
Synthesis and Structure of Iron (II) Complexes of Functionalized 1,5-Diaza-3,7-Diphosphacyclooctanes

 , , and
, , and
Abstract
:1. Introduction
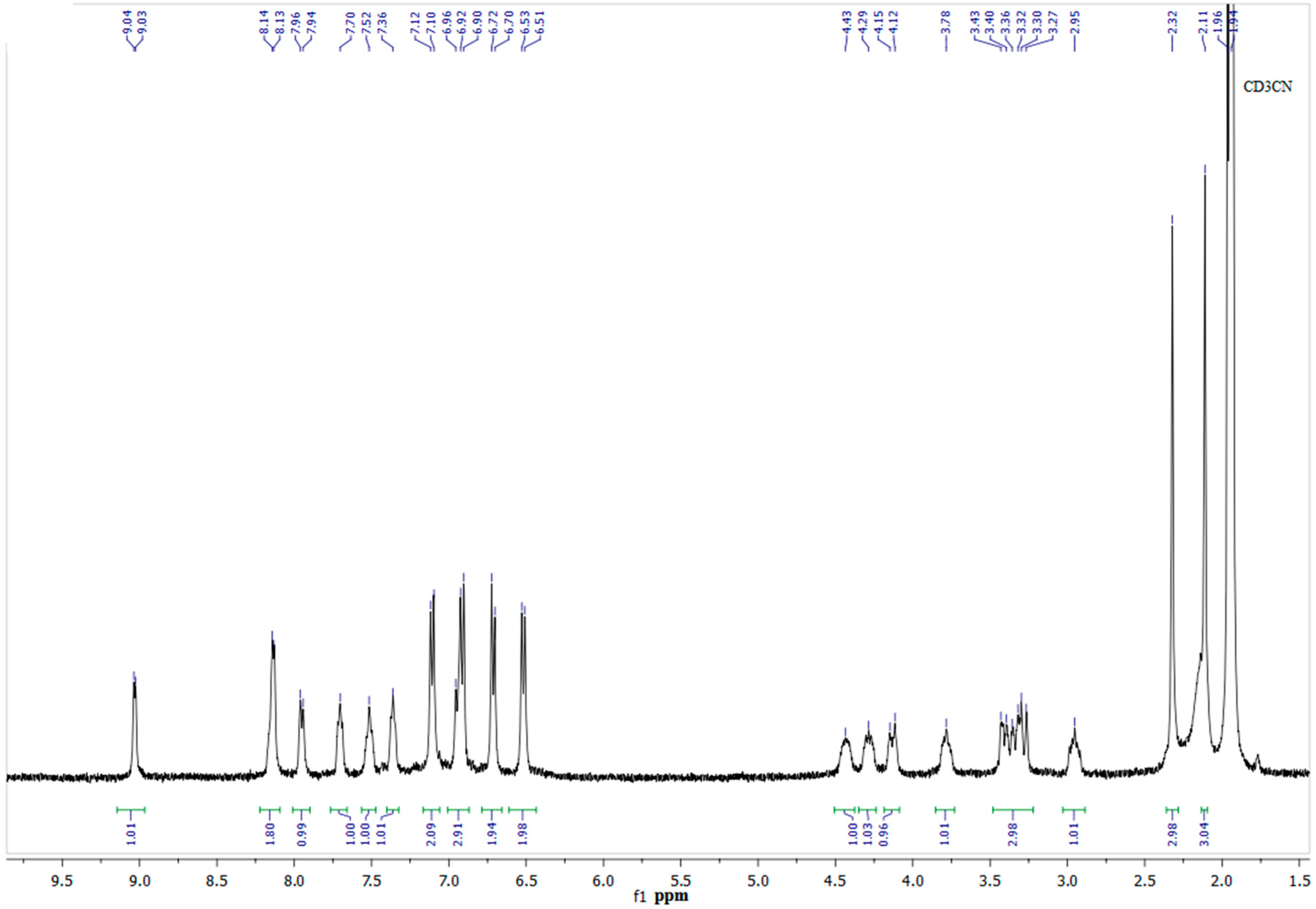
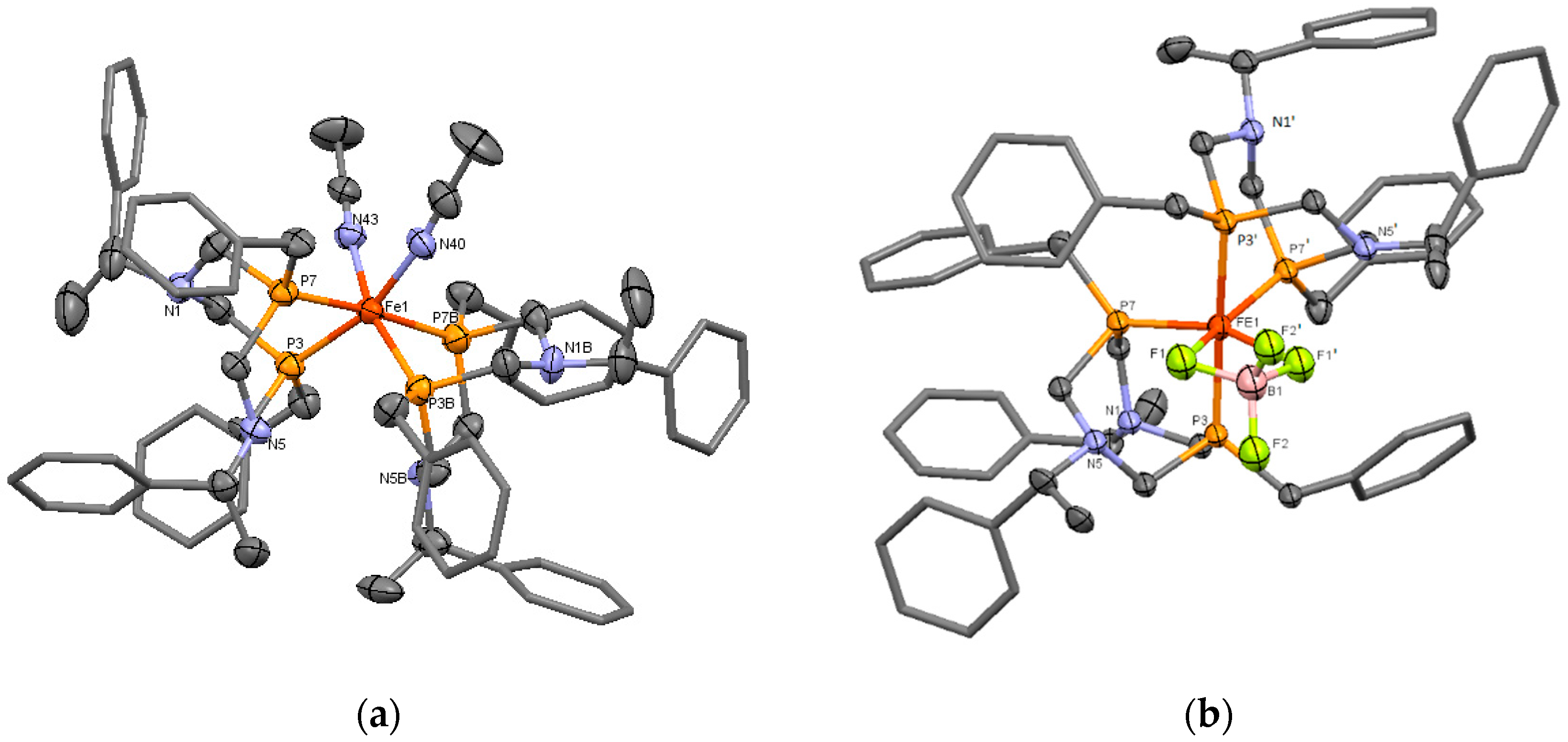
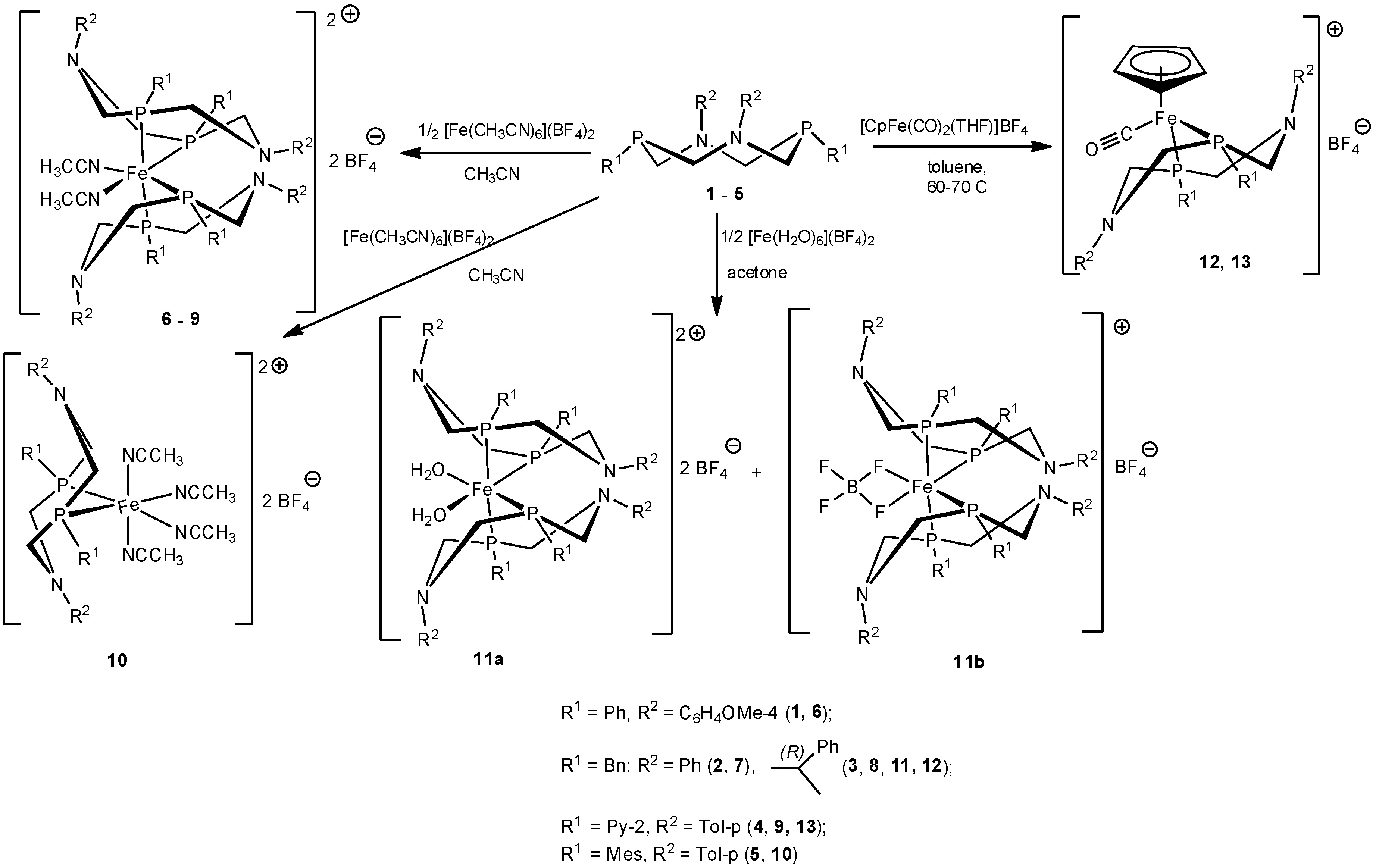
2. Results
3. Discussion
4. Materials and Methods
4.1. General
4.2. X-ray Crystallography Data
4.2.1. Crystal Data and Structure Refinement for Compound 8
4.2.2. Crystal Data and Structure Refinement for Compound 11b
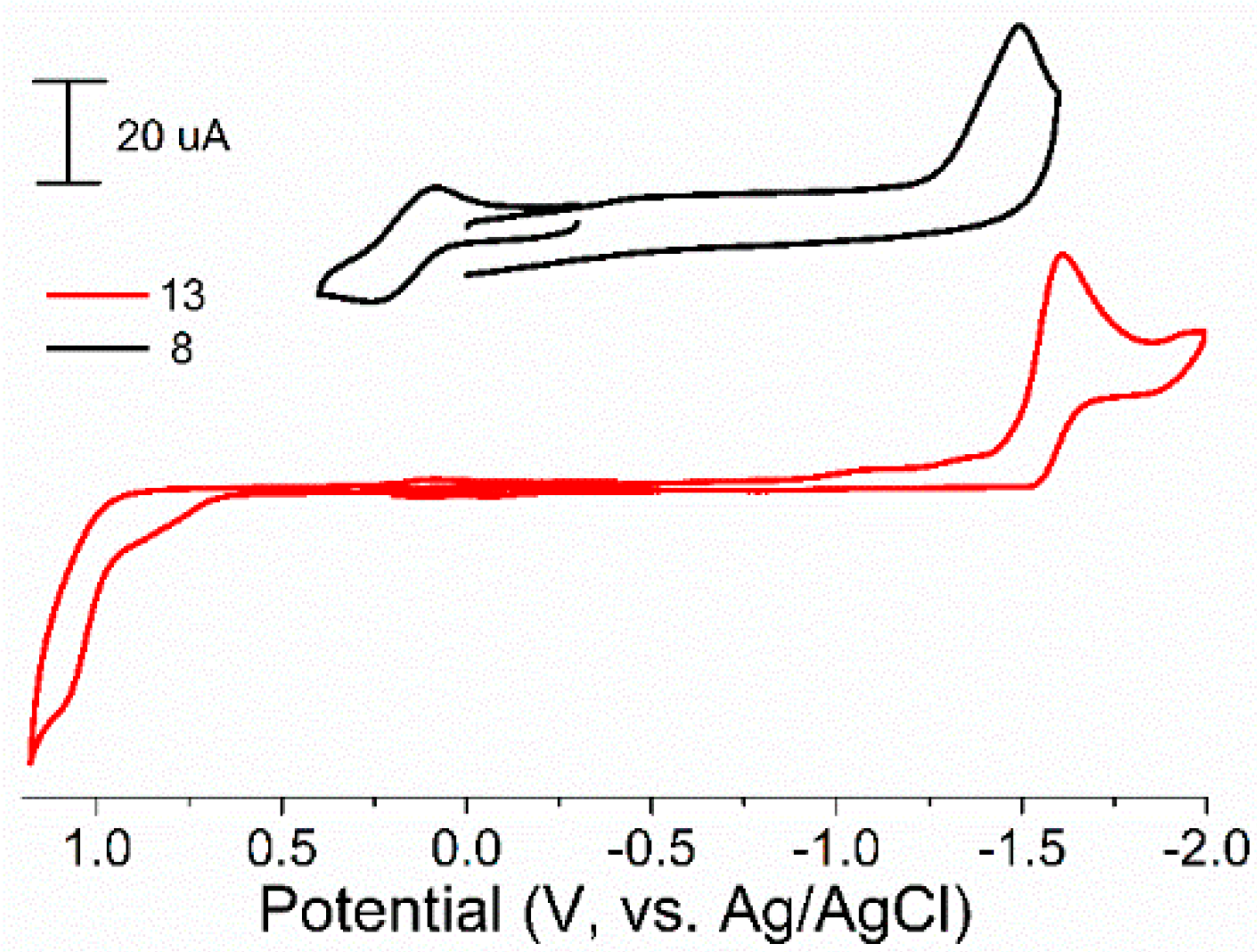
4.3. Electrochemical Measurements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DuBois, D.L.; Bullock, R.M. Molecular Electrocatalysts for the Oxidation of Hydrogen and the Production of Hydrogen—The Role of Pendant Amines as Proton Relays. Eur. J. Inorg. Chem. 2011, 2011, 1017–1027. [Google Scholar] [CrossRef]
- Musina, E.I.; Khrizanforova, V.V.; Strelnik, I.D.; Valitov, M.I.; Spiridonova, Y.S.; Krivolapov, D.B.; Litvinov, I.A.; Kadirov, M.K.; Lönnecke, P.; Hey-Hawkins, E.; et al. New Functional Cyclic Aminomethylphosphine Ligands for the Construction of Catalysts for Electrochemical Hydrogen Transformations. Chem. Eur. J. 2014, 20, 3169–3182. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.M.; Helm, M.L. Molecular Electrocatalysts for Oxidation of Hydrogen Using Earth-Abundant Metals: Shoving Protons Around with Proton Relays. Acc. Chem. Res. 2015, 48, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Kadirov, M.K.; Karasik, A.A.; Nizameev, I.R.; Strelnik, I.D.; Kholin, K.V.; Kadirov, D.M.; Ismaev, T.I.; Budnikova, Y.G.; Sinyashin, O.G. Organometallic Polymer Electrolyte Membrane Fuel Cell Bis-Ligand Nickel(II) Complex of 1,5-Di-P-Tolyl-3,7-Dipyridine-1,5,3,7-Diazadiphosphacyclooctane Catalyst. Energy Technol. 2018, 6, 1088–1095. [Google Scholar] [CrossRef]
- Prokopchuk, D.E.; Wiedner, E.S.; Walter, E.D.; Popescu, C.V.; Piro, N.A.; Kassel, W.S.; Bullock, R.M.; Mock, M.T. Catalytic N2 Reduction to Silylamines and Thermodynamics of N2 Binding at Square Planar Fe. J. Am. Chem. Soc. 2017, 139, 9291–9301. [Google Scholar] [CrossRef]
