
Development of Resistance to Endoplasmic Reticulum Stress-Inducing Agents in Mouse Leukemic L1210 Cells

 and
and
Abstract
:1. Introduction
2. Results
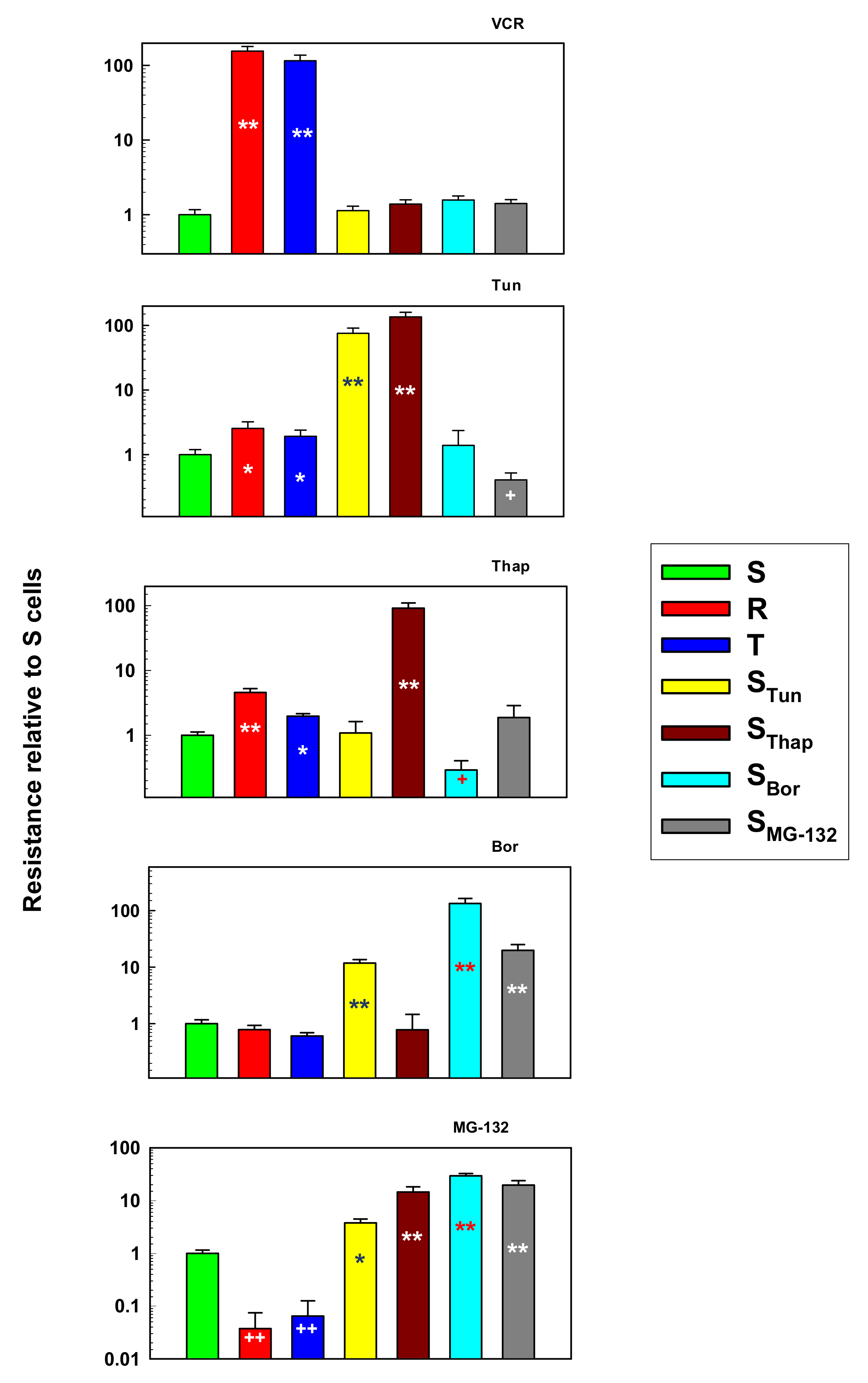
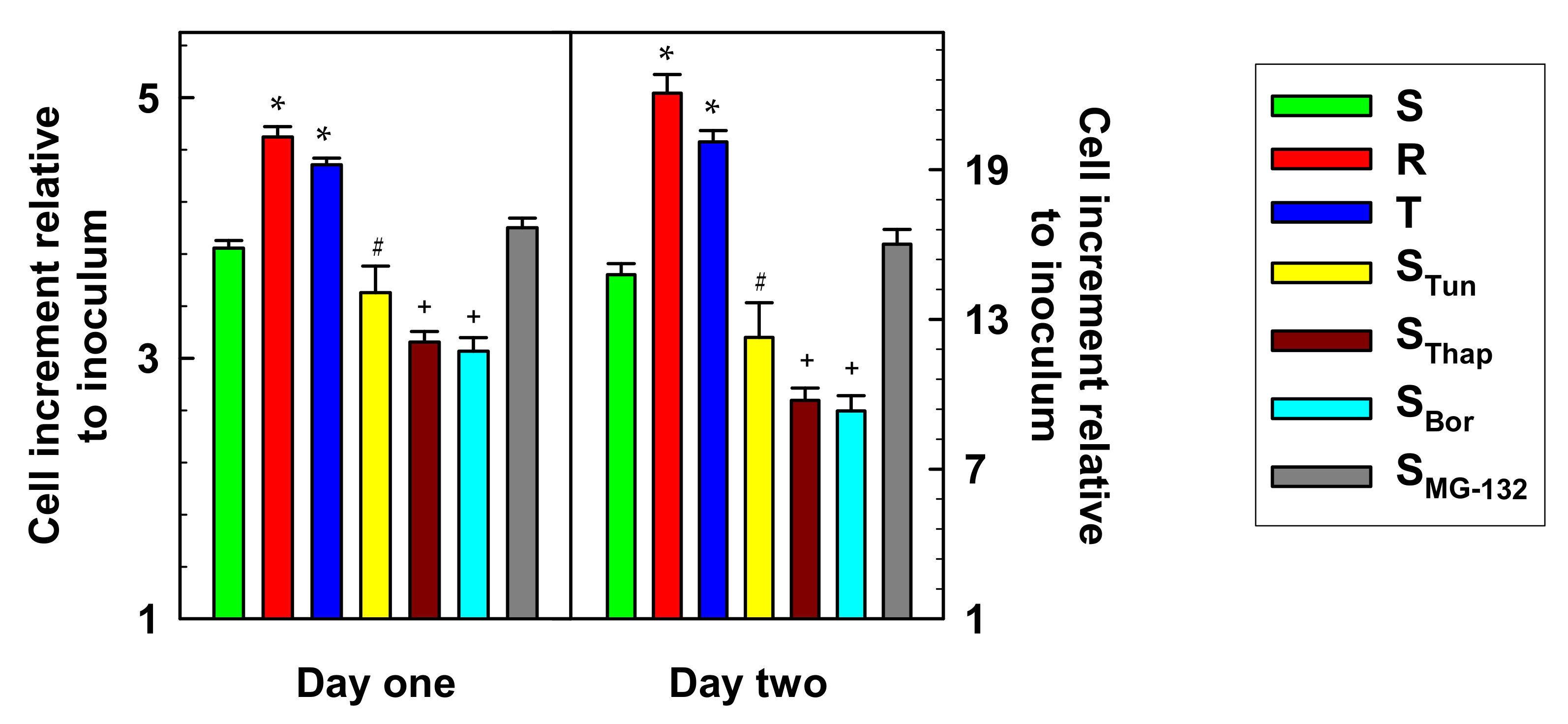
2.1. Characterization of L1210 Cell Variants Resistant to Tun, Thap, Bor and MG-132 and their Comparison with S, R and T Cells.
