
Optimization and a Kinetic Study on the Acidic Hydrolysis of Dialkyl α-Hydroxybenzylphosphonates



Abstract
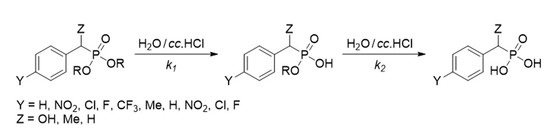
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Use of the 31P NMR Spectra in Quantitative Analysis
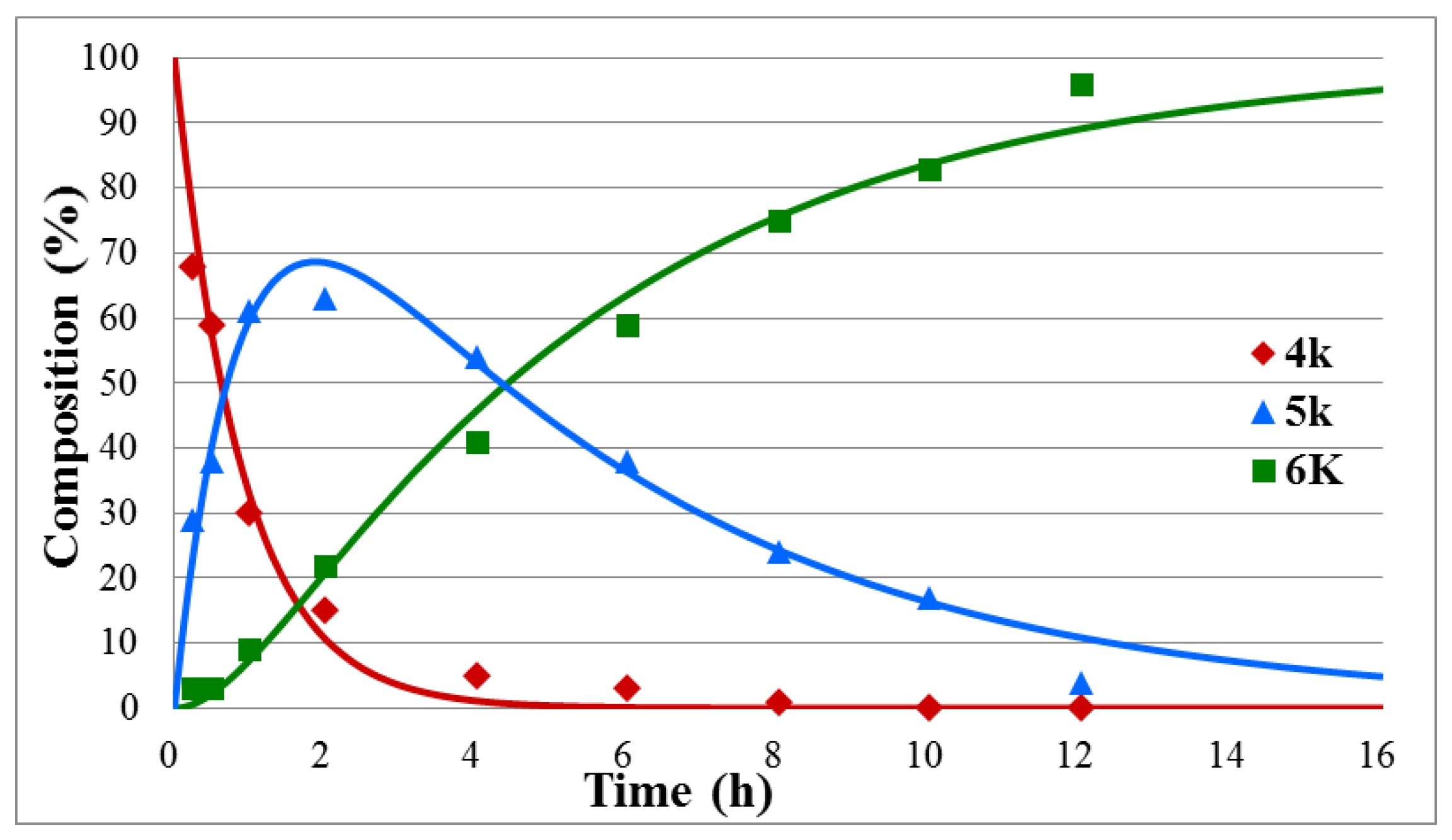
3.3. Curve Fitting on the Time–Relative Quantity Data Pairs
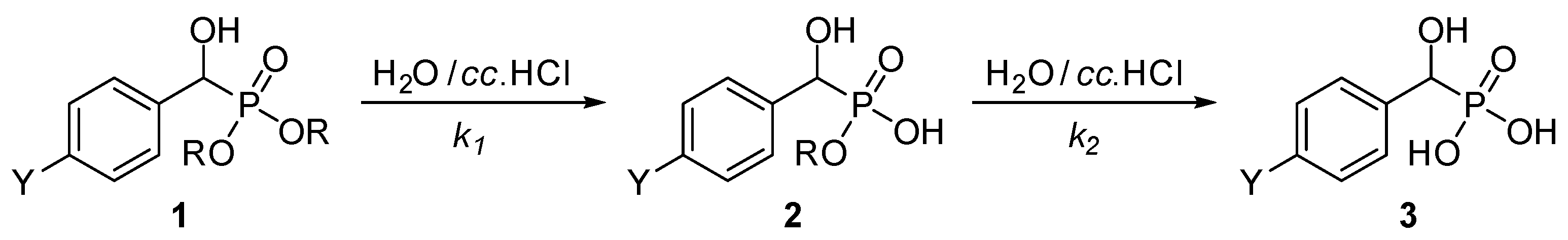
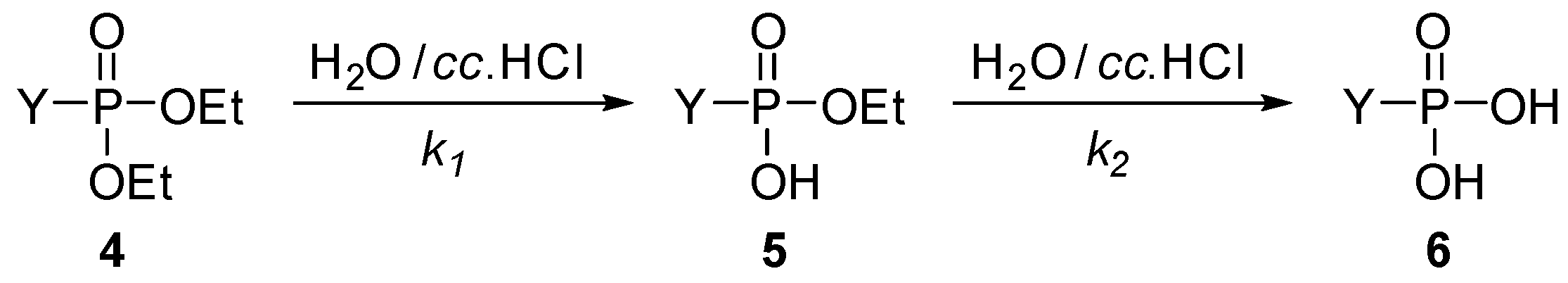
3.4. General Procedure for the Hydrolysis of Phosphonates (1a–j, 4k–m)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Desai, J.; Wang, Y.; Wang, K.; Malwal, S.R.; Oldfield, E. Isoprenoid biosynthesis inhibitors targeting bacterial cell growth. Chem. Med. Chem. 2016, 11, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Tcarkova, K.V.; Artyushin, O.I.; Bondarenko, N.A. Synthetic routes to bis(3-aminophenyl) phosphinic acid. Phosphorus Sulfur Silicon 2016, 191, 1520–1522. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Bölcskei, A.; Drahos, L.; Kraszni, M.; Balogh, G.T. Synthesis and proton dissociation properties of arylphosphonates; A microwave-assisted catalytic Arbuzov reaction with aryl bromides. Heteroat. Chem. 2012, 23, 574–582. [Google Scholar] [CrossRef]
- Gavande, N.; Yamamoto, I.; Salam, N.K.; Ai, T.