- Burgess, S.A.; Grubel, K.; Appel, A.M.; Wiedner, E.S.; Linehan, J.C. Hydrogenation of CO2 at Room Temperature and Low Pressure with a Cobalt Tetraphosphine Catalyst. Inorg. Chem. 2017, 56, 8580–8589. [Google Scholar] [CrossRef]
- Karasik, A.A.; Balueva, A.S.; Musina, E.I.; Sinyashin, O.G. Chelating cyclic aminomethylphosphines and their transition metal complexes as a promising basis of bioinspired mimetic catalysts. Mendeleev Commun. 2013, 23, 237–248. [Google Scholar] [CrossRef]
- Shaw, W.J.; Helm, M.L.; DuBois, D.L. A modular, energy-based approach to the development of nickel containing molecular electrocatalysts for hydrogen production and oxidation. Biochim. Biophys. Acta. 2013, 1827, 1123–1139. [Google Scholar] [CrossRef] [Green Version]
- Bullock, R.M.; Das, A.K.; Appel, A.M. Surface Immobilization of Molecular Electrocatalysts for Energy Conversion. Chem. Eur. J. 2017, 23, 7626–7641. [Google Scholar] [CrossRef]
- Klug, C.M.; Cardenas, A.J.P.; Bullock, R.M.; O’Hagan, M.; Wiedner, E.S. Reversing the Tradeoff Between Rate and Overpotential in Molecular Electrocatalysts for H 2 Production. ACS Catal. 2018, 8, 3286–3296. [Google Scholar] [CrossRef]
- Lense, S.; Dutta, A.; Roberts, J.A.S.; Shaw, W.J. A proton channel allows a hydrogen oxidation catalyst to operate at a moderate overpotential with water acting as a base. Chem. Commun. 2014, 50, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Klug, C.M.; Dougherty, W.G.; Kassel, W.S.; Wiedner, E.S. Electrocatalytic Hydrogen Production by a Nickel Complex Containing a Tetradentate Phosphine Ligand. Organometallics 2019, 38, 1269–1279. [Google Scholar] [CrossRef]
- Khrizanforova, V.V.; Musina, E.I.; Khrizanforov, M.N.; Gerasimova, T.P.; Katsyuba, S.A.; Spiridonova, Y.S.; Islamov, D.R.; Kataeva, O.N.; Karasik, A.A.; Sinyashin, O.G.; et al. Unexpected ligand effect on the catalytic reaction rate acceleration for hydrogen production using biomimetic nickel electrocatalysts with 1,5-diaza-3,7-diphosphacyclooctanes. J. Organomet. Chem. 2015, 789–790, 14–21. [Google Scholar] [CrossRef]
