
Research Progress of Small Molecule VEGFR/c-Met Inhibitors as Anticancer Agents (2016–Present)



Abstract
:
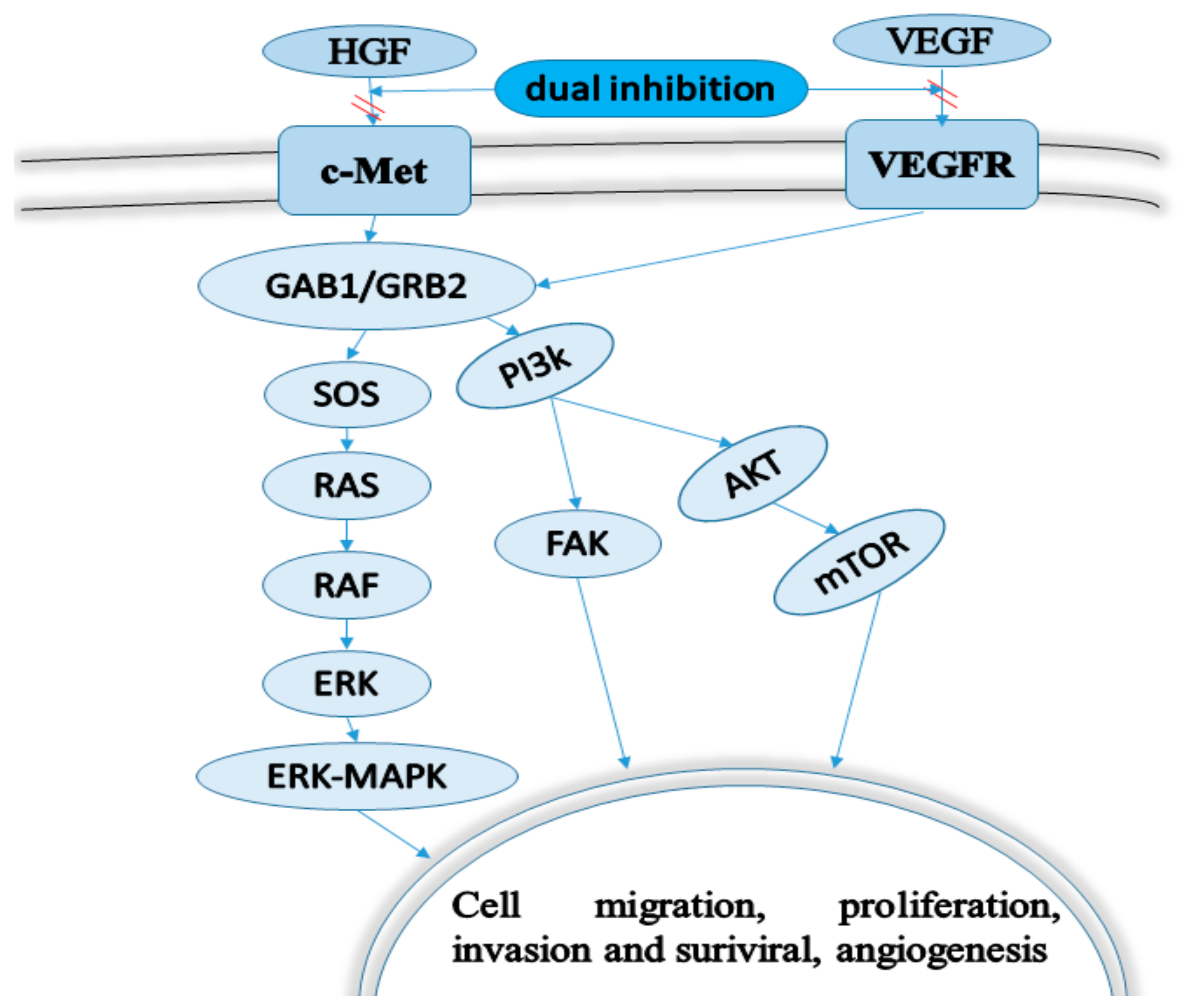
1. Introduction
2. Chemical Design Strategies of Dual-Target VEGFR/c-Met Kinase Anticancer Inhibitors
3. Dual VEGFR and c-Met Small Molecule Inhibitors
3.1. Pyridine Derivatives
3.2. Quinoline Derivatives
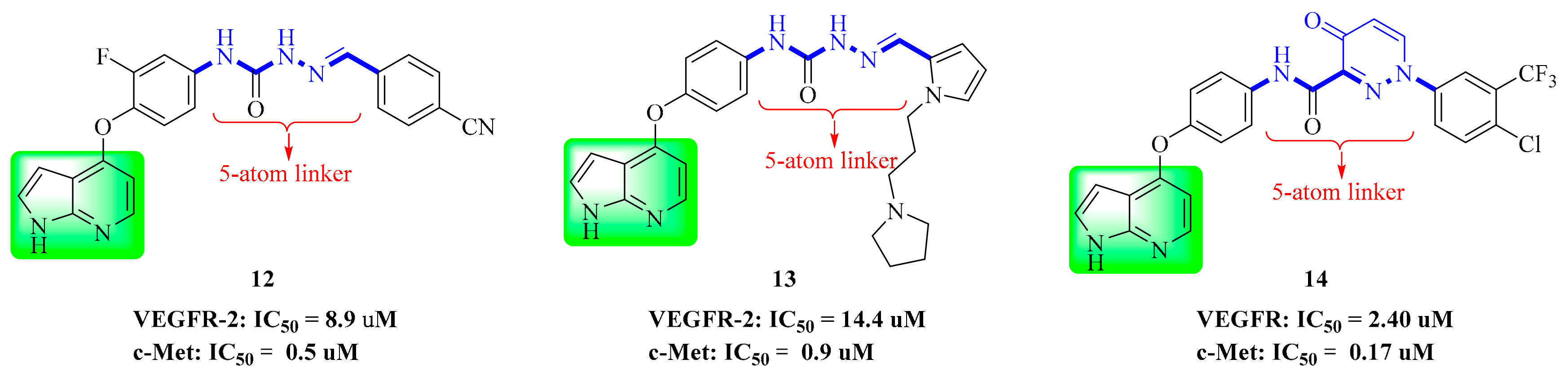
3.3. Pyrrolopyridine Derivatives
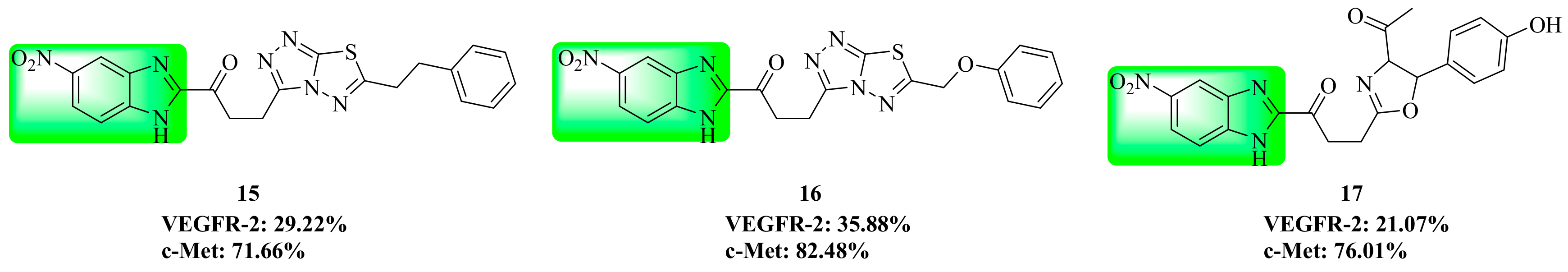
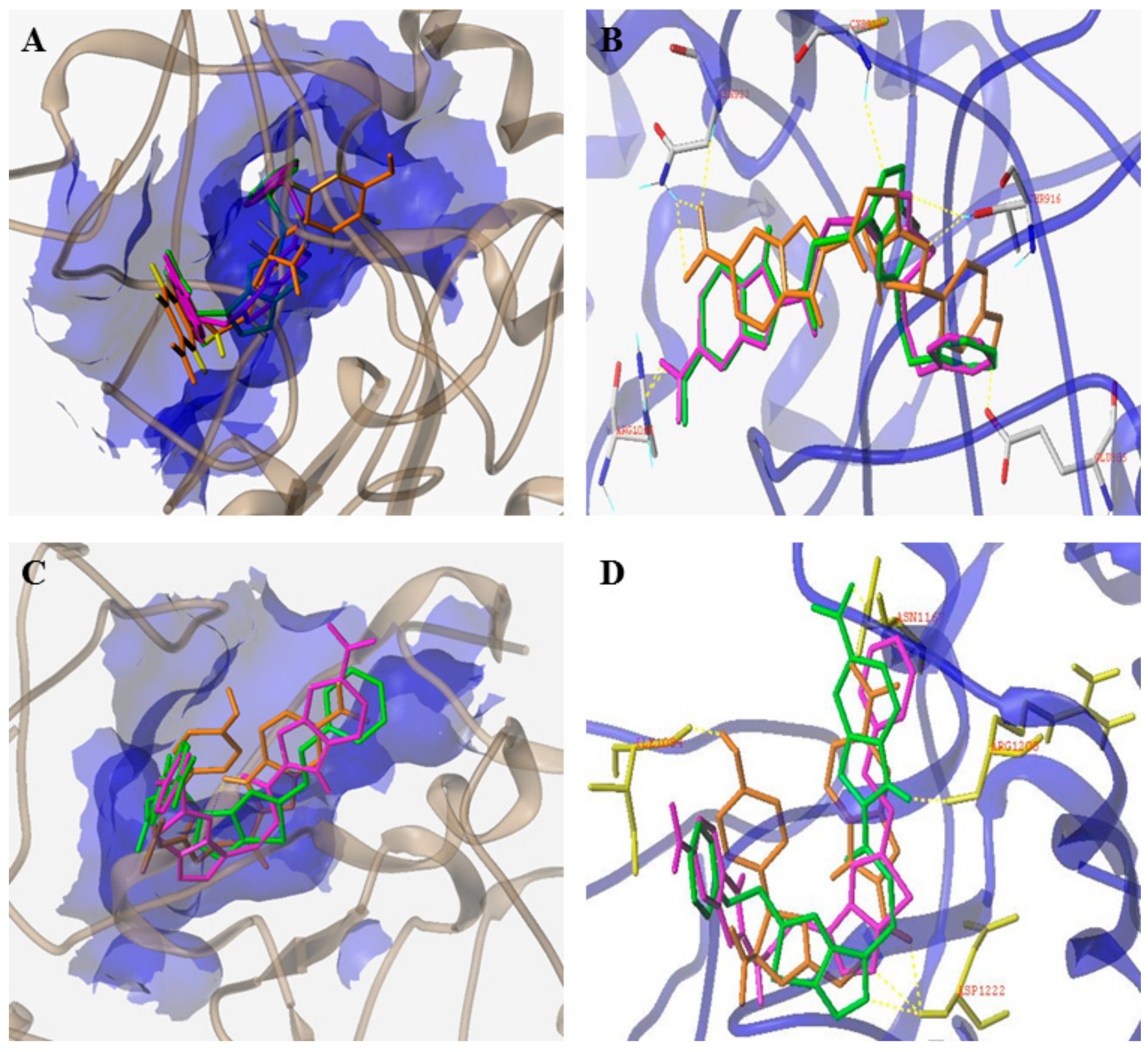
3.4. Benzimidazole Derivatives
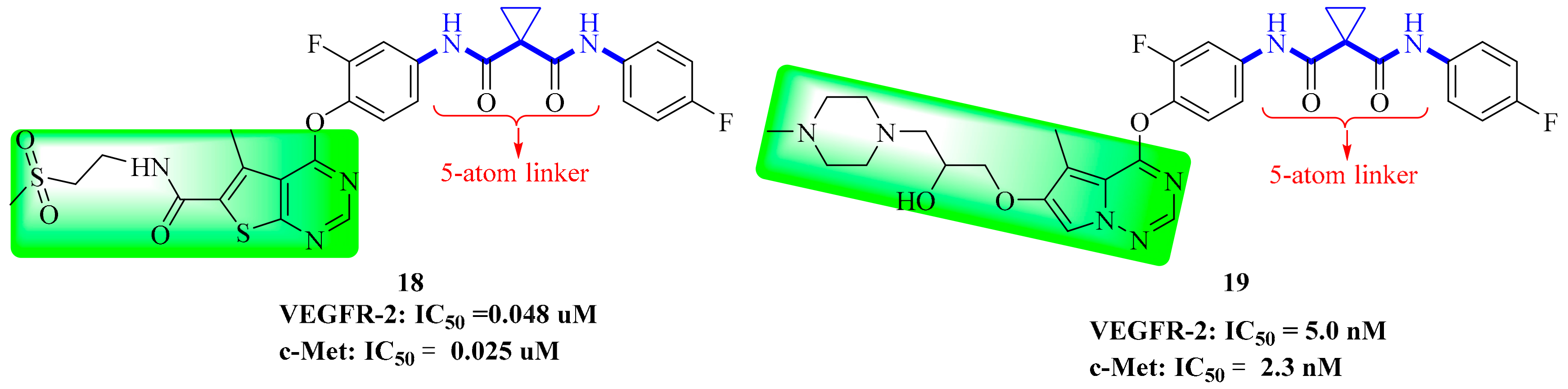
3.5. Thienopyrimidine Derivatives
3.6. Pyrrolotriazine Derivatives
3.7. Quinazoline Derivatives
3.8. Diazepine Derivatives
3.9. Pyrazolopyrimidine Derivatives
3.10. Naphthyridinone Derivatives
3.11. Triazine Derivatives
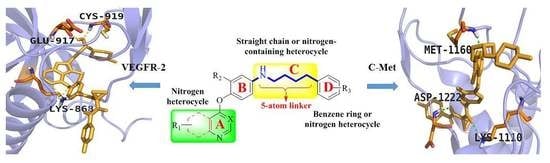
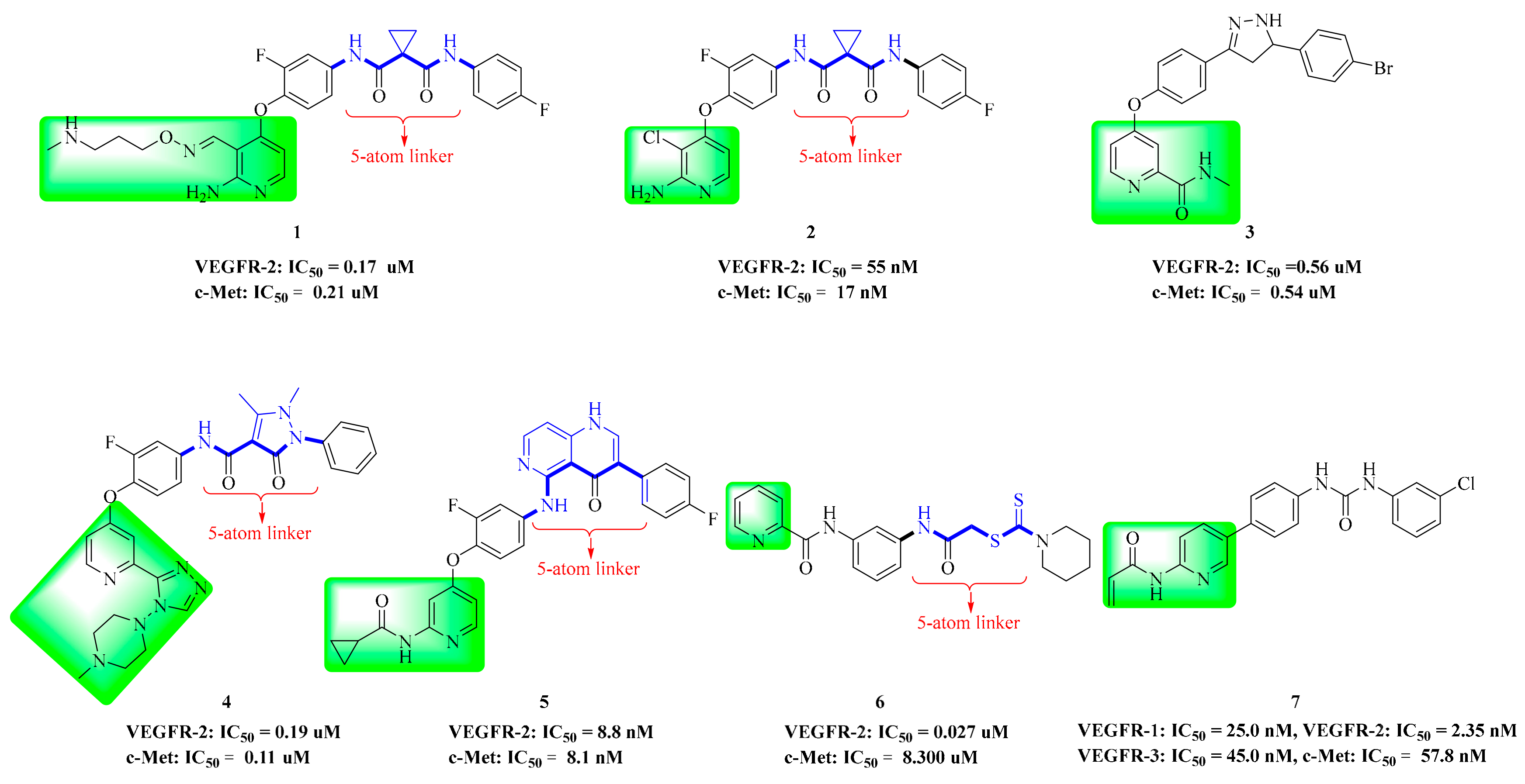
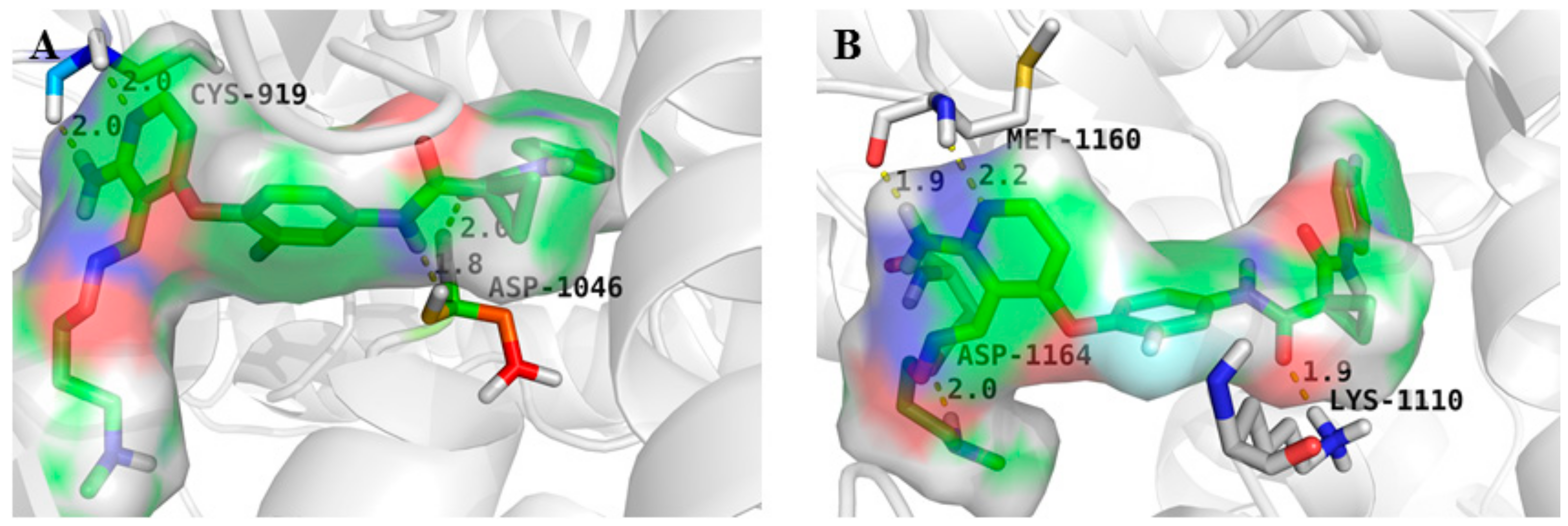
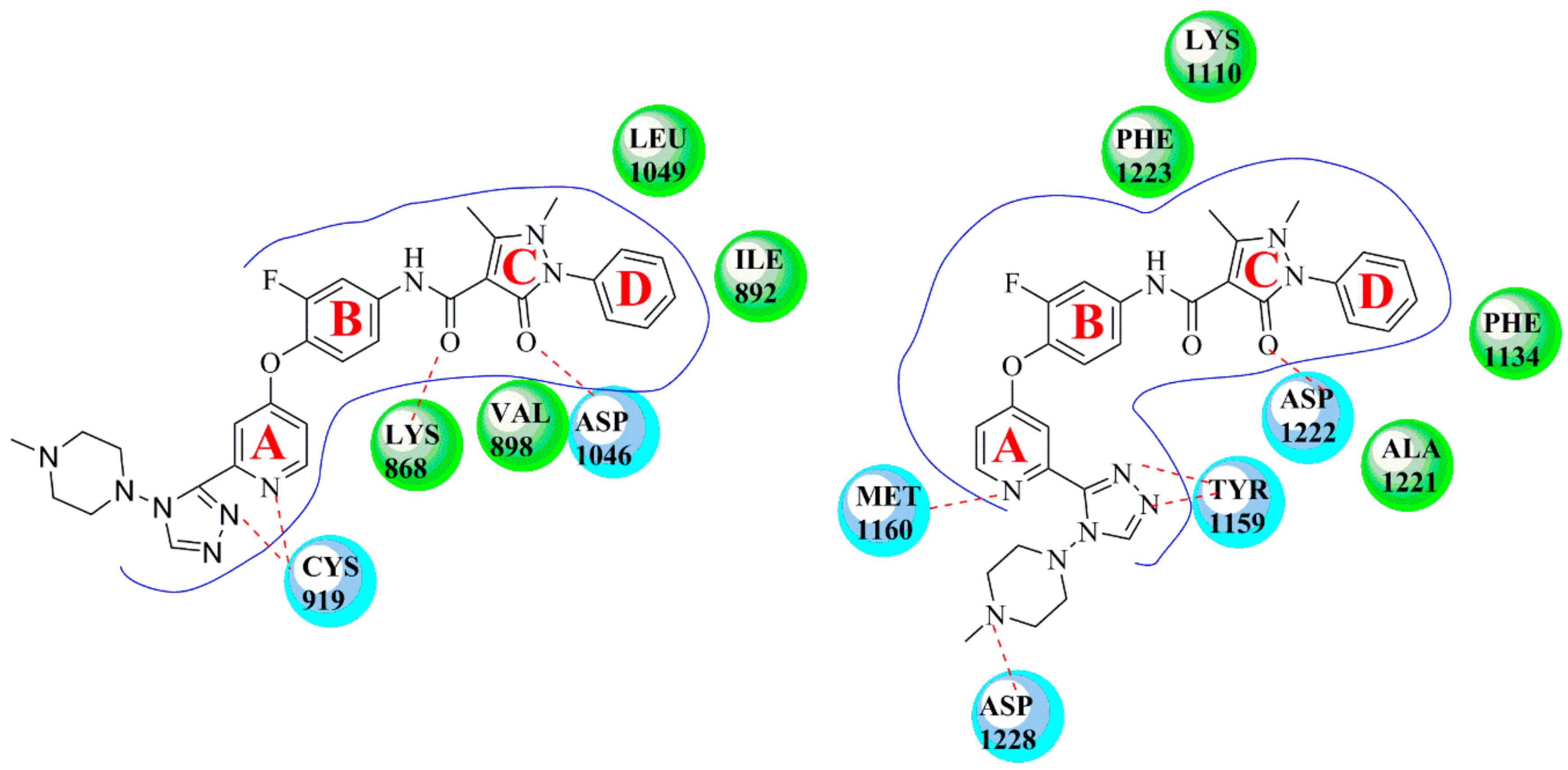
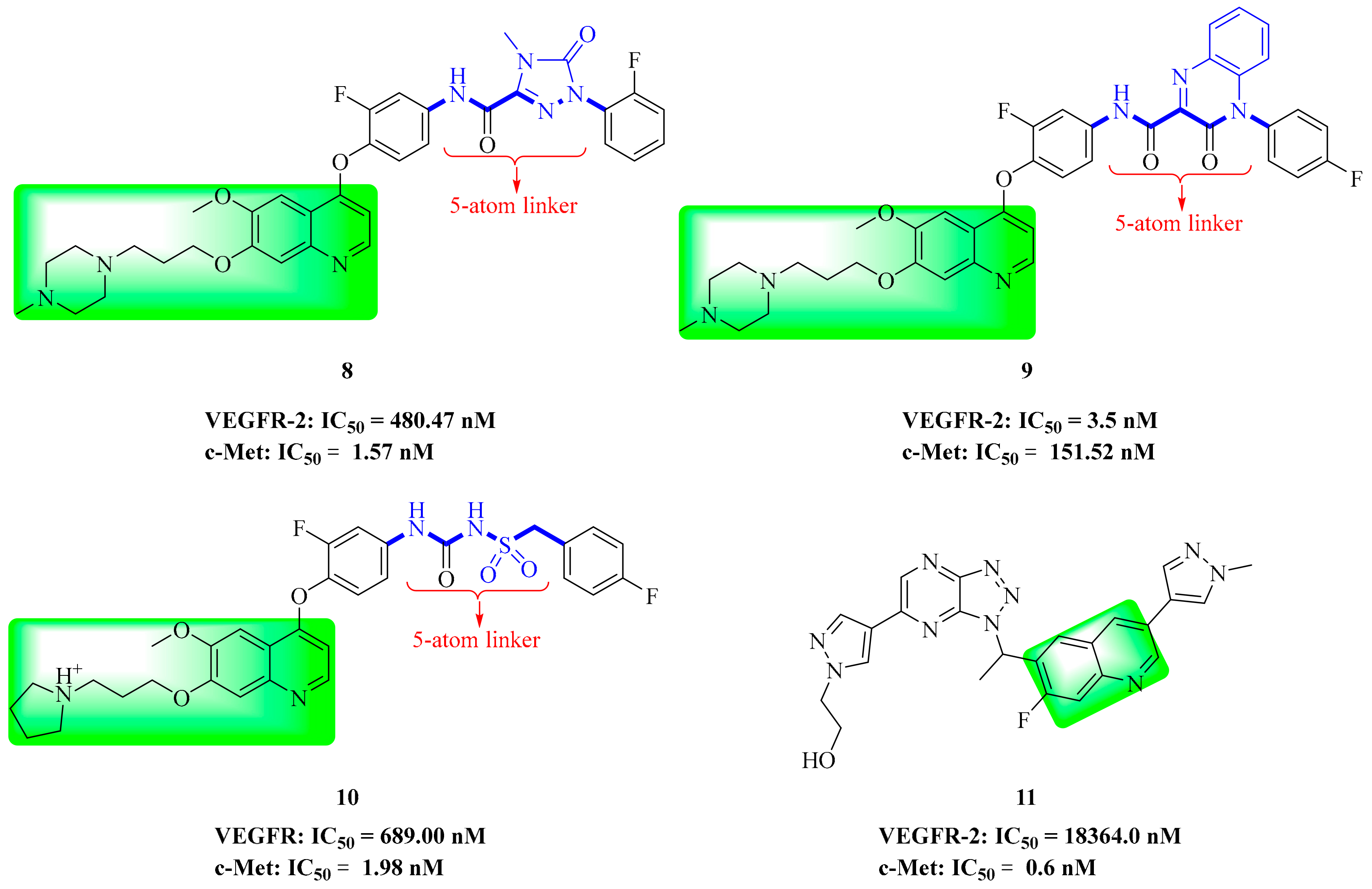
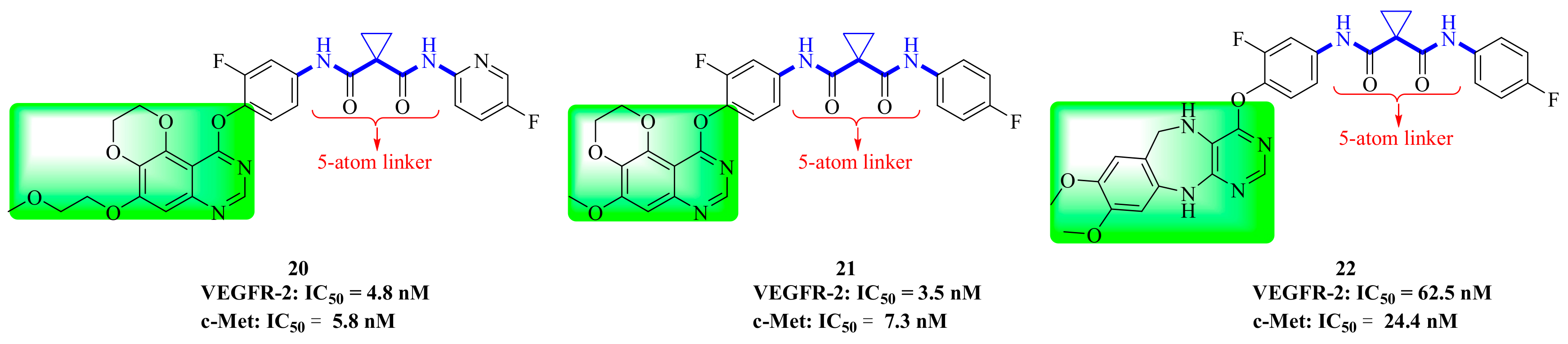
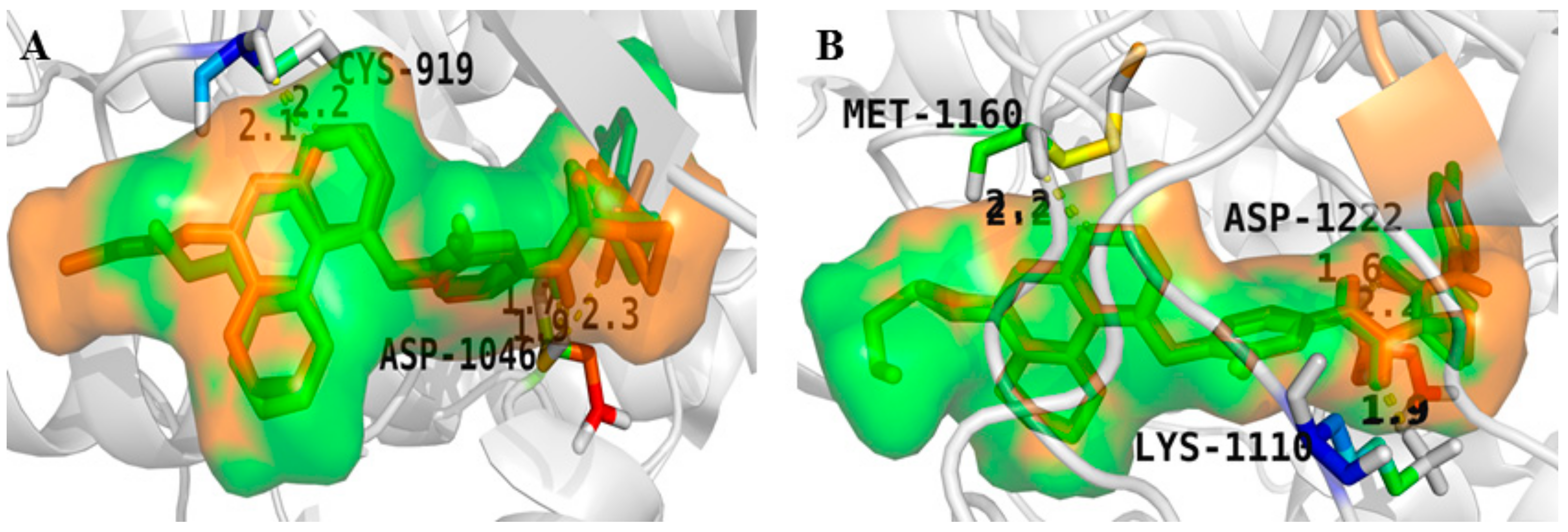
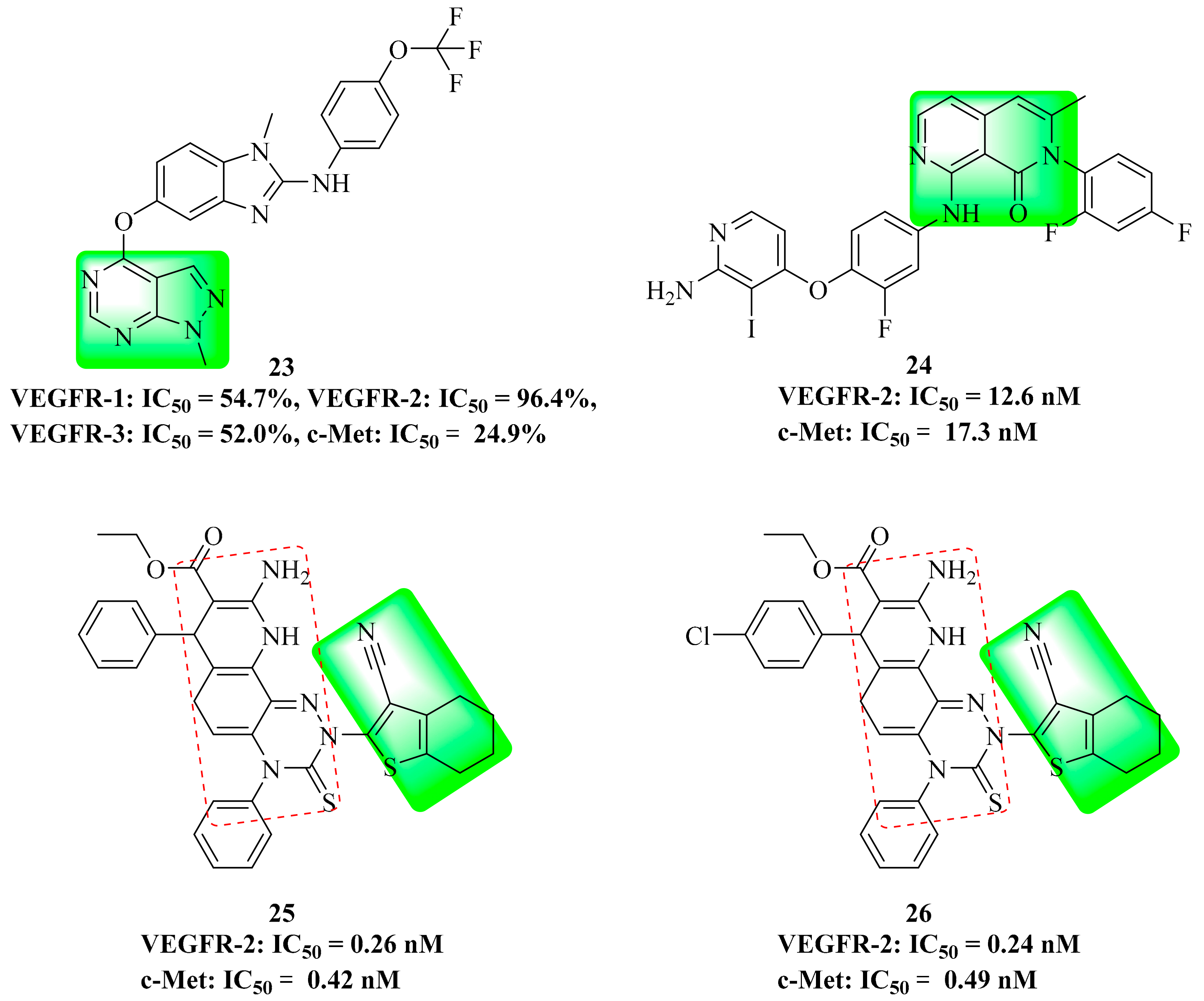
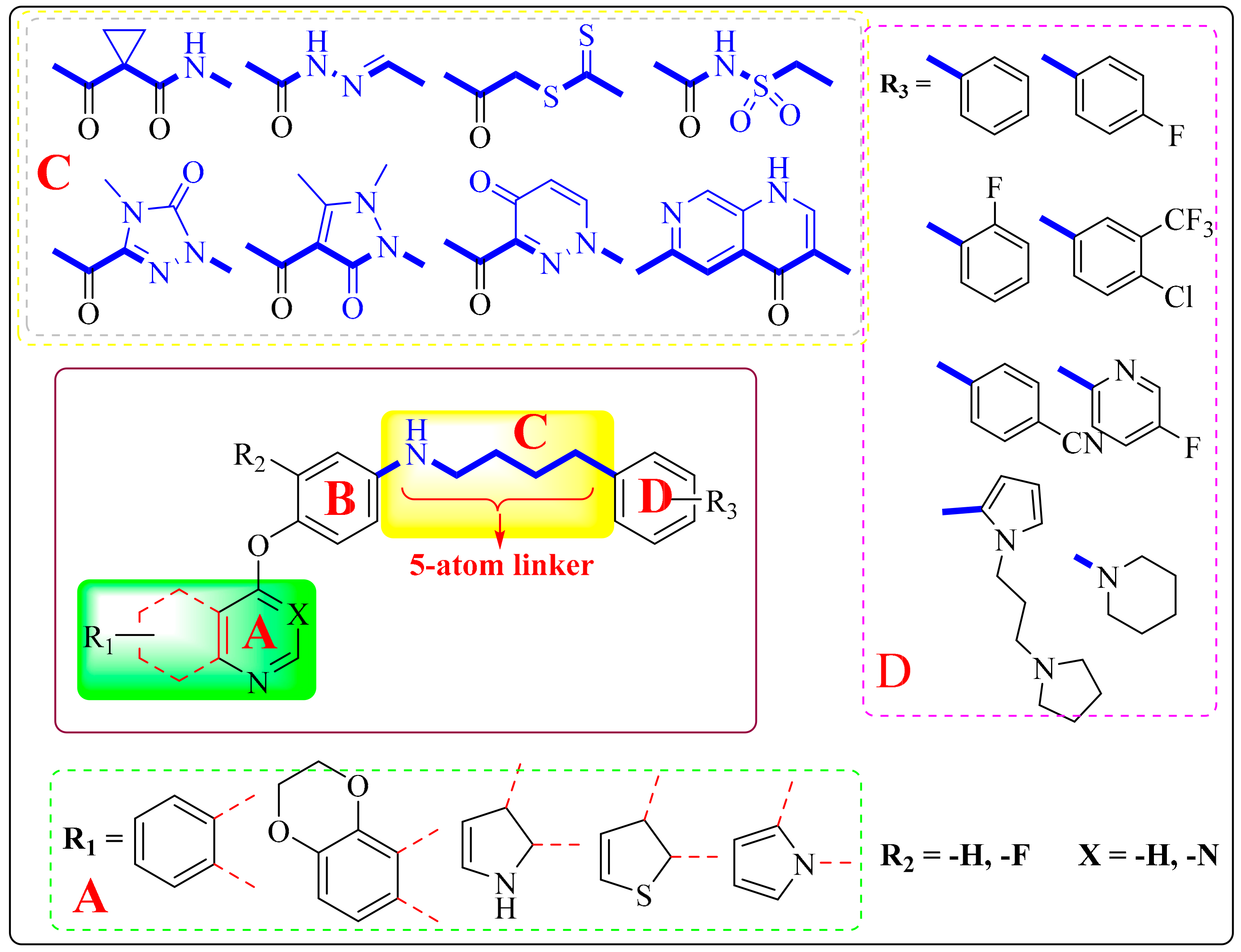
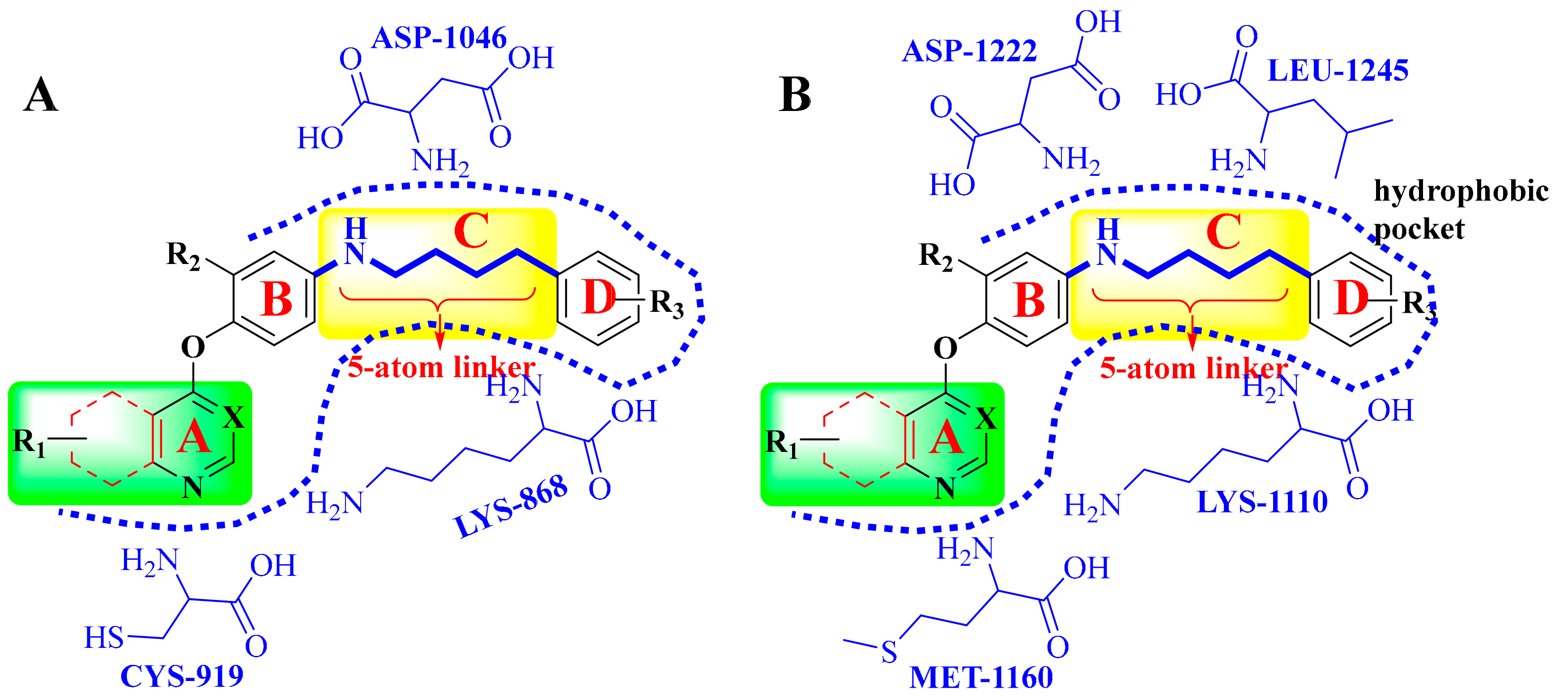
4. Structure–Activity Relationship
5. Conclusion and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dokla, E.M.; Fang, C.-S.; Abouzid, K.A.; Chen, C.S. 1,2,4-Oxadiazole derivatives targeting EGFR and c-Met degradation in TKI resistant NSCLC. Eur. J. Med. Chem. 2019, 182, 111607. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.-S.; Xu, H.; Wang, M.-S.; Zhao, X.-E.; Ming, Z.-H.; Zhu, X.-L.; Huang, W.; Yang, G.-F. 2,7-naphthyridinone-based MET kinase inhibitors: A promising novel scaffold for antitumor drug development. Eur. J. Med. Chem. 2019, 178, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Lin, S.-F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulgumnalli, M.R.; Divya, P.; Sundeep, K.; Akhila, M. Dual or Multi-Targeting Inhibitors: The Next Generation Anticancer Agents. Euro J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar]
- Gu, W.; Dai, Y.; Qiang, H.; Shi, W.; Liao, C.; Zhao, F.; Huang, W.; Qian, H. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorganic Chem. 2017, 72, 116–122. [Google Scholar] [CrossRef]
- Arpita, D.; Eric, J.S. Treatment of Advanced Renal Cell Carcinoma Patients with Cabozantinib, an Oral Multityrosine Kinase Inhibitor of MET, AXL and VEGF Receptors. Future Oncol. 2019, 15, 2337–2348. [Google Scholar]
- Sun, W.; Hu, S.; Fang, S.; Yan, H. Design, synthesis and biological evaluation of pyrimidine-based derivatives as VEGFR-2 tyrosine kinase inhibitors. Bioorganic Chem. 2018, 78, 393–405. [Google Scholar] [CrossRef]
