
How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids?


Abstract
:1. Introduction
2. Nature of the Biomolecular Target for Structure-Based Methods
2.1. Experimentally Determined Structure
2.2. Computational Methods for Structure Prediction
3. “40 Years of Protein Docking Highlights and Advancements”
3.1. An Evolutionary Perspective: From Rigid to Flexible Docking
3.2. Molecular Docking as a Crossing Tool for Multiple Scopes
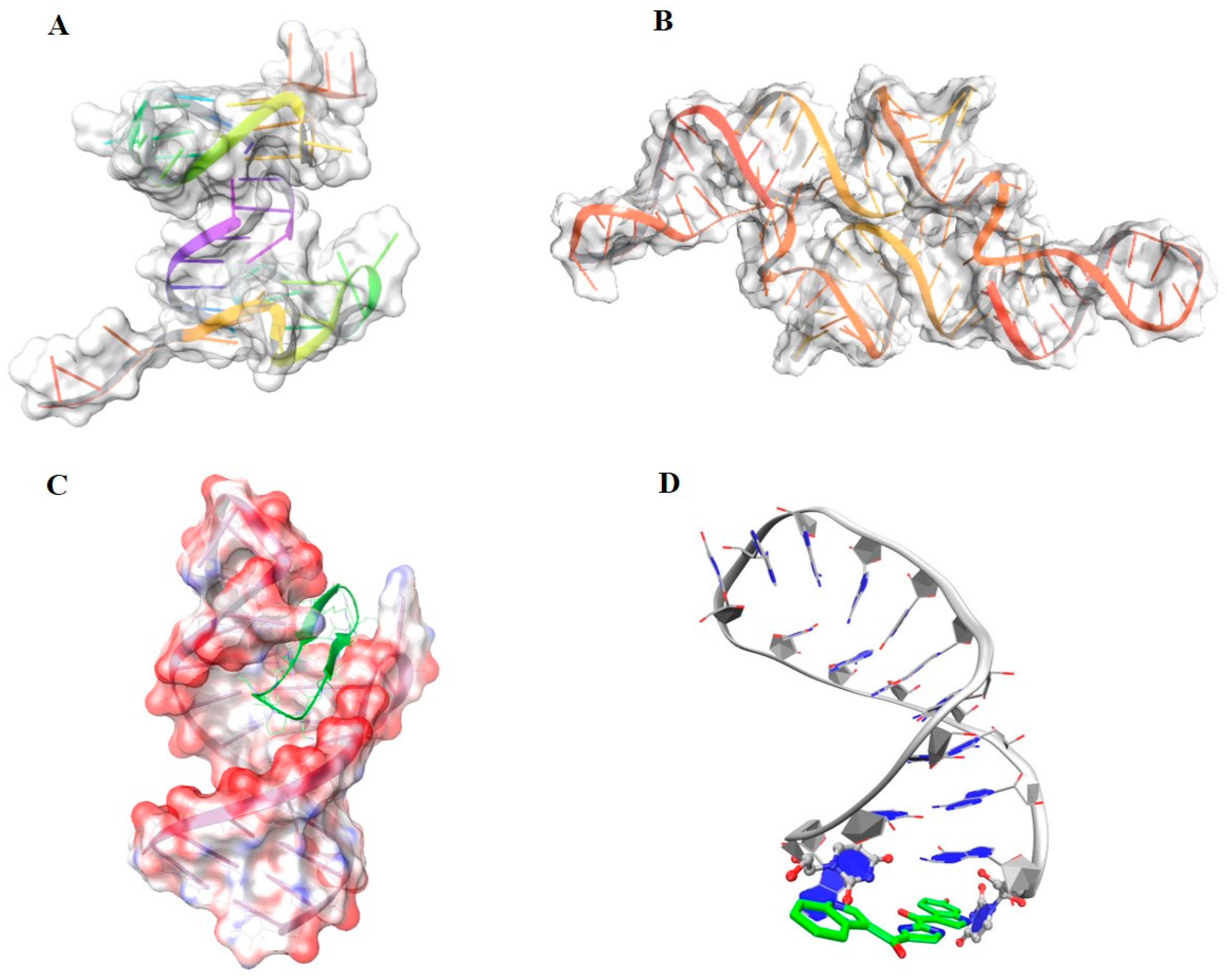
4. Nucleic Acids as Emerging Therapeutic Targets
4.1. DNA-Targeting for Cancer and Antimicrobial Therapy
4.2. RNA as Antiviral and Antibacterial Target
4.3. mRNA Triggering Splicing Machinery
5. Current ‘Protein Docking Algorithms’ Applied to Nucleic Acids: Challenges, Solutions, and Pitfalls
5.1. Challenges of Nucleic Acid Docking
5.1.1. Structural and Active Site Features:
5.1.2. Charge Distribution Effect on Ligand-Nucleic Acids Interactions:
5.1.3. Metal Ions and Solvation:
5.1.4. Target Flexibility
5.2. Benchmark of Docking Programs Applied to Nucleic Acids
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kendrew, J.C.; Dickerson, R.E.; Strandberg, B.E.; Hart, R.G.; Davies, D.R.; Phillips, D.C.; Shore, V. Structure of myoglobin: A three-dimensional Fourier synthesis at 2 Å. resolution. Nature 1960, 185, 422. [Google Scholar] [CrossRef] [PubMed]
- Herzik, M.A.; Wu, M.; Lander, G.C. High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat. Commun. 2019, 10, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)-Round XII. Proteins 2018, 86 (Suppl. 1), 7–15. [Google Scholar] [CrossRef] [PubMed]
- Somody, J.C.; MacKinnon, S.S.; Windemuth, A. Structural coverage of the proteome for pharmaceutical applications. Drug Discov. Today 2017, 22, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Hillisch, A.; Pineda, L.F.; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar] [CrossRef]
- Mukherjee, B.; Tessaro, F.; Vahokoski, J.; Kursula, I.; Marq, J.B.; Scapozza, L.; Soldati-Favre, D. Modeling and resistant alleles explain the selectivity of antimalarial compound 49c towards apicomplexan aspartyl proteases. EMBO J. 2018, 37, e98047. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Kuntz, I.D.; Bodian, D.L. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. II. Ber. Der Dtsch. Chem. Ges. 1894, 27, 3479–3483. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, B.L.; Koshland, D.E., Jr. Prediction of the structure of a receptor-protein complex using a binary docking method. Nature 1992, 358, 774–776. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Docking flexible ligands to macromolecular receptors by molecular shape. J. Med. Chem. 1986, 29, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins 1992, 13, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Aided Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541. [Google Scholar] [CrossRef]