- Mock, M.T.; Chen, S.; O’Hagan, M.; Rousseau, R.; Dougherty, W.G.; Kassel, W.S.; Bullock, R.M. Dinitrogen Reduction by a Chromium (0) Complex Supported by a16-Membered Phosphorus Macrocycle. J. Am. Chem. Soc. 2013, 135, 11493–11496. [Google Scholar] [CrossRef] [PubMed]
- Strelnik, I.D.; Dayanova, I.R.; Poryvaev, T.M.; Gerasimova, T.P.; Litvinov, I.A.; Katsyuba, S.A.; Musina, E.I.; Karasik, A.A.; Sinyashin, O.G. Rearrangement of two 8-membered 1,5-diaza-3,7-diphosphacyclooctane rings into 16-membered P4N4 ligand on the gold(I) template. Mendeleev Commun. 2020, 30, 40–42. [Google Scholar] [CrossRef]
- Strelnik, I.D.; Gurzhiy, V.V.; Sizov, V.V.; Musina, E.I.; Karasik, A.A.; Tunik, S.P.; Grachova, E.V. A stimuli-responsive Au(I) complex based on an aminomethylphosphine template: Synthesis, crystalline phases and luminescence properties. Cryst. Eng. Comm. 2016, 18, 7629–7635. [Google Scholar] [CrossRef] [Green Version]
- Elistratova, J.; Strelnik, I.; Brylev, K.; Shestopalov, M.; Gerasimova, T.; Babaev, V.; Kholin, K.; Dobrynin, A.; Musina, E.; Katsyuba, S.; et al. Novel water soluble cationic Au(I) complexes with cyclic PNNP ligand as building blocks for heterometallic supramolecular assemblies with anionic hexarhenium cluster units. J. Lumin. 2018, 196, 485–491. [Google Scholar] [CrossRef]
- Prokopchuk, D.E.; Chambers, G.M.; Walter, E.D.; Mock, M.T.; Bullock, R.M. H2 Binding, Splitting, and Net Hydrogen Atom Transfer at a Paramagnetic Iron Complex. J. Am. Chem. Soc. 2019, 141, 1871–1876. [Google Scholar] [CrossRef]
- Liu, T.; DuBois, D.L.; Bullock, R.M. An iron complex with pendent amines as a molecular electrocatalyst for oxidation of hydrogen. Nat. Chem. 2013, 5, 228–233. [Google Scholar] [CrossRef]
- Liu, T.; Liao, Q.; O’Hagan, M.; Hulley, E.B.; DuBois, D.L.; Bullock, R.M. Iron Complexes Bearing Diphosphine Ligands with Positioned Pendant Amines as Electrocatalysts for the Oxidation of H2. Organometallics 2015, 34, 2747–2764. [Google Scholar] [CrossRef]
