; Miscioscia, T.F.; Leonetti, F.; Muncipinto, G.; Carotti, A. Screening of matrix metalloproteinases available from the protein data bank: Insights into biological functions, domain organization, and zinc binding groups. J. Chem. Inf. Model. 2007, 47, 2439–2448. [Google Scholar]
- Nicolotti, O.; Catto, M.; Giangreco, I.; Barletta, M.; Leonetti, F.; Stefanachi, A.; Pisani, L.; Cellamare, S.; Tortorella, P.; Loiodice, F.; et al. Design, synthesis and biological evaluation of 5-hydroxy, 5-substituted-pyrimidine-2,4,6-triones as potent inhibitors of gelatinases MMP-2 and MMP-9. Eur. J. Med. Chem. 2012, 58, 368–376. [Google Scholar]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A New Approach for Drug Target and Bioactivity Prediction: The Multifingerprint Similarity Search Algorithm (MuSSeL). J. Chem. Inf. Model. 2019, 59, 586–596. [Google Scholar]
- Trisciuzzi, D.; Alberga, D.; Mansouri, K.; Judson, R.; Cellamare, S.; Catto, M.; Carotti, A.; Benfenati, E.; Novellino, E.; Mangiatordi, G.F.; et al. Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data. Future Med. Chem. 2015, 7, 1921–1936. [Google Scholar]
- Pellicani, R.Z.; Stefanachi, A.; Niso, M.; Carotti, A.; Leonetti, F.; Nicolotti, O.; Perrone, R.; Berardi, F.; Cellamare, S.; Colabufo, N.A. Potent Galloyl-Based Selective Modulators Targeting Multidrug Resistance Associated Protein 1 and P-glycoprotein. J. Med. Chem. 2012, 55, 424–436. [Google Scholar]
- Song, K.; Liu, X.; Huang, W.; Lu, S.; Shen, Q.; Zhang, L.; Zhang, J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017, 57, 2358–2363. [Google Scholar]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar]
- Yueh, C.; Rettenmaier, J.; Xia, B.; Hall, D.R.; Alekseenko, A.; Porter, K.A.; Barkovich, K.; Keseru, G.; Whitty, A.; Wells, J.A.; et al. Kinase Atlas: Druggability Analysis of Potential Allosteric Sites in Kinases. J. Med. Chem. 2019, 62, 6512–6524. [Google Scholar]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar]
- Singh, V.K.; Coumar, M.S. Ensemble-based virtual screening: Identification of a potential allosteric inhibitor of Bcr-Abl. J. Mol. Model. 2017, 23, 218. [Google Scholar]
- Fallacara, A.L.; Tintori, C.; Radi, M.; Schenone, S.; Botta, M. Insight into the allosteric inhibition of Abl kinase. J. Chem. Inf. Model. 2014, 54, 1325–1338. [Google Scholar]