-H.; Burden, P.M.; Johnston, G.A.R.; Hanrahan, J.R.; Chebib, M. Novel cyclic phosphinic acids as GABAC ρ receptor antagonists: Design, synthesis, and pharmacology. ACS Med. Chem. Lett. 2011, 2, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahil, J.; Haake, P. Rates and mechanism of the alkaline-hydrolysis of a sterically hindered phosphinate ester—Partial reaction by nucleophilic-attack at carbon. J. Org. Chem. 1981, 46, 3048–3052. [Google Scholar] [CrossRef]
- Cook, R.D.; Farah, S.; Ghawi, L.; Itani, A.; Rahil, J. The influence of the changing of P=O to P=S and P-O-R to P-S-R on the reactivity of phosphinate esters under alkaline-hydrolysis conditions. Can. J. Chem. 1986, 64, 1630–1637. [Google Scholar] [CrossRef]
- Wróblewski, A.E.; Verkade, J.G. 1-oxo-2-oxa-1-phosphabicyclo [2.2.2]octane: A new mechanistic probe for the basic hydrolysis of phosphate esters. J. Am. Chem. Soc. 1996, 118, 10168–10174. [Google Scholar] [CrossRef]
- Cevasco, G.; Thea, S. The quest for carbanion-promoted dissociative pathways in the hydrolysis of aryl phosphinates. J. Chem. Soc. Perkin Trans. 2 1993, 1103–1106. [Google Scholar] [CrossRef]
- Salomon, C.J.; Breuer, E. Efficient and selective dealkylation of phosphonate diisopropyl esters using ME3SiBr. Tetrahedron Lett. 1995, 36, 6759–6760. [Google Scholar] [CrossRef]
- Tulsi, N.S.; Downey, A.M.; Cairo, C.W. A protected L-bromophosphonomethylphenylalanine amino acid derivative (BrPmp) for synthesis of irreversible protein tyrosine phosphatase inhibitors. Bioorg. Med. Chem. 2010, 18, 8679–8686. [Google Scholar] [CrossRef]