- Jacobsen, G.M.; Shoemaker, R.K.; McNevin, M.J.; Rakowski DuBois, M.; DuBois, D.L. Syntheses and Structural Characterizations of Iron (II) Complexes Containing Cyclic Diphosphine Ligands with Positioned Pendant Nitrogen Bases. Organometallics 2007, 26, 5003–5009. [Google Scholar] [CrossRef]
- Liu, T.; Chen, S.; O’Hagan, M.J.; Rakowski DuBois, M.; Bullock, R.M.; DuBois, D.L. Synthesis, Characterization, and Reactivity of Fe Complexes Containing Cyclic Diazadiphosphine Ligands: The Role of the Pendant Base in Heterolytic Cleavage of H2. J. Am. Chem. Soc. 2012, 134, 6257–6272. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Liu, T.; Johnson, S.I.; Klug, C.M.; Wiedner, E.S.; Bullock, R.M.; DuBois, D.L. Evaluation of attractive interactions in the second coordination sphere of iron complexes containing pendant amines. Dalton Trans. 2019, 48, 4867–4878. [Google Scholar] [CrossRef] [PubMed]
- Orthaber, A.; Karnahl, M.; Tschierlei, S.; Streich, D.; Stein, M.; Ott, S. Coordination and conformational isomers in mononuclear iron complexes with pertinence to the [FeFe] hydrogenase active site. Dalton Trans. 2014, 43, 4537–4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Wang, X.; Hoffmann, C.; DuBois, D.L.; Bullock, R.M. Heterolytic Cleavage of Hydrogen by an Iron Hydrogenase Model: An Fe-H···H-N Dihydrogen Bond Characterized by Neutron Diffraction. Angew. Chem. Int. Ed. 2014, 53, 5300–5304. [Google Scholar] [CrossRef]
- Kumar, N.; Darmon, J.M.; Weiss, C.J.; Helm, M.L.; Raugei, S.; Bullock, R.M. Outer Coordination Sphere Proton Relay Base and Proximity Effects on Hydrogen Oxidation with Iron Electrocatalysts. Organometallics 2019, 38, 1391–1396. [Google Scholar] [CrossRef]
- Karasik, A.A.; Naumov, R.N.; Balueva, A.S.; Spiridonova, Y.S.; Golodkov, O.N.; Novikova, H.V.; Belov, G.P.; Katsyuba, S.A.; Vandyukova, E.E.; Lönnecke, P.; et al. Synthesis, Structure, and Transition Metal Complexes of Amphiphilic 1,5-Diaza-3,7-diphosphacyclooctanes. Heteroat. Chem. 2006, 17, 499–513. [Google Scholar] [CrossRef]