- Jiang, F.; Kim, S.H. “Soft docking”: Matching of molecular surface cubes. J. Mol. Biol. 1991, 219, 79–102. [Google Scholar] [CrossRef]
- Leach, A.R. Ligand docking to proteins with discrete side-chain flexibility. J. Mol. Biol. 1994, 235, 345–356. [Google Scholar] [CrossRef]
- Knegtel, R.M.A.; Kuntz, I.D.; Oshiro, C.M. Molecular docking to ensembles of protein structures11Edited by B. Honig. J. Mol. Biol. 1997, 266, 424–440. [Google Scholar] [CrossRef] [Green Version]
- Claussen, H.; Buning, C.; Rarey, M.; Lengauer, T. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 2001, 308, 377–395. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, O.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Wenying, S.; Xuanyi, L.; Hequan, Y.; Kejiang, L. Convolutional Neural Network Based Virtual Screening. Curr. Med. Chem. 2020, 27, 1–15. [Google Scholar]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Sharma, R.; Kumar, A. Docking techniques in pharmacology: How much promising? Comput. Biol. Chem. 2018, 76, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavasotto, C.N.; Adler, N.S.; Aucar, M.G. Quantum Chemical Approaches in Structure-Based Virtual Screening and Lead Optimization. Front. Chem. 2018, 6, 188. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Hurley, L.H.; Boyd, F.L. DNA as a target for drug action. Trends Pharmacol. Sci. 1988, 9, 402–407. [Google Scholar] [CrossRef]
- Wang, M.; Yu, Y.; Liang, C.; Lu, A.; Zhang, G. Recent Advances in Developing Small Molecules Targeting Nucleic Acid. Int. J. Mol. Sci. 2016, 17, 779. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Bhattacharya, S. DNA binders in clinical trials and chemotherapy. Bioorg. Med. Chem. 2014, 22, 4506–4521. [Google Scholar] [CrossRef]
- Kohn, K.W. Beyond DNA cross-linking: History and prospects of DNA-targeted cancer treatment--fifteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1996, 56, 5533–5546. [Google Scholar] [PubMed]
- Dutta, D.; Debnath, M.; Müller, D.; Paul, R.; Das, T.; Bessi, I.; Schwalbe, H.; Dash, J. Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex. Nucleic Acids Res. 2018, 46, 5355–5365. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, A.; Aldrich-Wright, J.R. DNA as a target for antimicrobials. Bioorg. Chem. 2014, 55, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Cimino-Reale, G.; Zaffaroni, N.; Folini, M. Emerging Role of G-quadruplex DNA as Target in Anticancer Therapy. Curr. Pharm. Des. 2016, 22, 6612–6624. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Gemmell, C.G.; Suckling, C.J. Minor groove binders as anti-infective agents. Pharm. Ther. 2013, 139, 12–23. [Google Scholar] [CrossRef]