- Jansa, P.; Hradil, O.; Baszczyňski, O.; Dračínský, M.; Janeba, Z. An efficient microwave-assisted synthesis and biological properties of polysubstituted pyrimidinyl- and 1,3,5-triazinylphosphonic acids. Tetrahedron 2012, 68, 865–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N.Z. A study on the acidic hydrolysis of cyclic phosphinates: 1-Alkoxy-3-phospholene 1-oxides, 1-ethoxy-3-methylphospholane 1-oxide, and 1-ethoxy-3-methyl-1,2,3,4,5,6-hexahydrophosphinine 1-oxide. Heteroat. Chem. 2017, 28, e21394. [Google Scholar] [CrossRef]
- Harsági, N.; Rádai, Z.; Kiss, N.Z.; Szigetvári, Á.; Keglevich, G. Two-step acidic hydrolysis of dialkyl arylhosphonates. Mendeleev Commun. 2020, 30, 38–39. [Google Scholar] [CrossRef]
- Rádai, Z.; Keglevich, G. Synthesis and reactions of α-hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, N.Z.; Rádai, Z.; Mucsi, Z.; Keglevich, G. Synthesis of aminophosphonates from α-hydroxyphosphonates; a theoretical study. Heteroat. Chem. 2016, 27, 260–268. [Google Scholar] [CrossRef]
- Rádai, Z.; Hodula, V.; Kiss, N.Z.; Kóti, J.; Keglevich, G. Phosphorylation of (1-aryl-1-hydroxymethyl)phosphonates. Mendeleev Commun. 2019, 29, 153–154. [Google Scholar] [CrossRef]
- Rádai, Z.; Szabó, R.; Szigetvári, Á.; Kiss, N.Z.; Mucsi, Z.; Keglevich, G. A study on the rearrangement of dialkyl 1-aryl-1-hydroxymethylphosphonates to benzyl phosphates. Curr. Org. Chem. 2020, 24, 465–471. [Google Scholar] [CrossRef]
- Rádai, Z.; Szeles, P.; Kiss, N.Z.; Hegedűs, L.; Windt, T.; Nagy, V.; Keglevich, G. Green synthesis and cytotoxic activity of dibenzyl α-hydroxyphosphonates and α-hydroxyphosphonic acids. Heteroat. Chem. 2018, 29, e21436. [Google Scholar] [CrossRef] [Green Version]