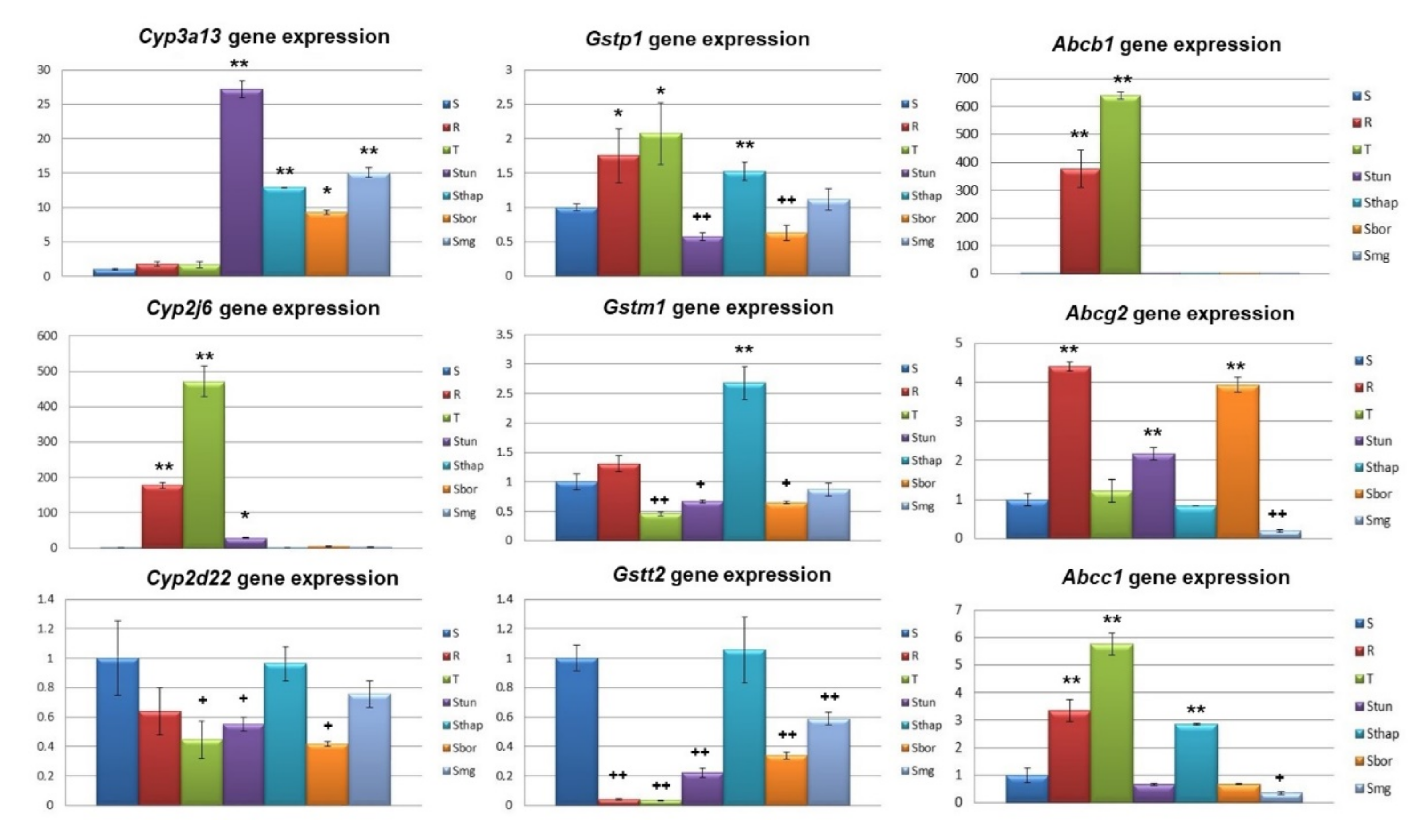
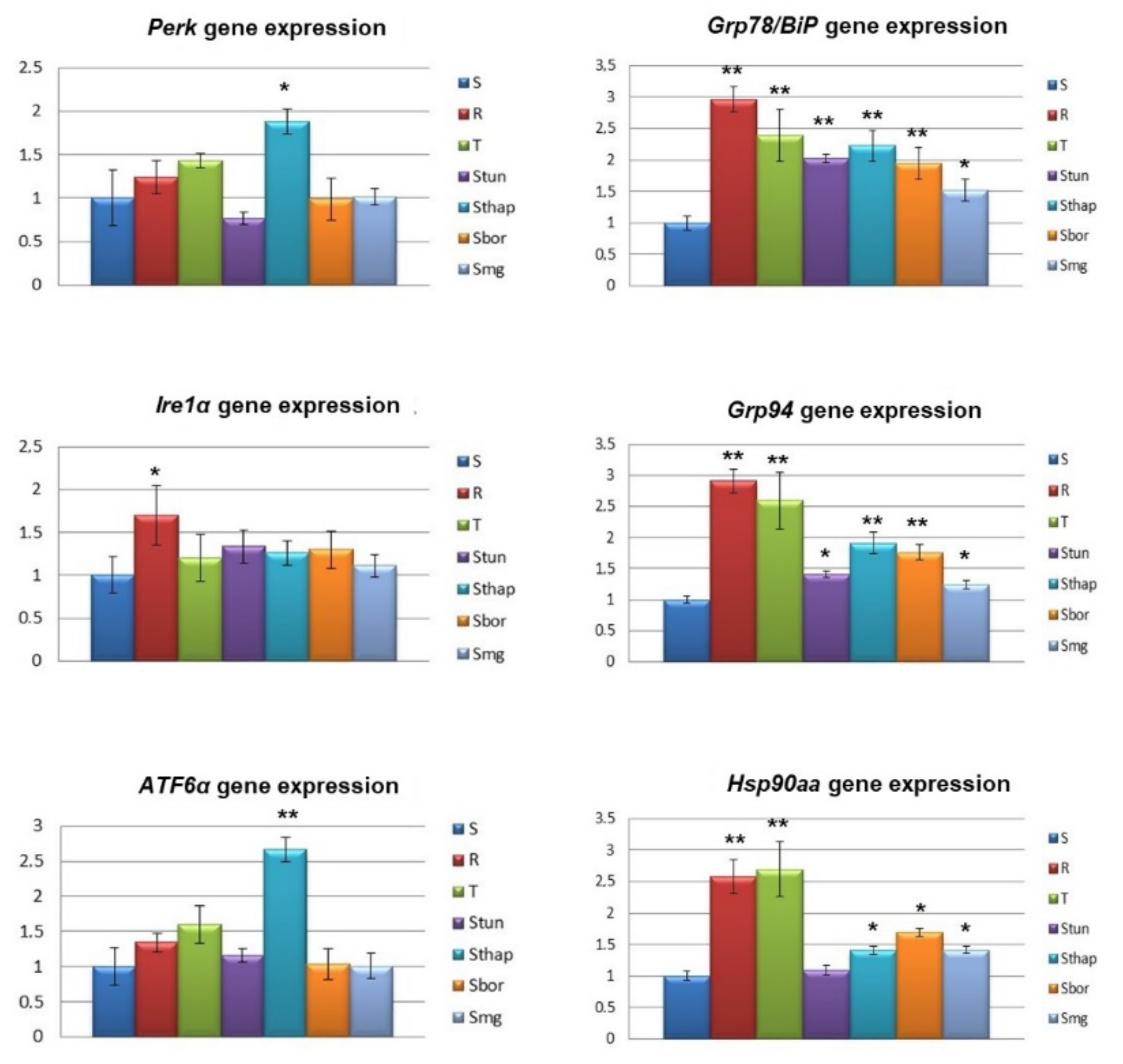
2.2. Altered Expression of Genes Known to be Involved in Drug Metabolism/Elimination and in the ER Stress Pathway
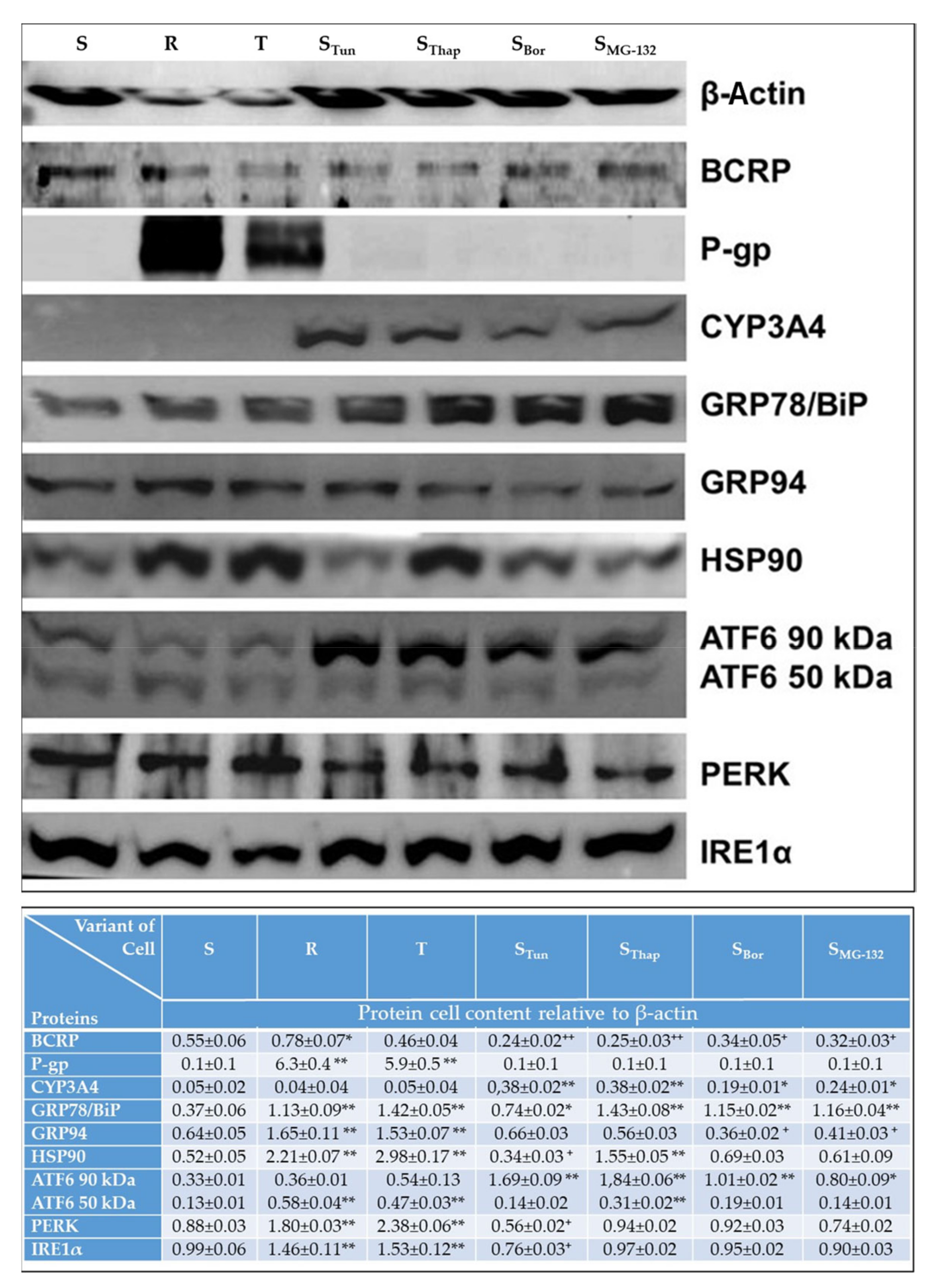
2.3. Altered Cell Content of Proteins Known to be Involved in Drug Metabolism/Elimination and in the ER Stress Pathway
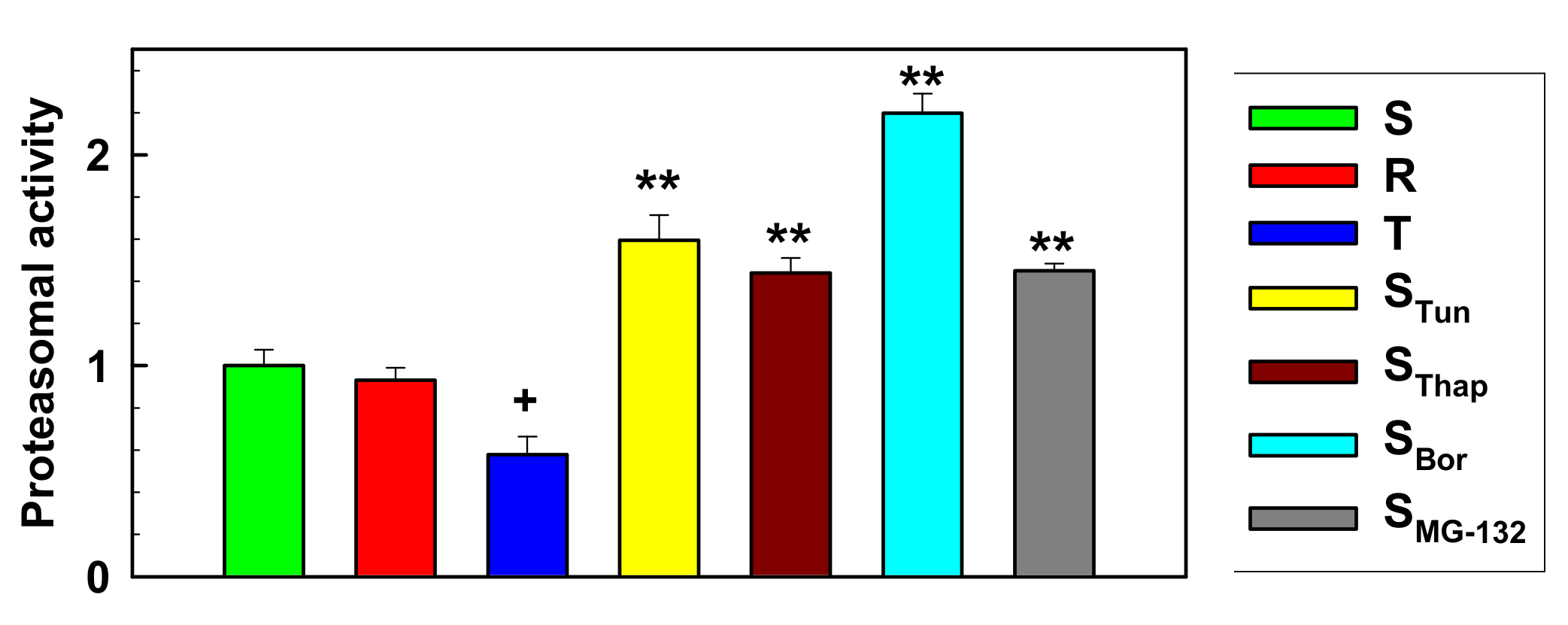
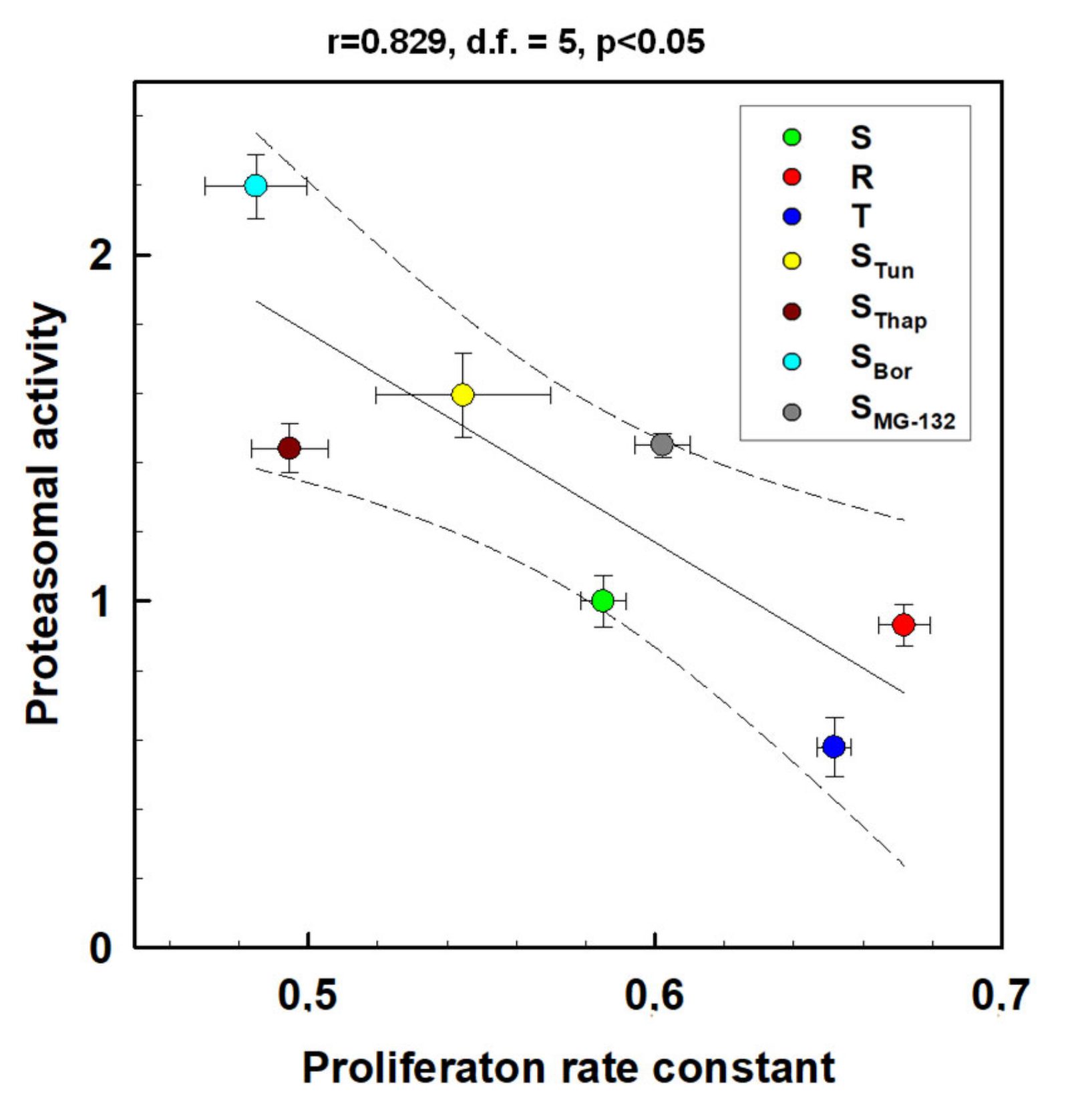
2.4. Proteasome Activity in L1210 Cell Variants
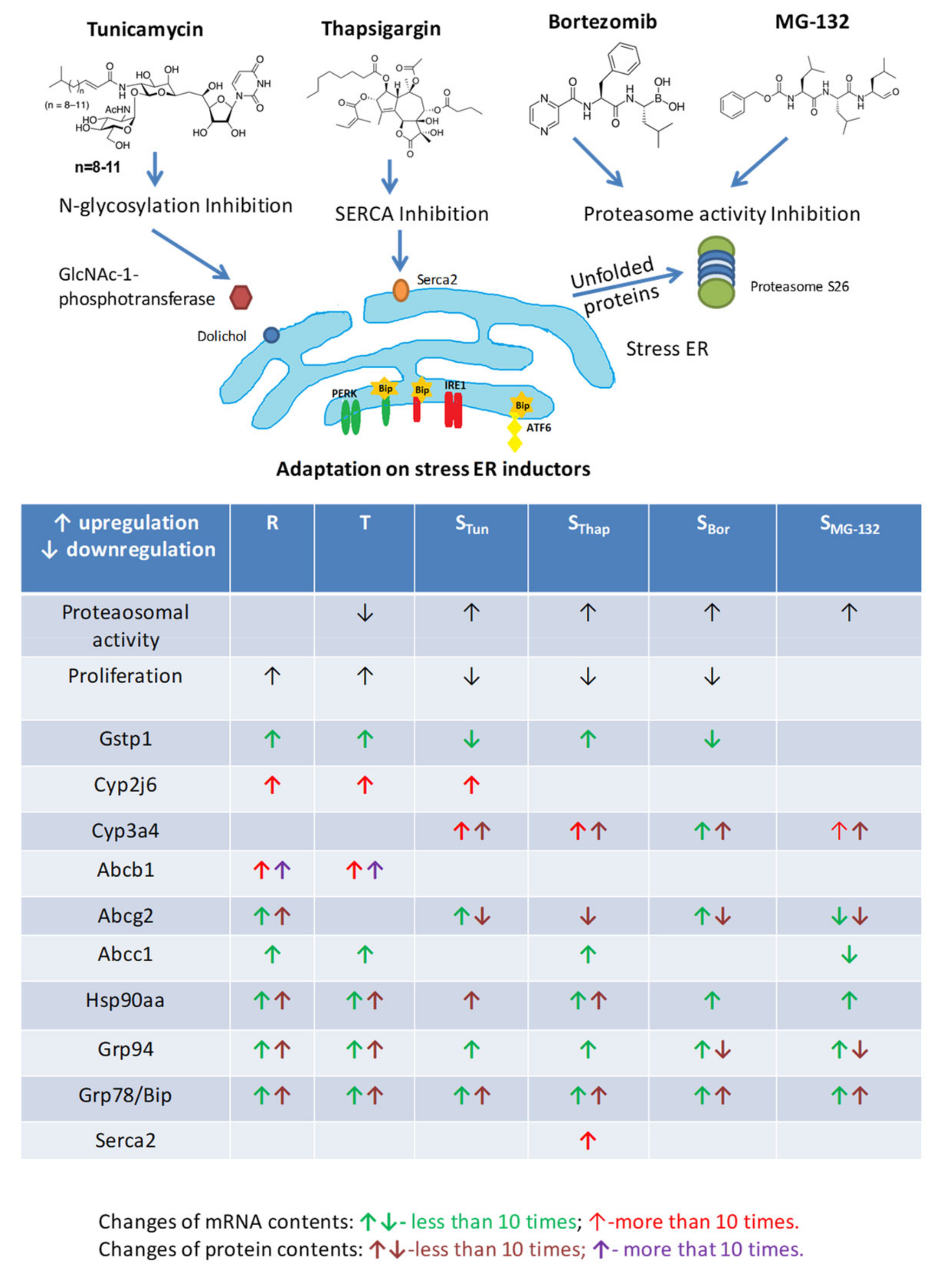
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Cultivation Conditions
4.3. Cell Viability Assay Using MTT
4.4. Cell Proliferation Assay
4.5. Real-time RT-PCR Conditions
4.6. Western Blotting
4.7. Proteasomal Activity Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ABCB1 | 1st member of ABCB gene subfamily |
| ABCC1 | 1st member of ABCC gene subfamily |
| ABCG2 | 2nd member of ABCG gene subfamily |
| ATF6α | activating transcription factor 6α |
| BCRP | breast cancer resistance protein |
| Bor | Bortezomib |
| CYP | cytochrome P-450 |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum-associated degradation |
| GRP78/BiP | glucose-regulated protein 78/binding immunoglobulin protein |
| GRP94 | glucose-regulated protein 94 |
| GST | glutathione S-transferase |
| HSP90 | heat shock protein 90 |
| MDR | multidrug resistance |
| MG-132 | N-Benzyloxycarbonyl-l-leucyl-l-leucyl-l-leucinal |
| MRP | multidrug resistance associated protein |
| MTT | [3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyltetrazolium bromide] |
| PERK | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| P-gp | P-glycoprotein |
| R | P-gp-positive L1210 cells induced for resistance by vincristine |
| S | P-gp negative drug sensitive parental L1210 cells |
| SBor | P-gp negative L1210 cells induced for resistance by bortezomib |
| SMG-132 | P-gp negative L1210 cells induced for resistance by MG-132 |
| SThap | P-gp negative L1210 cells induced for resistance by thapsigargin |
| STun | P-gp negative L1210 cells induced for resistance by tunicamycin |