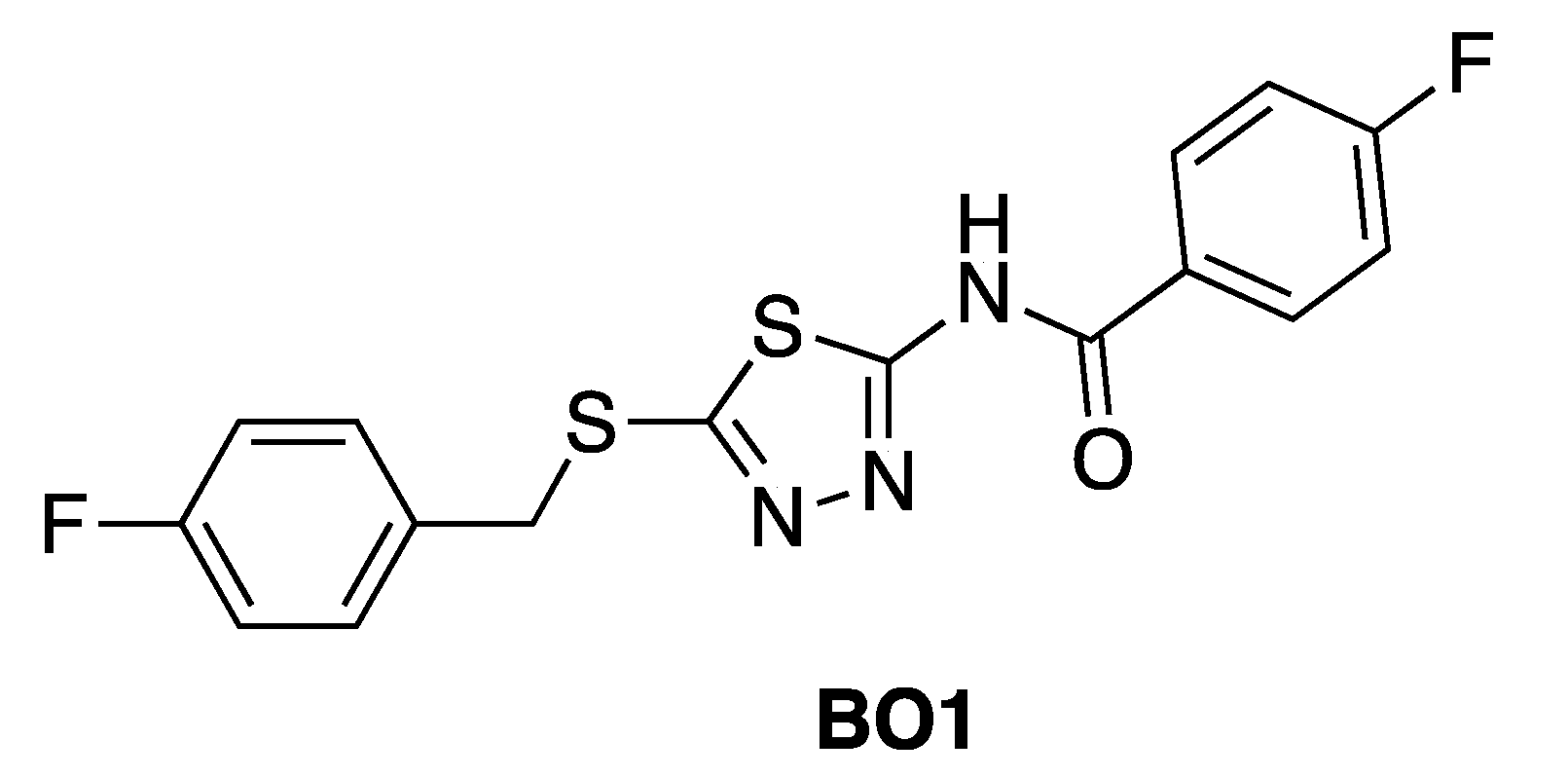
- Radi, M.; Crespan, E.; Botta, G.; Falchi, F.; Maga, G.; Manetti, F.; Corradi, V.; Mancini, M.; Santucci, M.A.; Schenone, S.; et al. Discovery and SAR of 1,3,4-thiadiazole derivatives as potent Abl tyrosine kinase inhibitors and cytodifferentiating agents. Bioorg. Med. Chem. Lett. 2008, 18, 1207–1211. [Google Scholar]
- Radi, M.; Crespan, E.; Falchi, F.; Bernardo, V.; Zanoli, S.; Manetti, F.; Schenone, S.; Maga, G.; Botta, M. Design and Synthesis of Thiadiazoles and Thiazoles Targeting the Bcr-Abl T315I Mutant: from Docking False Positives to ATP-Noncompetitive Inhibitors. ChemMedChem 2010, 5, 1226–1231. [Google Scholar]
- Crespan, E.; Radi, M.; Zanoli, S.; Schenone, S.; Botta, M.; Maga, G. Dual Src and Abl inhibitors target wild type Abl and the AblT315I Imatinib-resistant mutant with different mechanisms. Bioorg. Med. Chem. 2010, 18, 3999–4008. [Google Scholar]
- Zhan, J.-Y.; Ma, J.; Zheng, Q.-C. Molecular dynamics investigation on the Asciminib resistance mechanism of I502L and V468F mutations in BCR-ABL. J. Mol. Graph. Model. 2019, 89, 242–249. [Google Scholar]
- Banavath, H.N.; Sharma, O.P.; Kumar, M.S.; Baskaran, R. Identification of novel tyrosine kinase inhibitors for drug resistant T315I mutant BCR-ABL: a virtual screening and molecular dynamics simulations study. Sci. Rep. 2014, 4, 6948. [Google Scholar]
- Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar]
- Miljković, F.; Rodríguez-Pérez, R.; Bajorath, J. Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J. Med. Chem. 2019. [Google Scholar]
- Alberga, D.; Gambacorta, N.; Trisciuzzi, D.; Ciriaco, F.; Amoroso, N.; Nicolotti, O. De novo drug design of targeted chemical libraries based on artificial intelligence and pair based multiobjective optimization. J. Chem. Inf. Model. 2020. [Google Scholar]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins 2015, 83, 517–532. [Google Scholar]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar]
- Schrödinger Release 2020-2; Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2016; Impact, Schrödinger, LLC, New York, NY, 2016; Prime; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-2; LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar]
