
Bottom-Up Design Approach for OBOC Peptide Libraries


Abstract
:
1. Introduction
2. Results and Discussion
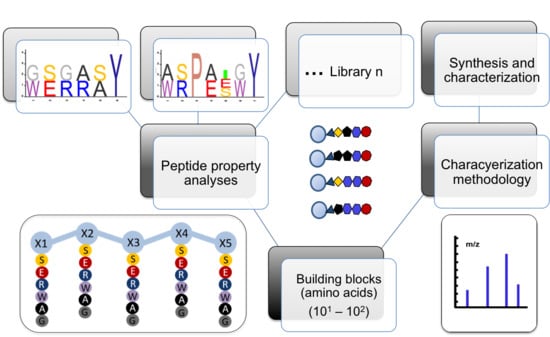
2.1. Bottom-Up Experimental Approach for the Design of OBOC Peptide Libraries
- (a)
- Library 1 (L1): {G, w} for x1, {s, e} for x2, {G, r} for x3, {a, r} for x4 and {s, a} for x5 and {y} for x6 to obtain a 32-component library (sequence logo in Figure 1a), where all the sequences are unique by mass;
- (b)
- Library 2 (L2): {a, w} for x1, {s, r} for x2, {p} for x3, {a, e} for x4, {i, e, s} for x5, {G, w} for x6 and {y} for x7 to obtain a 48-component library (sequence logo in Figure S1b,), where 47 permutations are unique by mass and one shows mass overlapping.
2.2. Theoretical Evaluation of Library Properties
2.3. Library Synthesis and Characterization by UPLC-MS, High-Resolution Mass Spectrometry and Nano LC-MS/MS
3. Conclusions
4. Materials and Methods
4.1. General
4.2. GA-Assisted Library Design
4.3. Solid-Phase Peptide Synthesis (SPPS) Procedure
4.4. Quantification of Resin Loading Capacity
4.5. The Spilt and Mix Method
4.6. PackageR Peptide Property Calculations
4.7. Ultra-High-Performance Liquid Chromatography-Quadrupole-Mass Spectrometry (UPLC-MS)
4.8. High-Resolution Mass Spectrometry
4.9. NanoLC-MS/MS Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meldal, M. The one-bead two-compound assay for solid phase screening of combinatorial libraries. Biopolym.-Pept. Sci. Sect. 2002, 66, 93–100. [Google Scholar] [CrossRef]
- Christensen, C.; Groth, T.; Schiødt, C.B.; Foged, N.T.; Meldal, M. Automated Sorting of Beads from a “One-Bead-Two-Compounds” Combinatorial Library of Metalloproteinase Inhibitors. QSAR Comb. Sci. 2003, 22, 737–744. [Google Scholar] [CrossRef]
- Madsen, D.; Jørgensen, F.P.; Palmer, D.; Roux, M.; Olsen, J.V.; Bols, M.; Schoffelen, S.; Diness, F.; Meldal, M. Design and Combinatorial Development of Shield-1 Peptide Mimetics Binding to Destabilized FKBP12. ACS Comb. Sci. 2020, 22, 156–164. [Google Scholar] [CrossRef]
- Hu, H.; Nikitin, S.; Berthelsen, A.B.; Diness, F.; Schoffelen, S.; Meldal, M. Sustainable Flow Synthesis of Encoded Beads for Combinatorial Chemistry and Chemical Biology. ACS Comb. Sci. 2018, 20, 492–498. [Google Scholar] [CrossRef]
- Meldal, M. “One bead two compound libraries” for detecting chemical and biochemical conversions. Curr. Opin. Chem. Biol. 2004, 8, 238–244. [Google Scholar] [CrossRef]
- Li, M.; Hoeck, C.; Schoffelen, S.; Gotfredsen, C.H.; Meldal, M. Specific Electrostatic Molecular Recognition in Water. Chem.-A Eur. J. 2016, 22, 7206–7214. [Google Scholar] [CrossRef] [Green Version]
- Meldal, M. Pega: A flow stable polyethylene glycol dimethyl acrylamide copolymer for solid phase synthesis. Tetrahedron Lett. 1992, 33, 3077–3080. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “clicking” by CuAAC reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Hansen, J.; Diness, F.; Meldal, M. C-Terminally modified peptides via cleavage of the HMBA linker by O-, N- or S-nucleophiles. Org. Biomol. Chem. 2016, 14, 3238–3245. [Google Scholar] [CrossRef] [Green Version]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar]
- Meldal, M.; Christensen, S.F. Microparticle matrix encoding of beads. Angew. Chemie-Int. Ed. 2010, 49, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Xiao, W.; Lam, K.S. Tumor-targeting peptides from combinatorial libraries. Adv. Drug Deliv. Rev. 2017, 110, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ceron, M.C.; Giudicessi, S.L.; Saavedra, S.L.; Gurevich-Messina, J.M.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; Camperi, S.A. Latest Advances in OBOC Peptide Libraries. Improvements in Screening Strategies and Enlarging the Family From Linear to Cyclic Libraries. Curr. Pharm. Biotechnol. 2016, 17, 449–457. [Google Scholar]