| Suc-LLVY-AMC | 4-[[(2S)-1-[[(2S)-1-hydroxyphenyl)-1-[(4-methyl-2-oxochromen-7-yl) amino]-1-oxopropan-2-yl] amino]-3-methyl-1-oxobutan-2-yl] amino]-4 methyl-1-oxopentan-2-yl] amino]-4-methyl-1-oxopentan-2-yl] amino]-4-oxobutanoic acid) |
| T | drug-resistant P-gp-positive L1210 cells obtained by transfection with the human P-gp gene |
| Thap | Thapsigargin |
| Tun | Tunicamycin |
| UPR | unfolded protein response |
| VCR | Vincristine |
References
- De Moraes, A.C.; Maranho, C.K.; Rauber, G.S.; Santos-Silva, M.C. Importance of detecting multidrug resistance proteins in acute leukemia prognosis and therapy. J. Clin. Lab. Anal. 2013, 27, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.Q.; Smith, P.G. Drug efflux transporters and multidrug resistance in acute leukemia: Therapeutic impact and novel approaches to mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anti-Cancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar]
- Hano, M.; Tomasova, L.; Seres, M.; Pavlikova, L.; Breier, A.; Sulova, Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules 2018, 23, 337. [Google Scholar] [CrossRef] [Green Version]
- Hodges, R.E.; Minich, D.M. Modulation of Metabolic Detoxification Pathways Using Foods and Food-Derived Components: A Scientific Review with Clinical Application. J. Nutr. Metab. 2015, 2015, 760689. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Kanagaratham, C.; Youssef, M.; Radzioch, D. New Frontiers in Cancer Chemotherapy—Targeting Cell Death Pathways. In Cell Biology—New Insights; Najman, S., Ed.; Intech Open: Rijeka, Croatia, 2016; pp. 93–139. [Google Scholar]
- Salehan, M.R.; Morse, H.R. DNA damage repair and tolerance: A role in chemotherapeutic drug resistance. Br. J. Biomed. Sci. 2013, 70, 31–40. [Google Scholar] [CrossRef]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Seres, M.; Pavlikova, L.; Bohacova, V.; Kyca, T.; Borovská, I.; Lakatos, B.; Breier, A.; Sulova, Z. Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum. Cells 2020, 9, 890. [Google Scholar]