- Wilson, W.D.; Tanious, F.A.; Mathis, A.; Tevis, D.; Hall, J.E.; Boykin, D.W. Antiparasitic compounds that target DNA. Biochimie 2008, 90, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lopez, A.; Tessaro, F.; Jonker, H.R.A.; Wacker, A.; Richter, C.; Comte, A.; Berntenis, N.; Schmucki, R.; Hatje, K.; Petermann, O.; et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat. Commun. 2018, 9, 2032. [Google Scholar] [CrossRef]
- McCown, P.J.; Corbino, K.A.; Stav, S.; Sherlock, M.E.; Breaker, R.R. Riboswitch diversity and distribution. RNA 2017, 23, 995–1011. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [Green Version]
- Shortridge, M.D.; Wille, P.T.; Jones, A.N.; Davidson, A.; Bogdanovic, J.; Arts, E.; Karn, J.; Robinson, J.A.; Varani, G. An ultra-high affinity ligand of HIV-1 TAR reveals the RNA structure recognized by P-TEFb. Nucleic Acids Res. 2019, 47, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wires Rna 2013, 4, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Marquis, J.; Meyer, K.; Angehrn, L.; Kämpfer, S.S.; Rothen-Rutishauser, B.; Schümperli, D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol. Ther. 2007, 15, 1479–1486. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Campagne, S.; Boigner, S.; Rüdisser, S.; Moursy, A.; Gillioz, L.; Knörlein, A.; Hall, J.; Ratni, H.; Cléry, A.; Allain, F.H.T. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat. Chem. Biol. 2019, 15, 1191–1198. [Google Scholar] [CrossRef]
- Detering, C.; Varani, G. Validation of automated docking programs for docking and database screening against RNA drug targets. J. Med. Chem. 2004, 47, 4188–4201. [Google Scholar] [CrossRef]
- Morley, S.D.; Afshar, M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. J. Comput. Aided Mol. Des. 2004, 18, 189–208. [Google Scholar] [CrossRef]
- Giambaşu, G.M.; Case, D.A.; York, D.M. Predicting Site-Binding Modes of Ions and Water to Nucleic Acids Using Molecular Solvation Theory. J. Am. Chem. Soc. 2019, 141, 2435–2445. [Google Scholar] [CrossRef]
- Moitessier, N.; Westhof, E.; Hanessian, S. Docking of Aminoglycosides to Hydrated and Flexible RNA. J. Med. Chem. 2006, 49, 1023–1033. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA–small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, P.; Gohlke, H. DrugScoreRNAKnowledge-Based Scoring Function to Predict RNA−Ligand Interactions. J. Chem. Inf. Model. 2007, 47, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Luo, J.; Waldispühl, J.; Moitessier, N. Predicting Positions of Bridging Water Molecules in Nucleic Acid–Ligand Complexes. J. Chem. Inf. Model. 2019, 59, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef]