- Gurevich-Messina, J.M.; Giudicessi, S.L.; Martínez-Ceron, M.C.; Acosta, G.; Erra-Balsells, R.; Cascone, O.; Albericio, F.; Camperi, S.A. A simple protocol for combinatorial cyclic depsipeptide libraries sequencing by matrix-assisted laser desorption/ionisation mass spectrometry. J. Pept. Sci. 2015, 21, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ceron, M.C.; Giudicessi, S.L.; Marani, M.M.; Albericio, F.; Cascone, O.; Erra-Balsells, R.; Camperi, S.A. Sample preparation for sequencing hits from one-bead-one-peptide combinatorial libraries by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 2010, 400, 295–297. [Google Scholar] [CrossRef]
- Marani, M.M.; Ceron, M.C.M.; Giudicessi, S.L.; De Oliveira, E.; Côté, S.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; Camperi, S.A. Screening of one-bead-one-peptide combinatorial library using red fluorescent dyes. Presence of positive and false positive beads. J. Comb. Chem. 2009, 11, 146–150. [Google Scholar] [CrossRef]
- Giudicessi, S.L.; Gurevich-Messina, J.M.; Martínez-Ceron, M.C.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; Camperi, S.A. Friendly strategy to prepare encoded one bead-one compound cyclic peptide library. ACS Comb. Sci. 2013, 15, 525–529. [Google Scholar] [CrossRef]
- Bononi, F.C.; Luyt, L.G. Synthesis and Cell-Based Screening of One-Bead-One- Compound Peptide Libraries. In Peptide Libraries: Methods and Protocols, Methods in Molecular Biology; Derda, R., Ed.; 2015; Volume 1248, pp. 223–237. ISBN 9781493920204. [Google Scholar]
- Liu, R.; Marik, J.; Lam, K.S. A novel peptide-based encoding system for “one-bead one-compound” peptidomimetic and small molecule combinatorial libraries. J. Am. Chem. Soc. 2002, 124, 7678–7680. [Google Scholar] [CrossRef]
- Cho, C.-F.; Behnam Azad, B.; Luyt, L.G.; Lewis, J.D. High-throughput screening of one-bead-one-compound peptide libraries using intact cells. ACS Comb. Sci. 2013, 15, 393–400. [Google Scholar] [CrossRef]
- Marani, M.M.; Oliveira, E.; Côte, S.; Camperi, S.A.; Albericio, F.; Cascone, O. Identification of protein-binding peptides by direct matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis of peptide beads selected from the screening of one bead-one peptide combinatorial libraries. Anal. Biochem. 2007, 370, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, H.; Xiao, W.; Kumaresan, P.R.; Shi, C.; Pan, C.X.; Aina, O.H.; Lam, K.S. Rainbow beads: A color coding method to facilitate high-throughput screening and optimization of one-bead one-compound combinatorial libraries. J. Comb. Chem. 2008, 10, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Bononi, F.C.; Townsend, J.; Li, Y.; Liu, R.; Lam, K.S. Immobilized OBOC combinatorial bead array to facilitate multiplicative screening. Comb. Chem. High Throughput Screen 2013, 16, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, C.F.; Amadei, G.A.; Breadner, D.; Luyt, L.G.; Lewis, J.D. Discovery of novel integrin ligands from combinatorial libraries using a multiplex “beads on a bead” approach. Nano Lett. 2012, 12, 5957–5965. [Google Scholar] [CrossRef] [PubMed]
- Guixer, B.; Arroyo, X.; Belda, I.; Sabidó, E.; Teixidó, M.; Giralt, E. Chemically synthesized peptide libraries as a new source of BBB shuttles. Use of mass spectrometry for peptide identification. J. Pept. Sci. 2016, 22, 577–591. [Google Scholar]
- Wright, T.; Gillet, V.J.; Green, D.V.S.; Pickett, S.D. Optimizing the size and configuration of combinatorial libraries. J. Chem. Inf. Comput. Sci. 2003, 43, 381–390. [Google Scholar] [CrossRef]
- Zhao, P.L.; Nachbar, R.B.; Bolognese, J.A.; Chapman, K. Two new criteria for choosing sample size in combinatorial chemistry. J. Med. Chem. 1996, 39, 350–352. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Mauša, G.; Todorovski, T.; Giralt, E. Algorithm-supported, mass and sequence diversity-oriented random peptide library design. J. Cheminform. 2019, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Osorio, M.D. Package ‘Peptides’ 2018. Available online: https://github.com/dosorio/Peptides/ (accessed on 28 November 2019).