- Sulova, Z.; Ditte, P.; Kurucova, T.; Polakova, E.; Rogozanova, K.; Gibalova, L.; Seres, M.; Skvarkova, L.; Sedlak, J.; Pastorek, J.; et al. The presence of P-glycoprotein in L1210 cells directly induces down-regulation of cell surface saccharide targets of concanavalin A. Anticancer Res. 2010, 30, 3661–3668. [Google Scholar]
- Seres, M.; Polakova, E.; Krizanova, O.; Hudecova, S.; Klymenko, S.V.; Breier, A.; Sulova, Z. Overexpression of P-glycoprotein in L1210/VCR cells is associated with changes in several endoplasmic reticulum proteins that may be partially responsible for the lack of thapsigargin sensitivity. Gen. Physiol. Biophys. 2008, 27, 211–221. [Google Scholar]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in L1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, L.; Seres, M.; Hano, M.; Bohacova, V.; Sevcikova, I.; Kyca, T.; Breier, A.; Sulova, Z. L1210 Cells Overexpressing ABCB1 Drug Transporters Are Resistant to Inhibitors of the N- and O-glycosylation of Proteins. Molecules 2017, 22, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses P-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in L1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Moller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca(2+)-ATPase by thapsigargin analogs induces cell death via ER Ca(2+) depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Hoseki, J.; Ushioda, R.; Nagata, K. Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 2010, 147, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Fribley, A.; Wang, C.Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol. Ther. 2006, 5, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xiao, J.; Zhang, X.; Bian, X. MicroRNAs as key mediators of hepatic detoxification. Toxicology 2016, 368–369, 80–90. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Graner, A.N.; Hellwinkel, J.E.; Lencioni, A.M.; Madsen, H.J.; Harland, T.A.; Marchando, P.; Nguyen, G.J.; Wang, M.; Russell, L.M.; Bemis, L.T.; et al. HSP90 inhibitors in the context of heat shock and the unfolded protein response: Effects on a primary canine pulmonary adenocarcinoma cell line. Int. J. Hyperth. 2017, 33, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Yoo, J.; Mashalidis, E.H.; Kuk, A.C.Y.; Yamamoto, K.; Kaeser, B.; Ichikawa, S.; Lee, S.Y. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat. Struct. Mol. Biol. 2018, 25, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Khandelwal, N.; Kumar, R.; Chander, Y.; Rawat, K.D.; Chaubey, K.K.; Sharma, S.; Singh, S.V.; Riyesh, T.; Tripathi, B.N.; et al. Inhibitor of Sarco/Endoplasmic Reticulum Calcium-ATPase Impairs Multiple Steps of Paramyxovirus Replication. Front. Microbiol. 2019, 10, 209. [Google Scholar] [CrossRef]
- Pitcher, D.S.; de Mattos-Shipley, K.; Tzortzis, K.; Auner, H.W.; Karadimitris, A.; Kleijnen, M.F. Bortezomib Amplifies Effect on Intracellular Proteasomes by Changing Proteasome Structure. EBioMedicine 2015, 2, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.J.; Walker, B.; Ovaa, H.; Chauhan, D.; Anderson, K.C.; Morris, T.C.; Irvine, A.E. Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO, PS-341, and MG-132. Cancer Res. 2006, 66, 6379–6386. [Google Scholar] [CrossRef] [Green Version]
- Kubickova, J.; Elefantova, K.; Pavlikova, L.; Cagala, M.; Seres, M.; Safar, P.; Marchalin, S.; Durisova, K.; Bohacova, V.; Sulova, Z.; et al. Screening of Phenanthroquinolizidine Alkaloid Derivatives for Inducing Cell Death of L1210 Leukemia Cells with Negative and Positive P-glycoprotein Expression. Molecules 2019, 24, 2127. [Google Scholar] [CrossRef] [Green Version]
- Pavlikova, L.; Seres, M.; Imrichova, D.; Hano, M.; Rusnak, A.; Zamorova, M.; Katrlik, J.; Breier, A.; Sulova, Z. The expression of P-gp in leukemia cells is associated with cross-resistance to protein N-glycosylation inhibitor tunicamycin. Gen. Physiol. Biophys. 2016, 35, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Seres, M.; Ditte, P.; Breier, A.; Sulova, Z. Effect of thapsigargin on P-glycoprotein-negative and P-glycoprotein-positive L1210 mouse leukaemia cells. Gen. Physiol. Biophys. 2010, 29, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Sulova, Z.; Seres, M.; Barancik, M.; Gibalova, L.; Uhrik, B.; Polekova, L.; Breier, A. Does any relationship exist between P-glycoprotein-mediated multidrug resistance and intracellular calcium homeostasis. Gen. Physiol. Biophys. 