- Lasdon, L.S.; Waren, A.D.; Jain, A.; Ratner, M. Design and testing of a generalized reduced gradient code for nonlinear programming. ACM T Math. Softw. 1978, 4, 34–50. [Google Scholar] [CrossRef]
- Keglevich, G.; Tóth, V.R.; Drahos, L. Microwave-assisted synthesis of α-hydroxy-benzylphosphonates and -benzylphosphine oxides. Heteroat. Chem. 2011, 22, 15–17. [Google Scholar] [CrossRef]
- Seven, O.; Polat-Cakir, S.; Hossain, M.S.; Emrullahoglu, M. Reactions of acyl phosphonates with organoaluminum reagents: A new method for the synthesis of secondary and tertiary alpha-hydroxy phosphonates. Tetrahedron 2011, 67, 3464–3469. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.-H.; Du, G.-F.; He, L.; Gu, C.-Z.; Dai, B. N-Heterocyclic carbene catalyzed hydrophosphonylation of aldehydes. Synthesis 2011, 2073–2078. [Google Scholar] [CrossRef]
- de Noronha, R.G.; Costa, P.J.; Romano, C.C.; Calhorda, M.J.; Fernandes, A.C. MoO2Cl2 as a novel catalyst for C–P bond formation and for hydrophosphonylation of aldehydes. Organometallics 2009, 28, 6206–6212. [Google Scholar] [CrossRef]
- Kedrowski, S.M.A.; Dougherty, D.A. Room-temperature alternative to the Arbuzov reaction: The reductive deoxygenation of acyl phosphonates. Org. Lett. 2010, 12, 3990–3993. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Chen, T.; Han, L.-B. Oxidative dephosphorylation of benzylic phosphonates with dioxygen generating symmetrical trans-stilbenes. J. Org. Chem. 2018, 83, 2959–2965. [Google Scholar] [CrossRef]