- Masabumi, S. VEGF-VEGFR System as a Target for Suppressing Inflammation and Other Diseases. Endocr. Metab. Immune Disord. Drug Targets. 2015, 2, 135–144. [Google Scholar]
- Yuan, X.; Yang, Q.; Liu, T.; Li, K.; Liu, Y.; Zhu, C.; Zhang, Z.; Li, L.; Zhang, C.; Xie, M.; et al. Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase. Eur. J. Med. Chem. 2019, 179, 147–165. [Google Scholar] [CrossRef]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Mohsen, H.T.; Omar, M.A.; El Kerdawy, A.M.; Mahmoud, A.E.; Ali, M.M.; El Diwani, H.I. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur. J. Med. Chem. 2019, 179, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin. Ther. Patents 2017, 27, 987–1004. [Google Scholar] [CrossRef]
- Fan, H.; Wei, D.; Zheng, K.; Qin, X.; Yang, L.; Yang, Y.; Duan, Y.; Xu, Y.; Hu, L. Discovery of Dioxino[2,3-f]quinazoline derivative VEGFR-2 inhibitors exerting significant antipro-liferative activity in HUVECs and mice. Eur. J. Med. Chem. 2019, 175, 349–356. [Google Scholar] [CrossRef]
- Lu, D.; Yan, J.; Wang, L.; Liu, H.; Zeng, L.; Zhang, M.; Duan, W.; Ji, Y.; Cao, J.; Geng, M.; et al. Design, Synthesis, and Biological Evaluation of the First c-Met/HDAC Inhibitors Based on Pyridazinone Derivatives. ACS Med. Chem. Lett. 2017, 8, 830–834. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Jiang, Y.-F.; Li, H.-J.; Wang, J.-H.; Wu, Y.-C. Design, synthesis and evaluation of sulfonylurea-containing 4-phenoxyquinolines as highly selective c-Met kinase inhibitors. Bioorganic Med. Chem. 2019, 27, 2801–2812. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Zhuang, R.; He, R.; Xi, J.; Pan, X.; Shao, Y.; Pan, J.; Sun, J.; Cai, Z.; et al. Synthesis and evaluation of a series of pyridine and pyrimidine derivatives as type II c-Met inhibitors. Bioorganic Med. Chem. 2017, 25, 3195–3205. [Google Scholar] [CrossRef]
- Zhai, X.; Bao, G.; Wang, L.; Cheng, M.; Zhao, M.; Zhao, S.; Zhou, H.; Gong, P. Design, synthesis and biological evaluation of novel 4-phenoxy-6,7-disubstituted quinolines possessing (thio)semicarbazones as c-Met kinase inhibitors. Bioorganic Med. Chem. 2016, 24, 1331–1345. [Google Scholar] [CrossRef]
- Maria, B.; Jonathan, Z.; Moshe, E.; Marina, W.; Vadim, E.F. c-Met as a New Marker of Cellular Senescence. Aging 2019, 9, 2889–2897. [Google Scholar] [CrossRef]
- Liu, J.; Yang, D.; Yang, X.; Nie, M.; Wu, G.; Wang, Z.; Li, W.; Liu, Y.; Gong, P. Design, synthesis and biological evaluation of novel 4-phenoxyquinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moiety as c-Met kinase inhibitors. Bioorganic Med. Chem. 2017, 25, 4475–4486. [Google Scholar] [CrossRef] [PubMed]