- Ignatieva, S.N.; Balueva, A.S.; Karasik, A.A.; Latypov, S.K.; Nikonova, A.G.; Naumova, O.E.; Lönnecke, P.; Hey-Hawkins, E.; Sinyashin, O.G. First Representative of Optically Active P-L-Menthyl-Substituted (Aminomethyl)phosphine and Its Borane and Metal Complexes. Inorg. Chem. 2010, 49, 5407–5412. [Google Scholar] [CrossRef]
- Latypov, S.K.; Strelnik, A.G.; Ignatieva, S.N.; Hey-Hawkins, E.; Balueva, A.S.; Karasik, A.A.; Sinyashin, O.G. Structure and Dynamics of P, N-Containing Heterocycles and Their Metal Complexes in Solution. J. Phys. Chem. A 2012, 116, 3182–3193. [Google Scholar] [CrossRef]
- Burrows, A.D.; Dodds, D.; Kirk, A.S.; Lowe, J.P.; Mahon, M.F.; Warren, J.E.; Whittlesey, M.K. Substitution and derivatization reactions of a water soluble iron (II) complex containing a self-assembled tetradentate phosphine ligand. Dalton Trans. 2007, 570–580. [Google Scholar] [CrossRef]
- Burrows, A.D.; Harrington, R.W.; Kirk, A.S.; Mahon, M.F.; Marken, F.; Warren, J.E.; Whittlesey, M.K. Synthesis, Characterization, and Electrochemistry of a Series of Iron(II) Complexes Containing Self-Assembled 1,5-Diaza-3,7-diphosphabicyclo[3.3.1]nonane Ligands. Inorg. Chem. 2009, 48, 9924–9935. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.E.; Morris, R.H.; Lough, A.J. Acidic Dicationic Iron (II) Dihydrogen Complexes and Compounds Related by H2 Substitution. Inorg. Chem. 1999, 38, 6060–6068. [Google Scholar] [CrossRef] [PubMed]
- Halfen, J.A.; Moore, H.L.; Fox, D.C. Synthetic Models of the Reduced Active Site of Superoxide Reductase. Inorg. Chem. 2002, 41, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.; Smith, J.M.; Yu, Y.; Ketterer, N.A.; Flaschenriem, C.J.; Lachicotte, R.J.; Holland, P.L. Synthesis and Reactivity of Low-Coordinate Iron (II) Fluoride Complexes and Their Use in the Catalytic Hydrodefluorination of Fluorocarbons. J. Am. Chem. Soc. 2005, 127, 7857–7870. [Google Scholar] [CrossRef] [PubMed]
- Quesada, M.; de la Peña-O’Shea, V.A.; Aromí, G.; Geremia, S.; Massera, C.; Roubeau, O.; Gamez, P.; Reedijk, J. A Molecule-Based Nanoporous Material Showing Tuneable Spin-Crossover Behavior near Room Temperature. Adv. Mater. 2007, 19, 1397–1402. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, L.; Deng, L. Dinuclear Iron−Imido Complexes with N-Heterocyclic Carbene Ligation: Synthesis, Structure, and Redox Reactivity. Organometallics 2012, 31, 4537–4543. [Google Scholar] [CrossRef]