2009, 28, F89–F95. [Google Scholar]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.I.; Sumbilla, C.; Lee, M.; Natesavelalar, C.; Klein, M.G.; Ross, D.D.; Inesi, G.; Hussain, A. Mechanisms of resistance and adaptation to thapsigargin in androgen-independent prostate cancer PC3 and DU145 cells. Arch. Biochem. Biophys. 2007, 464, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Tong, X.; Ye, X. Cyclin D1 promotes cell cycle progression through enhancing NDR1/2 kinase activity independent of cyclin-dependent kinase 4. J. Biol. Chem. 2013, 288, 26678–26687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, A.; Zelivianski, S.; Jeruss, J.S. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010, 9, 4900–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Zhao, H.; Zhang, S.; Lee, M.Y.; Lee, E.Y.; Zhang, Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21WAF1, Cdt1 and the p12 subunit of DNA polymerase delta revealed in individual cells by cytometry. Oncotarget 2015, 6, 11735–11750. [Google Scholar] [CrossRef]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef]
- Winter, S.S.; Ricci, J.; Luo, L.; Lovato, D.M.; Khawaja, H.M.; Serna-Gallegos, T.; Debassige, N.; Larson, R.S. ATP Binding Cassette C1 (ABCC1/MRP1)-mediated drug efflux contributes to disease progression in T-lineage acute lymphoblastic leukemia. Health 2013, 5, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volk, E.L.; Farley, K.M.; Wu, Y.; Li, F.; Robey, R.W.; Schneider, E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002, 62, 5035–5040. [Google Scholar] [PubMed]
- Lee, C.M.; Kumar, V.; Riley, R.I.; Morgan, E.T. Metabolism and action of proteasome inhibitors in primary human hepatocytes. Drug Metab. Dispos. 2010, 38, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cizkova, K.; Konieczna, A.; Erdosova, B.; Lichnovska, R.; Ehrmann, J. Peroxisome proliferator-activated receptors in regulation of cytochromes P450: New way to overcome multidrug resistance? J. Biomed. Biotechnol. 2012, 2012, 656428. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Elraghy, O.; Baldwin, W.S. Repression of multiple CYP2D genes in mouse primary hepatocytes with a single siRNA construct. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depeille, P.; Cuq, P.; Passagne, I.; Evrard, A.; Vian, L. Combined effects of GSTP1 and MRP1 in melanoma drug resistance. Br. J. Cancer 2005, 93, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Peklak-Scott, C.; Smitherman, P.K.; Townsend, A.J.; Morrow, C.S. Role of glutathione S-transferase P1-1 in the cellular detoxification of cisplatin. Mol. Cancer Ther. 2008, 7, 3247–3255. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Schewe, D.M.; Aguirre-Ghiso, J.A. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009, 69, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Pastan, I.; Gottesman, M.M.; Ueda, K.; Lovelace, E.; Rutherford, A.V.; Willingham, M.C. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4486–4490. [Google Scholar] [CrossRef] [Green Version]
- Gerlier, D.; Thomasset, N. Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are commercially available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | LC50 nM | SD nM |
|---|---|---|
| Vincristine | 1.0 | 0.2 |
| Tunicamycin | 440.0 | 120.7 1 |
| Thapsigargin | 11.8 | 3.6 |
| Bortezomib | 6.1 | 1.5 |
| MG-132 | 238.4 | 72.5 |
| Gene | Gene alias | Primers | bp | |
|---|---|---|---|---|
| Gstm1 | Forward Reverse | 5ʹ-CCGTATGTTTGAGCCCAAGT-3ʹ 5ʹ-CTCCTAGTGAGTGCCCGTGT-3ʹ | 186 | |
| Gstp1 | Forward Revers | 5ʹ-TGCCACCATACACCATTGTC-3ʹ 5ʹ-GGTGAGGTCTCCATCCTCAA-3 | 185 | |
| Gstt2 | Forward Reverse | 5ʹ-GTACCAGGTGGCAGACCACT-3ʹ 5ʹ-GTTGCAGAACCAGGACCATT-3 | 203 | |
| Cyp2d22 | Forward Reverse | 5ʹ-CAGTGTCCAGAGATGGCAGA-3ʹ 5ʹ-AGGACAGGTTGGTGATGAGG-3ʹ | 175 | |
| Cyp2j6 | Forward Reverse | 5ʹ-GAAGGGTGCCCTTGTTGTTA-3ʹ 5ʹ-ACCAACAGAGTCCTGGGATG-3ʹ | 151 | |
| Cyp3a13 | Forward Reverse | 5ʹ-CAAGAATCGTCCCCAAGAAA-3ʹ 5ʹ-GAAAGGTGCAGCACACAAAA-3ʹ | 218 | |
| Abcb1 Mouse | Forward Reverse | 5ʹ-TGGGAACTCTGGCTGCTATT-3ʹ 5ʹ-GGCGTACGTGGTCATTTCTT-3ʹ | 179 | |
| ABCB1 Human | Forvard Reverse | 5′-GCAATGGAGGAGCAAAGAAG-3’ 5´-CCAAAGTTCCCACCACCATA-3´ | 150 | |