- Anestis, A.; Zoi, I.; Karamouzis, M.V. Current advances of targeting HGF/c-Met pathway in gastric cancer. Ann. Transl. Med. 2018, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Silakari, O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorganic Chem. 2018, 79, 163–170. [Google Scholar] [CrossRef]
- Fakhrudin, N.; Waltenberger, B.; Cabaravdic, M.; Atanasov, A.; Heiss, E.; Grzywacz, A.; Mihaly-Bison, J.; Breuss, J.; Rollinger, J.; Bochkov, V.; et al. Discovery of Plumericin as a novel potent NF-?B inhibitor from Himatanthus sucuuba. Planta Medica 2013, 79, 2328–2342. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Hou, Y.; Qin, M.; Gong, P.; Liu, J.; Zhao, Y. Design, synthesis, and biological evaluation of 4-phenoxyquinoline derivatives as potent c-Met kinase inhibitor. Bioorganic Med. Chem. Lett. 2019. [Google Scholar] [CrossRef]
- Dorsch, D.; Schadt, O.; Stieber, F.; Meyring, M.; Grädler, U.; Bladt, F.; Friese-Hamim, M.; Knühl, C.; Pehl, U.; Blaukat, A. Identification and optimization of pyridazinones as potent and selective c-Met kinase inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 1597–1602. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, L.; Tu, Y.; Zhu, W.; Luo, R.; Tu, Q.; Wang, P.; Wu, C.; Gong, P.; Zheng, P. Discovery of novel pyrrolo[2,3-b]pyridine derivatives bearing 1,2,3-triazole moiety as c-Met kinase inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 1680–1684. [Google Scholar] [CrossRef]
- Xu, D.; Wang, T.-L.; Sun, L.-P.; You, Q.-D. Recent progress of small molecular VEGFR inhibitors as anticancer agents. Mini-Rev. Med. Chem. 2011, 11, 18–31. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M. Small-Molecule Inhibitors of the Receptor Tyrosine Kinases: Promising Tools for Targeted Cancer Therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, E.M.; Khalil, N.A.; Taher, A.T.; Refaey, R.H.; Nissan, Y.M. Triazolopyridazine derivatives: Synthesis, cytotoxic evaluation, c-Met kinase activity and molecular docking. Bioorganic Chem. 2019, 92, 103272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shan, Y.; Pan, X.; He, L. Recent advances in antiangiogenic agents with VEGFR as target. Mini-Rev. Med. Chem. 2011, 11, 920–946. [Google Scholar] [CrossRef] [PubMed]
- Dejuan, S.; Yuqian, Z. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Euro J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Li, W.; Man, X.-Y.; Chen, J.-Q.; Zhou, J.; Cai, S.-Q.; Zheng, M. Targeting VEGF/VEGFR in the treatment of psoriasis. Discov. Med. 2014, 18, 97–104. [Google Scholar]