- St. Maurice, M.; Bearne, S.L. Reaction intermediate analogues for mandelate racemase: Interaction between Asn 197 and the α-hydroxyl of the substrate promotes catalysis. Biochemistry 2000, 39, 13324–13335. [Google Scholar] [CrossRef]
- Hamerschmidt, F.; Hanninger, A. Enantioselective deprotonation of benzyl phosphates by homochiral lithium amide bases—Configurational stability of benzyl carbanions with a dialkoxyphosphoryloxy substituent and their rearrangement to optically-active α-hydroxy phosphonates. Chem. Ber. 1995, 128, 823–830. [Google Scholar] [CrossRef]
- Mortier, J.; Gridnev, I.D.; Guénot, P. Reactions of phosphonates with organohaloboranes: New route to molecular borophosphonates. Organometallics 2000, 19, 4266–4275. [Google Scholar] [CrossRef]
- Eom, D.; Jeong, Y.; Kim, Y.R.; Lee, E.; Choi, W.; Lee, P.H. Palladium-catalyzed C(sp2 and sp3)-H activation/C-O bond formation: Synthesis of benzoxaphosphole 1- and 2-oxides. Org. Lett. 2013, 15, 5210–5213. [Google Scholar] [CrossRef]
- Galardy, R.E.; Kontoyiannidou-Ostrem, V.; Kortylewicz, Z.P. Inhibition of angiotensin converting enzyme by phosphonic amides and phosphonic-acids. Biochemistry 1983, 22, 1990–1995. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Maryashkin, A.V. Reaction of trimethylsilyl phosphites with functionalyzed aromatic aldehydes. Russ. J. Gen. Chem. 2005, 75, 1965–1967. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Zhang, Y.-J.; Wang, W.-M.; Yang, K.-W. Synthesis and inhibitory activity of acetamidophosphonic acids against metallo-β-lactamases. Phosphorus Sulfur Silicon 2017, 192, 14–18. [Google Scholar] [CrossRef]
- Safonova, T.Y.; Gulyukina, N.S.; Novakovskaya, Y.V.; Astaf’ev, E.A.; Bondarenko, G.N.; Petrii, O.A.; Tsirlina, G.A.; Beletskaya, I.P. Electrochemical hydrogenation of substituted alpha-phenylvinylphosphonic acids: General characteristics of the reaction layer and prediction of preparative electrolysis conditions. Russ. J. Electrochem. 2002, 38, 457–466. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Vonder Embse, R.A. The invention of radical reactions. Part 39. The reaction of white phosphorus with carbon-centered radicals. An improved procedure for the synthesis of phosphonic acids and further mechanistic insights. Tetrahedron 1998, 54, 12475–12496. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
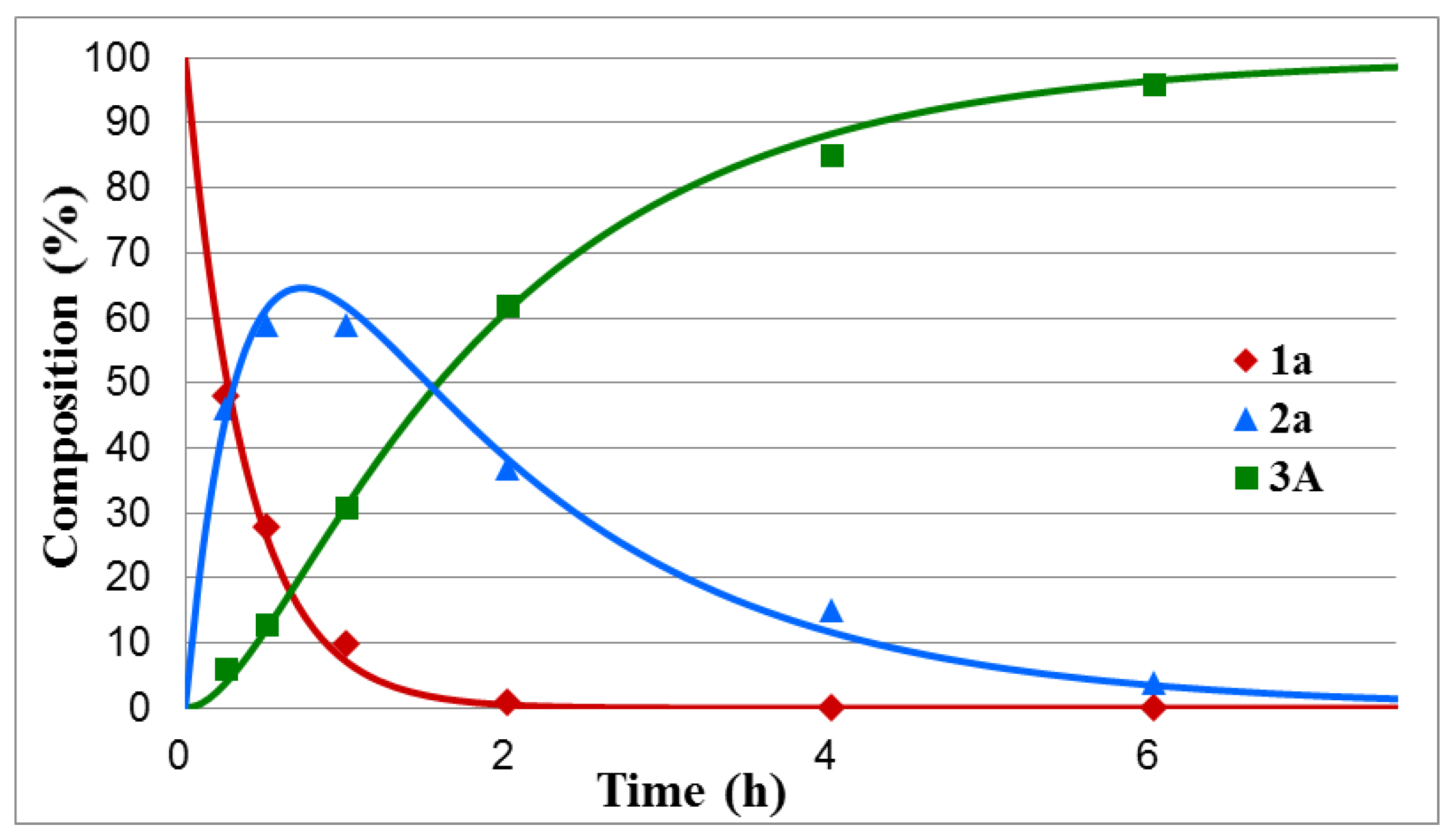
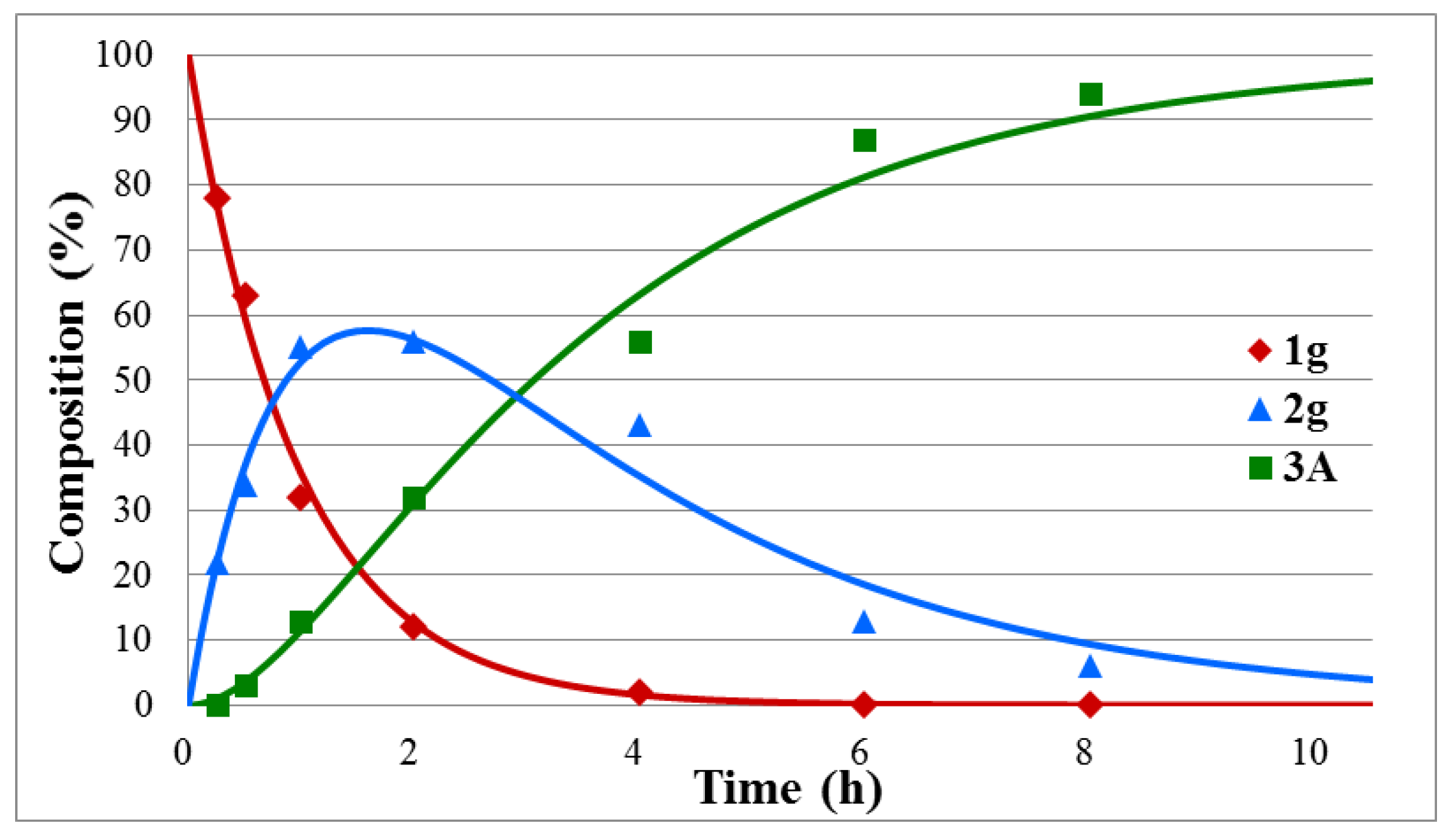
| Entry | Y | R | tmax (min) | tr (h) | k1 (h−1) | k2 (h−1) | R2 |
|---|---|---|---|---|---|---|---|
| 1 | H (a) | Me | 44 | 6.5 | 2.64 | 0.60 | 0.994 |
| 2 | NO2 (b) | Me | 22 | 2.5 | 5.18 | 1.24 | 0.989 |
| 3 | Cl (c) | Me | 34 | 5.5 | 3.36 | 0.79 | 0.987 |
| 4 | F (d) | Me | 32 | 6.0 | 3.93 | 0.67 | 0.965 |
| 5 | CF3 (e) | Me | 51 | 5.5 | 2.03 | 0.61 | 0.988 |
| 6 | Me (f) | Me | 76 | 8 | 1.64 | 0.31 | 0.962 |