- Ke, C.-H.; Chen, C.-H.; Tsai, M.-L.; Wang, H.-C.; Tsai, F.-T.; Chiang, Y.-W.; Shih, W.-C.; Bohle, D.S.; Liaw, W.-F. {Fe(NO)2}9 Dinitrosyl Iron Complex Acting as a Vehicle for the NO Radical. J. Am. Chem. Soc. 2017, 139, 67–70. [Google Scholar] [CrossRef]
- Hemming, E.B.; Chan, B.; Turner, P.; Corcilius, L.; Price, J.R.; Gardiner, M.G.; Masters, A.F.; Maschmeyer, T. [Fe(C5Ar5)(CO)2Br] complexes as hydrogenase mimics for the catalytic hydrogen evolution reaction. Appl. Catal. B Environ. 2018, 223, 234–241. [Google Scholar] [CrossRef]
- Narwane, M.; Chang, Y.-L.; Ching, W.-M.; Tsai, M.-L.; Hsu, S.C.N. Investigation on the coordination behaviors of tris(2-pyridyl) pyrazolyl borates iron (II) complexes. Inorg. Chim. Acta. 2019, 495, 118966. [Google Scholar] [CrossRef]
- Horn, E.; Snow, M.R.; Tiekink, E.R.T. A Polymeric Silver Lutidine Tetrafluoroborate Compound, [Ag(lut)2(µ4-BF4)]N, with Weak μ4-Tetrafluoroborate Coordination to Silver. Aust. J. Chem. 1987, 40, 761–765. [Google Scholar] [CrossRef]
- Blake, A.J.; Brooks, N.R.; Champness, N.R.; Cunningham, J.W.; Hubberstey, P.; Schröder, M. Thioether ligands as molecular rods in silver(I) coordination networks: 1,4-dithiane as an analogue of pyrazine. CrystEngComm 2000, 6, 41–45. [Google Scholar] [CrossRef]
- Fenton, H.; Tidmarsh, I.S.; Ward, M.D. Hierarchical self-assembly of heteronuclear co-ordination networks. Dalton Trans. 2010, 39, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Kilduff, B.; Pogozhev, D.; Baudron, S.A.; Hosseini, M.W. Heterometallic Architectures Based on the Combination of Heteroleptic Copper and Cobalt Complexes with Silver Salts. Inorg. Chem. 2010, 49, 11231–11239. [Google Scholar] [CrossRef] [PubMed]
- Poorters, L.; Armspach, D.; Matt, D.; Toupet, L.; Jones, P.G. A Metallocavitand Functioning as a Container for Anions: Formation of Noncovalent Linear Assemblies Mediated by a Cyclodextrin-Entrapped NO3 Ion. Angew. Chem. Int. Ed. 2007, 46, 2663–2665. [Google Scholar] [CrossRef] [PubMed]