- Hu, C.-T.; Wu, J.-R.; Cheng, C.-C.; Wu, W.-S. The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches. Cancers 2017, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Qiang, H.; Gu, W.; Huang, W.; Shi, W.; Qiu, Q.; Dai, Y.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-cabaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorganic Med. Chem. 2016, 24, 3353–3358. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.-M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018. [Google Scholar] [CrossRef] [Green Version]
- Rothenberger, N.J.; Stabile, L.P. Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers 2017, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Zillhardt, M.; Park, S.M.; Romero, I.L.; Sawada, K.; Montag, A. Foretinib (GSK1363089), an orally available multi-kinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin. Cancer Res. 2011, 17, 4042–4051. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Wu, T.-T.; Wang, Z.; Xue, J.-Y.; Xu, Y.-G. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorganic Med. Chem. 2014, 22, 4735–4744. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, X.; Jiang, Y.; Guo, M.; Zhang, S.; Li, J.; He, J.; Liu, J.; Wang, J.; Ouyang, L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016, 108, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Smith, B.D.; Zhou, Y.; Kaufman, M.D.; Godwin, A.K. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene 2013, 34, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Vijaykumar, V.E.; Natarajan, S.K.; Sengupta, S.; Sabbisetti, V.S. Sustained inhibition of cMET-VEGFR2 signaling using liposome-mediated delivery increases efficacy and reduces toxicity in kidney cancer. Nanomedicine 2016, 12, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Goltsov, A.A.; Fang, B.; Pandita, T.K.; Maru, D.; Swisher, S.G.; Hofstetter, W.L. HER2 Confers Resistance to Foretinib Inhibition of MET-Amplified Esophageal Adenocarcinoma Cells. Ann. Thorac. Surg. 2018, 105, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IS6-4 - Results of Phase 1 Studies of Golvatinib (E7050), A c-Met and EPH Receptor-Targeted Multi-Kinase Inhibitor. Ann. Oncol. 2012. [CrossRef]
- Semrad, T.J.; Kim, E.J.; Tanaka, M.S.; Sands, J.; Roberts, C.; Burich, R.A.; Li, Y.; Gandara, D.R.; Lara, P.; Mack, P.C. Phase II Study of Dovitinib in Patients Progressing on Anti-Vascular Endothelial Growth Factor Therapy. Cancer Treat. Res. Commun. 2017, 10, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Molina, A.M.; Hutson, T.E.; A Nosov, D.; Tomczak, P.; Lipatov, O.N.; Sternberg, C.N.; Motzer, R.J.; Eisen, T. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: crossover of a phase 3 study. Eur. J. Cancer 2018, 94, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Fargnoli, J.; Henley, B.; Wautlet, B.; Borzilleri, R. 106 Preclinical studies and characterization of BMS-794833, a small molecule inhibitor of Met and VEGFR-2 kinases. Eur. J. Cancer Suppl. 2010. [Google Scholar] [CrossRef]