| 7 | H (g) | Et | 90 | 9.5 | 1.03 | 0.35 | 0.986 |
| 8 | NO2 (h) | Et | 75 | 5.5 | 1.40 | 0.61 | 0.992 |
| 9 | Cl (i) | Et | 60 | 8.0 | 1.08 | 0.42 | 0.992 |
| 10 | F (j) | Et | 80 | 9.0 | 1.35 | 0.31 | 0.970 |
| Entry | Y | tmax (h) | tr (h) | k1 (h−1) | k2 (h−1) | R2 |
|---|---|---|---|---|---|---|
| 1 | PhCH2 (k) | 2 | 15 | 1.12 | 0.20 | 0.983 |
| 2 | PhCHMe (l) | 4 | 25 | 0.51 | 0.11 | 0.940 |
| 3 | Ph(CH2)2 (m) | 2.75 | 20 | 0.70 | 0.15 | 0.949 |
 | ||||||||
| tr (h) | 2.5–6.0 | 6.5 | 5.5–9.0 | 8 | 9.5 | 15 | 20 | 25 |
| k1 (h−1) | 3.36–5.18 | 2.64 | 1.08–1.40 | 1.64 | 1.03 | 1.12 | 0.70 | 0.51 |
| k2 (h−1) | 0.67–1.24 | 0.60 | 0.31–0.61 | 0.31 | 0.35 | 0.20 | 0.15 | 0.11 |
| δ 31P NMR | [M + H] | ||
|---|---|---|---|
| Found (DMSO) | Literature | ||
| 1a | 23.9 | 23.9 (CDCl3) [20] | 216.9 |
| 1b | 22.7 | 21.9 (CDCl3) [20] | 262.2 |
| 1c | 23.5 | 22.1 (CDCl3) [20] | 273.0 |
| 1d | 23.8 | 23.3 (CDCl3) [21] | 235.0 |
| 1e [22] a | 23.1 | – | 285.1 |
| 1f | 24.1 | 24.0 (CDCl3) [20] | 231 |
| 1g | 21.8 | 21.5 (CDCl3) [20] | 245.1 |
| 1h | 20.4 | 19.9 (CDCl3) [20] | 290.1 |
| 1i | 21.2 | 21.9 (CDCl3) [20] | 354.1 |
| 1j | 21.7 | 21.7 (CDCl3) [23] | 262.1 |
| 4k | 26.7 | 26.4 (CDCl3) [24] | 229.2 |
| 4l | 29.8 | 30.4 (CDCl3) [25] | 243.1 |
| 4m | 30.6 | 30.8 (CDCl3) [24] | 243.1 |
| 2a | 20.8 | 19.9 (D2O) [26] | 203.04675 (203.04677 c) |
| 2bb | 18.8 | 248.03164 (248.03185 c) | |
| 2cb | 20.3 | 237.00752 (237.00780 c) | |
| 2db | 20.4 | 221.03692 (221.03735 c) | |