- Cimadevilla, F.; García, E.M.; García-Vivó, D.; Ruiz, M.A.; Rueda, M.T.; Halut, S. Protonation reactions of the oxo complex cis-[Mo2(h5-C5H5)2(O)(μ-PPh2)2(CO)]. Hydroxo and tetrafluoroborate derivatives. J. Organomet. Chem. 2012, 699, 67–74. [Google Scholar] [CrossRef]
- Musina, E.I.; Karasik, A.A.; Balueva, A.S.; Strelnik, I.D.; Fesenko, T.I.; Dobrynin, A.B.; Gerasimova, T.P.; Katsyuba, S.A.; Kataeva, O.N.; Lönnecke, P.; et al. Synthesis and Stereoselective Interconversion of Chiral 1-Aza-3,6-diphosphacycloheptanes. Eur. J. Inorg. Chem. 2012, 2012, 1857–1866. [Google Scholar] [CrossRef]
- Kilgore, U.J.; Roberts, J.A.S.; Pool, D.H.; Appel, A.M.; Stewart, M.P.; Rakowski DuBois, M.; Dougherty, W.G.; Kassel, W.S.; Bullock, R.M.; DuBois, D.L. [Ni(PPh2NC6H4X2)2]2+ Complexes as Electrocatalysts for H2 Production: Effect of Substituents, Acids, and Water on Catalytic Rates. J. Am. Chem. Soc. 2011, 133, 5861–5872. [Google Scholar] [CrossRef]
- Kilgore, U.J.; Stewart, M.P.; Helm, M.L.; Dougherty, W.G.; Kassel, W.S.; Rakowski DuBois, M.; DuBois, D.L.; Bullock, R.M. Studies of a Series of [Ni(PR2NPh2)2(CH3CN)]2+ Complexes as Electrocatalysts for H2 Production: Substituent Variation at the Phosphorus Atom of the P2N2 Ligand. Inorg. Chem. 2011, 50, 10908–10918. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J.J. WinGX and ORTEP for Windows: An update. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6–13 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Complex 8 | Complex 11b | ||
|---|---|---|---|---|
| Bond Length (Type) | Bond | d, Å | Bond | d, Å |
| d Fe-Pa | Fe1-P7 | 2.232(3) | Fe1-P3 | 2.267(3) |
| Fe1-P7B | 2.242(3) | Fe1-P3′ | 2.267(3) | |
| d Fe-Peq | Fe1-P3 | 2.233(3) | Fe1-P7 | 2.209(4) |
| Fe1-P3B | 2.246(3) | Fe1-P7′ | 2.209(4) | |
| d Fe-Xeq | Fe1-N40 | 1.960(10) | Fe1-F1 | 2.050(8) |
| Fe1-N43 | 1.940(9) | Fe1-F1′ | 2.050(8) | |
| Bond Angle (Type) | Angle | ° | Angle | ° |