- Zhang, W.; Ai, J.; Shi, D.; Peng, X.; Ji, Y.; Liu, J.; Geng, M.-Y.; Li, Y. Discovery of novel type II c-Met inhibitors based on BMS-777607. Eur. J. Med. Chem. 2014, 80, 254–266. [Google Scholar] [CrossRef]
- Wang, Q.; Quan, H.; Zhao, J.; Xie, C.-Y.; Wang, L.; Lou, L.-G. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett. 2013, 340, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Cortes, J.; Massard, C.; Armand, J.-P.; De Andreis, D.; Ropert, S.; Lopez, E.; Catteau, A.; James, J.; Marier, J.-F.; et al. Phase I safety, pharmacokinetic and pharmacodynamic trial of BMS-599626 (AC480), an oral pan-HER receptor tyrosine kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2012, 23, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Jitesh, P.J.; Richard, S.F.; Mary, C.; Kevin, G.C.; Richard, D.C.; Nicolas, C.; Erling, O.E. Discovery and Pharmacologic Characterization of CP-724,714, a Selective ErbB2 Tyrosine Kinase Inhibitor. Cancer Res. 2007, 67, 9887–9893. [Google Scholar]
- Li, B.; Torossian, A.; Sun, Y.; Du, R.; Dicker, A.; Lu, B. Higher Levels of c-Met Expression and Phosphorylation Identify Cell Lines with Increased Sensitivity to AMG-458, a Novel Selective c-Met Inhibitor with Radiosensitizing Effects. Int. J. Radiat. Oncol. 2012, 84, e525–e531. [Google Scholar] [CrossRef]
- Zhan, Z.; Ai, J.; Liu, Q.; Ji, Y.; Chen, T.; Xu, Y.; Geng, M.; Duan, W. Discovery of Anilinopyrimidines as Dual Inhibitors of c-Met and VEGFR-2: Synthesis, SAR, and Cellular Activity. ACS Med. Chem. Lett. 2014, 5, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Qiang, H.; Huang, W.; Bi, X.; Huang, W.; Qian, H. Exploration of novel pyrrolo[2,1-f][1,2,4]triazine derivatives with improved anticancer efficacy as dual inhibitors of c-Met/VEGFR-2. Eur. J. Med. Chem. 2018, 158, 814–831. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting Receptor Tyrosine Kinase MET in Cancer: Small Molecule Inhibitors and Clinical Progress. J. Med. Chem. 2013, 57, 4427–4453. [Google Scholar] [CrossRef]