| Abcc1 | Forward Reverse | 5ʹ-ACCAGCAACCCCGACTTTAC-3ʹ 5ʹ-TGGTTTTGTTGAGGTGTGTCA-3ʹ | 151 | |
| Abcg2 | Forward Reverse | 5ʹ-CCACGTGTTAGTACCAATGTCG-3ʹ 5ʹ-TTTCCGGACTAGAAACCCACT-3ʹ | 151 | |
| Ccna1 | CycA1 | Forward Reverse | 5ʹ-ACACAGACCCAAGGCTCACT-3ʹ 5ʹ-ACAGGGTCTCTGTGCGAAGT-3ʹ | 122 |
| Ccnb1 | CycB1 | Forward Reverse | 5ʹ-GGTGACTTCGCCTTTGTGAC-3ʹ 5ʹ-CTACGGAGGAAGTGCAGAGG-3ʹ | 125 |
| Ccnd1 | CycD1 | Forward Reverse | 5ʹ-AGCAGAAGTGCGAAGAGGAG-3ʹ 5ʹ-CAAGGGAATGGTCTCCTTCA-3ʹ | 149 |
| Ccne1 | CycE1 | Forward Reverse | 5ʹ-GGAAAATCAGACCACCCAGA-3ʹ 5ʹ-AGGATGACGCTGCAGAAAGT-3ʹ | 131 |
| Actb | β-actin | Forward Reverse | 5ʹ-TCGCCATGGATGACGATA-3ʹ 5ʹ-CACGATGGAGGGGAATACAG-3ʹ | 110 |
| Atf6 | Atf6α | Forward Reverse | 5ʹ-GAGCCGCACAGCTACCTAAC-3ʹ 5ʹ-CCCATACTTCTGGTGGCACT-3ʹ | 121 |
| Eif2ak3 | Perk | Forward Reverse | 5ʹ-CTGCTGCTTCTGTTCCTGCT-3ʹ 5ʹ-CCCCTAAGCCAAACACTGTC-3ʹ | 106 |
| Ern1 | Ire1α | Forward Reverse | 5ʹ-TGCATGCTGTTAGCAAGAGG-3ʹ 5ʹ-GACTGCCATCATTGGGATCT-3ʹ | 120 |
| Hspa5 | Grp78/BiP | Forward Reverse | 5ʹ-TTTTCTGATGTATCCTCTTCACCAGT-3 5ʹ-TTCAGCCAATTATCAGCAAACTCT-3ʹ | 73 |
| Hsp90aa1 | Hsp90aa | Forward Reverse | 5ʹ-GGG AGC TCA TCT CCA ATT CA-3ʹ 5ʹ-ATTGATGTGCAGCTCCTTCC-3ʹ | 101 |
| Hsp90b1 | Grp94 | Forward Reverse | 5ʹ-GGGGAGGTCACCTTCAAGTC-3ʹ 5ʹ-TGAGGGGGAGATCATCGGAA-3ʹ | 199 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cagala, M.; Pavlikova, L.; Seres, M.; Kadlecikova, K.; Breier, A.; Sulova, Z. Development of Resistance to Endoplasmic Reticulum Stress-Inducing Agents in Mouse Leukemic L1210 Cells. Molecules 2020, 25, 2517. https://doi.org/10.3390/molecules25112517
Cagala M, Pavlikova L, Seres M, Kadlecikova K, Breier A, Sulova Z. Development of Resistance to Endoplasmic Reticulum Stress-Inducing Agents in Mouse Leukemic L1210 Cells. Molecules. 2020; 25(11):2517. https://doi.org/10.3390/molecules25112517
Chicago/Turabian StyleCagala, Martin, Lucia Pavlikova, Mario Seres, Karolina Kadlecikova, Albert Breier, and Zdena Sulova. 2020. "Development of Resistance to Endoplasmic Reticulum Stress-Inducing Agents in Mouse Leukemic L1210 Cells" Molecules 25, no. 11: 2517. https://doi.org/10.3390/molecules25112517