| 2eb | 19.6 | 271.03384 (271.03416 c) | |
| 2fb | 20.8 | 217.06177 (217.06242 c) | |
| 2g [27] a | 19.6 | 217.06193 (217.06242 c) | |
| 2hb | 18.1 | 262.04707 (262.04750 c) | |
| 2ib | 19.0 | 251.02306 (251.02345 c) | |
| 2jb | 19.5 | 235.05267 (235.05300 c) | |
| 5k | 23.7 | 25.5 (CD3OD) [28] | 201.1 |
| 5l | 27.0 | 32.3 (CDCl3) [29] | 215.1 |
| 5m [30] a | 27.5 | – | 215.1 |
| 3A | 18.9 | 19.4(D2O) [18] | 189.1 |
| 3B | 16.6 | 16.4 (D2O) [18] | 234.0 |
| 3C | 17.9 | 18.6 (D2O) [18] | 223.0 |
| 3D | 18.2 | 18.5 [31] | 207.0 |
| 3Eb | 17.4 | 257.0 | |
| 3F | 18.6 | 18.8 (D2O) [18] | 203.1 |
| 6K | 21.8 | 22.1 (DMSO) [32] | 173.0 |
| 6L | 25.7 | 27.8 (CD3OD) [33] | 187.0 |
| 6M | 26.0 | 26.7 (DMSO) [34] | 187.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harsági, N.; Rádai, Z.; Szigetvári, Á.; Kóti, J.; Keglevich, G. Optimization and a Kinetic Study on the Acidic Hydrolysis of Dialkyl α-Hydroxybenzylphosphonates. Molecules 2020, 25, 3793. https://doi.org/10.3390/molecules25173793
Harsági N, Rádai Z, Szigetvári Á, Kóti J, Keglevich G. Optimization and a Kinetic Study on the Acidic Hydrolysis of Dialkyl α-Hydroxybenzylphosphonates. Molecules. 2020; 25(17):3793. https://doi.org/10.3390/molecules25173793
Chicago/Turabian StyleHarsági, Nikoletta, Zita Rádai, Áron Szigetvári, János Kóti, and György Keglevich. 2020. "Optimization and a Kinetic Study on the Acidic Hydrolysis of Dialkyl α-Hydroxybenzylphosphonates" Molecules 25, no. 17: 3793. https://doi.org/10.3390/molecules25173793