- Kaya, M.; Cokakli, M.; Berk, A.T.; Yaman, A.; Yesilirmak, D. Associations of VEGF/VEGF-receptor and HGF/c-Met promoter poly-morphisms with progression/regression of retinopathy of prematurity. Curr. Eye Res. 2013, 38, 137–142. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Yang, L.; Zhao, L.; Si, L.; Zhang, H.; Liu, Q.; Zhou, J. Discovery of imidazopyridine derivatives as novel c-Met kinase inhibitors: Synthesis, SAR study, and biological activity. Bioorganic Chem. 2017, 70, 126–132. [Google Scholar] [CrossRef]
- Lai, S.; Chen, J.; Huang, H.; Zhang, X.; Jiang, H.; Li, W.; Wang, P.; Wang, J.; Liu, F.-N. Structure activity relationships of chrysoeriol and analogs as dual c-Met and VEGFR2 tyrosine kinase inhibitors. Oncol. Rep. 2018, 40, 405–422. [Google Scholar] [CrossRef]
- Wang, M.; Xu, S.; Lei, H.; Wang, C.; Xiao, Z.; Jia, S.; Zhi, J.; Zheng, P.; Zhu, W. Design, synthesis and antitumor activity of Novel Sorafenib derivatives bearing pyrazole scaffold. Bioorganic Med. Chem. 2017, 25, 5754–5763. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Zhuo, L.-S.; Yang, F.-P.; Wang, W.-J.; Huang, W.; Yang, G.-F. Synthesis and biological evaluation of new MET inhibitors with 1,6-naphthyridinone scaffold. Eur. J. Med. Chem. 2019, 185, 111803. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, M.A.; Mostafa, A.S.; Gomaa, R.M.; Abou-Zeid, L.A.; El-Mesery, M.; El-Sayed, M.A.-A.; Selim, K.B. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur. J. Med. Chem. 2019, 168, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shan, Y.; Sun, Y.; Si, R.; Liang, L.; Pan, X.; Wang, B.-H.; Zhang, J. Discovery of novel anti-angiogenesis agents. Part 7: Multitarget inhibitors of VEGFR-2, TIE-2 and EphB4. Eur. J. Med. Chem. 2017, 141, 506–518. [Google Scholar] [CrossRef]
- Liu, J.; Nie, M.; Wang, Y.; Hu, J.; Zhang, F.; Gao, Y.; Liu, Y.; Gong, P. Design, synthesis and structure-activity relationships of novel 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2016, 123, 431–446. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, L.-D.; Hao, Y.; Chen, N.; Bai, R.; Wang, Y.-J.; Zhang, C.-C.; Li, G.-S.; Hao, L.; Shi, C.; et al. Identification of 3-substituted-6-(1-(1H-[1,2,3]triazolo[4,5-b]pyrazin-1-yl)ethyl)quinoline derivatives as highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitors via metabolite profiling-based structural optimization. Eur. J. Med. Chem. 2017, 134, 147–158. [Google Scholar] [CrossRef]
- Wang, W.; Xu, S.; Duan, Y.; Liu, X.; Zhang, H.; Wang, C.; Zhao, B.; Zheng, P.; Zhu, W. Synthesis and bioevaluation and doking study of 1H-pyrrolo[2,3-b]pyridine derivatives bearing aromatic hydrazone moiety as c-Met inhibitors. Eur. J. Med. Chem. 2017, 145, 315–327. [Google Scholar] [CrossRef]