- Chen, Q.; Shafer, R.H.; Kuntz, I.D. Structure-Based Discovery of Ligands Targeted to the RNA Double Helix. Biochemistry 1997, 36, 11402–11407. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.E.; Du, Z.; Fujinaga, K.; Peterlin, B.M.; James, T.L. Structure-Based Computational Database Screening, In Vitro Assay, and NMR Assessment of Compounds that Target TAR RNA. Chem. Biol. 2002, 9, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Guilbert, C.; James, T.L. Docking to RNA via Root-Mean-Square-Deviation-Driven Energy Minimization with Flexible Ligands and Flexible Targets. J. Chem. Inf. Model. 2008, 48, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Rohs, R.; Bloch, I.; Sklenar, H.; Shakked, Z. Molecular flexibility in ab initio drug docking to DNA: Binding-site and binding-mode transitions in all-atom Monte Carlo simulations. Nucleic Acids Res. 2005, 33, 7048–7057. [Google Scholar] [CrossRef] [Green Version]
- Krüger, D.M.; Bergs, J.; Kazemi, S.; Gohlke, H. Target Flexibility in RNA−Ligand Docking Modeled by Elastic Potential Grids. ACS Med. Chem. Lett. 2011, 2, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, S.; Krüger, D.M.; Sirockin, F.; Gohlke, H. Elastic potential grids: Accurate and efficient representation of intermolecular interactions for fully flexible docking. ChemMedChem 2009, 4, 1264–1268. [Google Scholar] [CrossRef]
- Luo, J.; Wei, W.; Waldispuhl, J.; Moitessier, N. Challenges and current status of computational methods for docking small molecules to nucleic acids. Eur. J. Med. Chem. 2019, 168, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Deligkaris, C.; Ascone, A.T.; Sweeney, K.J.; Greene, A.J.Q. Validation of a computational docking methodology to identify the non-covalent binding site of ligands to DNA. Mol. Biosyst. 2014, 10, 2106–2125. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Calin, G.A.; Zhang, S. Novel insights of structure-based modeling for RNA-targeted drug discovery. J. Chem. Inf. Model. 2012, 52, 2741–2753. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, J.; Sun, X.; Li, W.; Liu, G.; Tang, Y. Accuracy Assessment of Protein-Based Docking Programs against RNA Targets. J. Chem. Inf. Model. 2010, 50, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J. SPA-LN: A scoring function of ligand-nucleic acid interactions via optimizing both specificity and affinity. Nucleic Acids Res. 2017, 45, e110. [Google Scholar] [CrossRef]
- McElfresh, G.W.; Deligkaris, C. A vibrational entropy term for DNA docking with autodock. Comput. Biol. Chem. 2018, 74, 286–293. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessaro, F.; Scapozza, L. How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids? Molecules 2020, 25, 2749. https://doi.org/10.3390/molecules25122749
Tessaro F, Scapozza L. How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids? Molecules. 2020; 25(12):2749. https://doi.org/10.3390/molecules25122749
Chicago/Turabian StyleTessaro, Francesca, and Leonardo Scapozza. 2020. "How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids?" Molecules 25, no. 12: 2749. https://doi.org/10.3390/molecules25122749