- Cruciani, G.; Baroni, M.; Carosati, E.; Clementi, M.; Valigi, R.; Clementi, S. Peptide studies by means of principal properties of amino acids derived from MIF descriptors. J. Chemom. 2004, 18, 146–155. [Google Scholar] [CrossRef]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. R J. 2015, 7, 4–14. [Google Scholar] [CrossRef]
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Akai, A. Thermostability and Aliphatic Index of Globular Proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.M.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Galiñanes Reyes, S.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Furka, Á.; Sebestyén, F.; Asgedom, M.; Dibó, G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Prot. Res. 1991, 37, 487–493. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color Test for Detection of Free Terminal Amino Groups in the Solid-Phase Synthesis of Peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Palmblad, M.; Drijfhout, J.W.; Deelder, A.M. High resolution mass spectrometry for rapid characterization of combinatorial peptide libraries. J. Comb. Chem. 2010, 12, 65–68. [Google Scholar] [CrossRef]
- Scott Youngquist, R.; Fuentes, G.R.; Lacey, M.P.; Keough, T.; Baillie, T.A. Matrix-assisted laser desorption ionization for rapid determination of the sequences of biologically active peptides isolated from support-bound combinatorial peptide libraries. Rapid Commun. Mass Spectrom. 1994, 8, 77–81. [Google Scholar] [CrossRef]
- Semmler, A.; Weber, R.; Przybylsk, M.; Wittmann, V. De Novo Sequencing of Peptides on SingleResin Beads by MALDI-FTICR Tandem MassSpectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, S.H.; Xiao, Q.; Ling, Y.; Gopishetty, B.; Pei, D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J. Am. Chem. Soc. 2006, 128, 13000–13009. [Google Scholar] [CrossRef] [PubMed]
- Paulick, M.G.; Hart, K.M.; Brinner, K.M.; Tjandra, M.; Charych, D.H.; Zuckermann, R.N. Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J. Comb. Chem. 2006, 8, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tholey, A.; Becker, A. Top-down proteomics for the analysis of proteolytic events-Methods, applications and perspectives. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evaluation Measure | Measurement Technique | ||
|---|---|---|---|
| Abbreviation | Formula | UPLC-MS | LTQ |
| MAE | 0.1917 | 0.0007 | |
| RMSE | 0.2054 | 0.0007 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalafatovic, D.; Mauša, G.; Rešetar Maslov, D.; Giralt, E. Bottom-Up Design Approach for OBOC Peptide Libraries. Molecules 2020, 25, 3316. https://doi.org/10.3390/molecules25153316
Kalafatovic D, Mauša G, Rešetar Maslov D, Giralt E. Bottom-Up Design Approach for OBOC Peptide Libraries. Molecules. 2020; 25(15):3316. https://doi.org/10.3390/molecules25153316
Chicago/Turabian StyleKalafatovic, Daniela, Goran Mauša, Dina Rešetar Maslov, and Ernest Giralt. 2020. "Bottom-Up Design Approach for OBOC Peptide Libraries" Molecules 25, no. 15: 3316. https://doi.org/10.3390/molecules25153316