| Pa—Fe-Pa | P7-Fe1-P7B | 175.4(1) | P3-Fe1-P3′ | 176.5(2) |
| Peq-Fe-Peq | P3-Fe1-P3B | 104.09(1) | P7-Fe1-P7′ | 101.1(2) |
| Pa—Fe-Peq | P7-Fe1-P3 | 79.1(1) | P3-Fe1-P7 | 79.96(13) |
| P7B-Fe1-P3B | 79.3(1) | P3′-Fe1-P7′ | 79.96(13) | |
| P7B-Fe1-P3 | 98.5(3) | P3-Fe1-P7′ | 102.28(14) | |
| P7-Fe1-P3B | 97.4(1) | P3′-Fe1-P7 | 102.28(14) | |
| Xeq-Fe-Xeq | N40-Fe1-N43 | 82.9(4) | F1-Fe1-F1′ | 70.1(4) |
| X-Fe-Pa | N40-Fe1-P7B | 93.5(3) | F1-Fe1-P3 | 85.4(3) |
| N43-Fe1-P7 | 94.0(3) | F1′-Fe1-P3′ | 85.4(3) | |
| N40-Fe1-P7 | 89.6(3) | F1-Fe1-P3′ | 91.7(3) | |
| N43-Fe1-P7B | 89.8(3) | F1′-Fe1-P3 | 91.7(3) | |
| X-Fe-Peq | N40-Fe1-P3 | 164.0(3) | F1-Fe1-P7′ | 159.5(2) |
| N43-Fe1-P3B | 165.7(3) | F1′-Fe1-P7 | 159.5(2) | |
| N40-Fe1-P3B | 88.6(3) | F1-Fe1-P7 | 95.9(2) | |
| N43-Fe1-P3 | 86.6(3) | F1′-Fe1-P7′ | 95.9(2) | |
| Complex | Reduction Potential, V | Oxidation Potential, V |
|---|---|---|
| 6 | −1.74 | 0.34 |
| 7 | −1.50 | 0.30 |
| 8 | −1.48 | 0.25 |
| 9 | −1.37 | 0.30 |
| 10 | −1.64 | 0.32 |
| 12 | −1.80 | 1.23 |
| 13 | −1.60 | 1.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiridonova, Y.S.; Nikolaeva, Y.A.; Balueva, A.S.; Musina, E.I.; Litvinov, I.A.; Strelnik, I.D.; Khrizanforova, V.V.; Budnikova, Y.G.; Karasik, A.A. Synthesis and Structure of Iron (II) Complexes of Functionalized 1,5-Diaza-3,7-Diphosphacyclooctanes. Molecules 2020, 25, 3775. https://doi.org/10.3390/molecules25173775
Spiridonova YS, Nikolaeva YA, Balueva AS, Musina EI, Litvinov IA, Strelnik ID, Khrizanforova VV, Budnikova YG, Karasik AA. Synthesis and Structure of Iron (II) Complexes of Functionalized 1,5-Diaza-3,7-Diphosphacyclooctanes. Molecules. 2020; 25(17):3775. https://doi.org/10.3390/molecules25173775
Chicago/Turabian StyleSpiridonova, Yulia S., Yulia A. Nikolaeva, Anna S. Balueva, Elvira I. Musina, Igor A. Litvinov, Igor D. Strelnik, Vera V. Khrizanforova, Yulia G. Budnikova, and Andrey A. Karasik. 2020. "Synthesis and Structure of Iron (II) Complexes of Functionalized 1,5-Diaza-3,7-Diphosphacyclooctanes" Molecules 25, no. 17: 3775. https://doi.org/10.3390/molecules25173775