- Wang, L.X.; Liu, X.; Xu, S.; Tang, Q.; Duan, Y.; Xiao, Z.; Zhi, J.; Jiang, L.; Zheng, P.; Zhu, W. Discovery of novel pyrrolo-pyridine/pyrimidine derivatives bearing pyridazinone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2017, 141, 538–551. [Google Scholar] [CrossRef]
- Ibrahim, H.A.; Awadallah, F.M.; Refaat, H.M.; Amin, K.M. Molecular docking simulation, synthesis and 3D pharmacophore studies of novel 2-substituted-5-nitro-benzimidazole derivatives as anticancer agents targeting VEGFR-2 and c-Met. Bioorganic Chem. 2018, 77, 457–470. [Google Scholar] [CrossRef]
- A Ibrahim, H.; Awadallah, F.M.; Refaat, H.M.; Amin, K.M. Design, synthesis and molecular modeling study for some new 2-substituted benzimidazoles as dual inhibitors for VEGFR-2 and c-Met. Futur. Med. Chem. 2018, 10, 493–509. [Google Scholar] [CrossRef]
- Li, J.; Gu, W.; Bi, X.; Li, H.; Liao, C.; Liu, C.; Huang, W.; Qian, H. Design, synthesis, and biological evaluation of thieno[2,3-d]pyrimidine derivatives as novel dual c-Met and VEGFR-2 kinase inhibitors. Bioorganic Med. Chem. 2017, 25, 6674–6679. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Fan, H.; Zheng, K.; Qin, X.; Yang, L.; Yang, Y.; Duan, Y.; Zhang, Q.; Zeng, C.; Hu, L. Synthesis and anti-tumor activity of[1,4]dioxino[2,3-f]quinazoline derivatives as dual inhibitors of c-Met and VEGFR-2. Bioorganic Chem. 2019, 88, 102916. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Huang, L.; Zhang, Q.-W.; Li, J. Synthesis and biological evaluation of novel 6,11-dihydro-5H-benzo[e]pyrimido-[5,4-b][1,4]diazepine derivatives as potential c-Met inhibitors. Eur. J. Med. Chem. 2017, 140, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wan, S.; Li, Z.; Fu, Y.; Wang, G.; Zhang, J.; Wu, X. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d]pyrimidine derivatives as BRAFV600E and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2018, 155, 210–228. [Google Scholar] [CrossRef] [PubMed]
- Abdo, N.Y.M.; Mohareb, R.M.; Halim, P.A. Uses of cyclohexane-1,3-dione for the synthesis of 1,2,4-triazine derivatives as anti-proliferative agents and tyrosine kinases inhibitors. Bioorganic Chem. 2020, 97, 103667. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Structural Formula | Target | Indication | Phase | Signaling Pathway | Research and Development Company |
|---|---|---|---|---|---|---|
| Foretinib [45] |  | c-Met, VEGFR-2 (KDR), Tie-2, VEGFR-3/FLT4 | Gastric cancer and head/neck cancer | II | Protein tyrosine kinase | Exelixis |
| Golvatinib [46] |  | c-Met, VEGFR-2 | Head and neck cancer, liver cancer | II | Angiogenesis; protein tyrosine kinase | Eisai |
| Dovitinib [47] |  | FLT3, c-Kit, FGFR-1/3, VEGFR1-4, EGFR, c-Met | Solid tumors | IV | Angiogenesis | Novartis |