- Schrödinger Release 2020-2; Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Nicolotti, O.; Giangreco, I.; Introcaso, A.; Leonetti, F.; Stefanachi, A.; Carotti, A. Strategies of multi-objective optimization in drug discovery and development. Expert Opin. Drug Discov. 2011, 6, 871–884. [Google Scholar]
- Cavalluzzi, M.M.; Imbrici, P.; Gualdani, R.; Stefanachi, A.; Mangiatordi, G.F.; Lentini, G.; Nicolotti, O. Human ether-à-go-go-related potassium channel: Exploring SAR to improve drug design. Drug Discov. Today 2020, 25, 344–366. [Google Scholar]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar]
- Pisani, L.; Catto, M.; Nicolotti, O.; Grossi, G.; Di Braccio, M.; Soto-Otero, R.; Mendez-Alvarez, E.; Stefanachi, A.; Gadaleta, D.; Carotti, A. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2013, 70, 723–739. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |
| Bcr-Abl.lib_01-10.704 a 3.791 b 3.977 c | Bcr-Abl.lib _02-10.338 a 3.743 b 2.970 c | Bcr-Abl.lib_03-10.305 a 3.737 b 2.810 c |
 |  |  |
| Bcr-Abl.lib_04-10.296 a 3.874 b 2.764 c | Bcr-Abl.lib_05-10.246 a 3.667 b 4.055 c | Bcr-Abl.lib_06-10.169 a 3.795 b 2.643 c |
 |  |  |
| Bcr-Abl.lib_07-10.158 a 3.914 b 2.475 c | Bcr-Abl.lib_08-10.139 a 2.998 b 2.898 c | Bcr-Abl.lib_09-10.104 a 3.909 b 2.689 c |
 |  |  |
| Bcr-Abl.lib_10-10.072 a 3.989 b 2.749 c | Bcr-Abl.lib_11-10.052 a 3.727 b 2.869 c | Bcr-Abl.lib_12-9.991 a 2.986 b 3.472 c |
 |  |  |
| Bcr-Abl.lib_13-9.939 a 3.506 b 2.501 c | Bcr-Abl.lib_14-9.916 a 3.437 b 2.668 c | Bcr-Abl.lib_15-9.915 a 3.979 b 2.735 c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carofiglio, F.; Trisciuzzi, D.; Gambacorta, N.; Leonetti, F.; Stefanachi, A.; Nicolotti, O. Bcr-Abl Allosteric Inhibitors: Where We Are and Where We Are Going to. Molecules 2020, 25, 4210. https://doi.org/10.3390/molecules25184210
Carofiglio F, Trisciuzzi D, Gambacorta N, Leonetti F, Stefanachi A, Nicolotti O. Bcr-Abl Allosteric Inhibitors: Where We Are and Where We Are Going to. Molecules. 2020; 25(18):4210. https://doi.org/10.3390/molecules25184210
Chicago/Turabian StyleCarofiglio, Francesca, Daniela Trisciuzzi, Nicola Gambacorta, Francesco Leonetti, Angela Stefanachi, and Orazio Nicolotti. 2020. "Bcr-Abl Allosteric Inhibitors: Where We Are and Where We Are Going to" Molecules 25, no. 18: 4210. https://doi.org/10.3390/molecules25184210