| Tivozanib [48] |  | VEGFR-1, VEGFR-2, VEGFR-3, c-Met, PDGFR, c-Kit | Advanced renal cell carcinoma | III | Angiogenesis | Aevo |
| BMS-794833 [49] |  | c-Met, VEGFR-2, Ron, Axl, FLT3 | Gastric cancer | I | Angiogenesis; protein tyrosine kinase | Bristol Myers Squibb |
| BMS-777607 [50] |  | c-Met, Axl, Ron, VEGFR-2, lck | Advanced solid tumors | II | Protein tyrosine kinase | Bristol Myers Squibb |
| MGCD-265 [51] |  | c-Met, Ron, VEGFR-1, VEGFR-2 | Non-small cell lung cancer | II | Angiogenesis; protein tyrosine kinase | MethylGene |
| AC480 [52] |  | HER1, HER2, HER4, VEGFR-2, c-Kit, Lck, MET | Advanced solid tumors | I | Angiogenesis | Ambit Biosciences |
| CP-724714 [53] |  | HER2/ErbB2, EGFR, VEGFR-2, c-Met | Advanced solid tumors | II | Protein tyrosine kinase | Pfizer |
| AMG-458 [54] |  | c-Met, VEGFR-2 | Solid tumors | Non-medicinal | Protein tyrosine kinase | Amgen |
| Compound | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| VEGFR-2 | c-Met | VEGFR-1 | VEGFR-3 | EGFR | IGF1-R | B-Raf | c-Kit | |
| 7 | 2.35 | 33.12 | 25.00 | 45.00 | 8.70 | 40.00 | 32.00 | 68.90 |
| Compound | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| c-Met | c-Kit | FLT3 | PDGFRα | Ron | VEGFR | EGFR | ALK | |
| 8 | 1.57 | 3.38 | 8.19 | 95.23 | 140.47 | 480.47 | >10000 | >10000 |
| 9 | 0.09 | 2.45 | 268.81 | 19.13 | 82.56 | 151.52 | 980.83 | 2840.72 |
| 10 | 1.98 | 380 | 400 | 242 | 375 | 689 | >10000 | >10000 |
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| A549 | HepG2 | MCF-7 | PC-3 | |
| 12 | 0.82 | 1.00 | 0.93 | 0.92 |
| 13 | 1.30 | 1.42 | 4.53 | 4.29 |
| 14 | 2.19 | 1.32 | 6.27 | 4.63 |
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| c-Met | VEGFR-2 | c-Kit | FLT3 | EGFR | |
| 12 | 0.5 | 8.9 | 2.6 | >100 | |
| 13 | 0.9 | 14.4 | 3.8 | 45.0 | |
| 14 | 0.073 | 2.4 | 2.2 | 0.77 | >10 |
| Compound | % Inhibition at 10 µM | |
|---|---|---|
| VEGFR-2 | c-Met | |
| 15 | 29.22 | 71.66 |
| 16 | 35.88 | 82.48 |
| 17 | 21.07 | 76.01 |
| AG-1478 | 73.73 | 33.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zheng, P.; Zhu, W. Research Progress of Small Molecule VEGFR/c-Met Inhibitors as Anticancer Agents (2016–Present). Molecules 2020, 25, 2666. https://doi.org/10.3390/molecules25112666
Zhang Q, Zheng P, Zhu W. Research Progress of Small Molecule VEGFR/c-Met Inhibitors as Anticancer Agents (2016–Present). Molecules. 2020; 25(11):2666. https://doi.org/10.3390/molecules25112666
Chicago/Turabian StyleZhang, Qian, Pengwu Zheng, and Wufu Zhu. 2020. "Research Progress of Small Molecule VEGFR/c-Met Inhibitors as Anticancer Agents (2016–Present)" Molecules 25, no. 11: 2666. https://doi.org/10.3390/molecules25112666