
Investigation of Potential Amorphisation and Co-Amorphisation Behaviour of the Benzene Di-Carboxylic Acids upon Cryo-Milling



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Single Component Samples
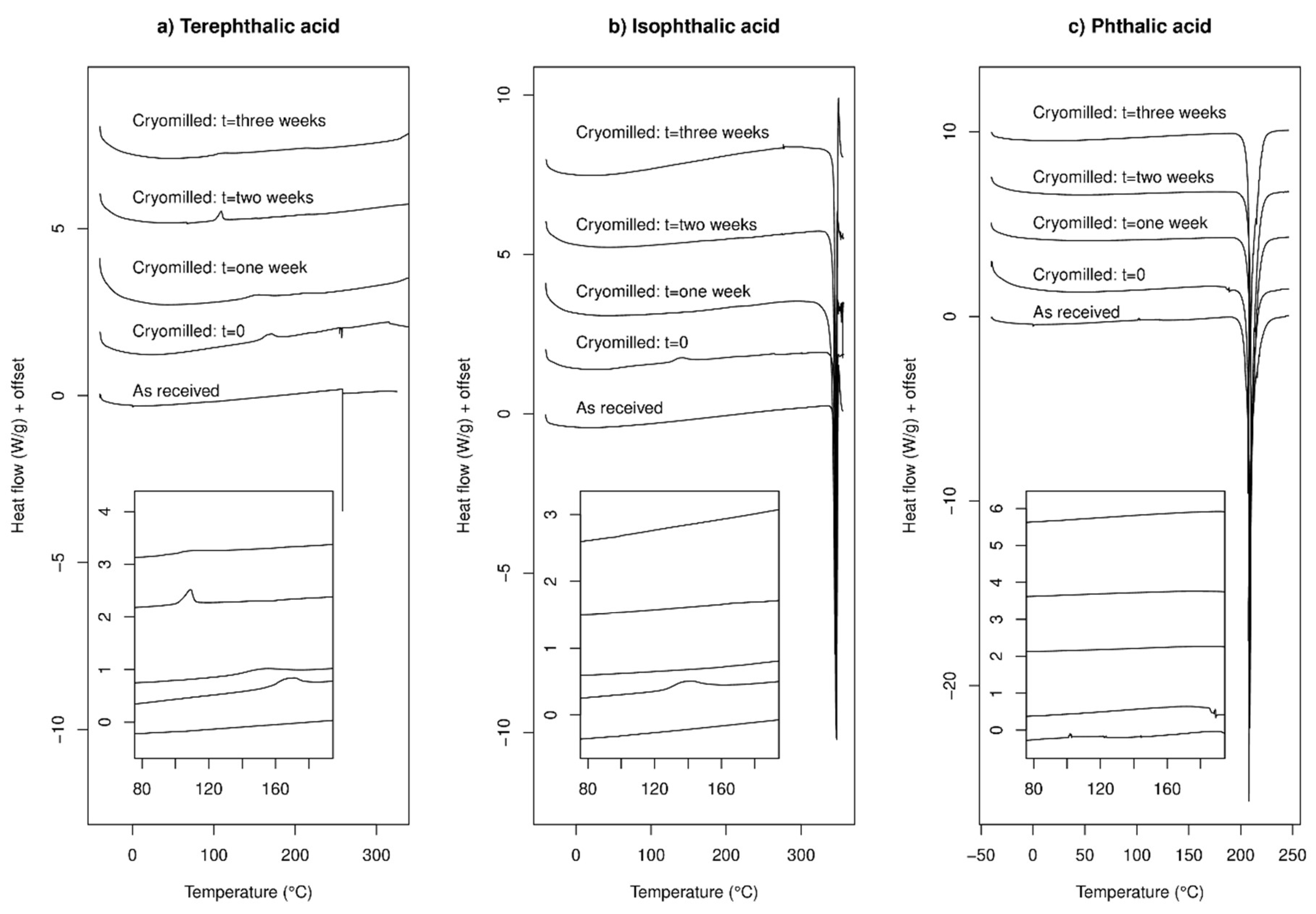
2.1.1. DSC Experiments
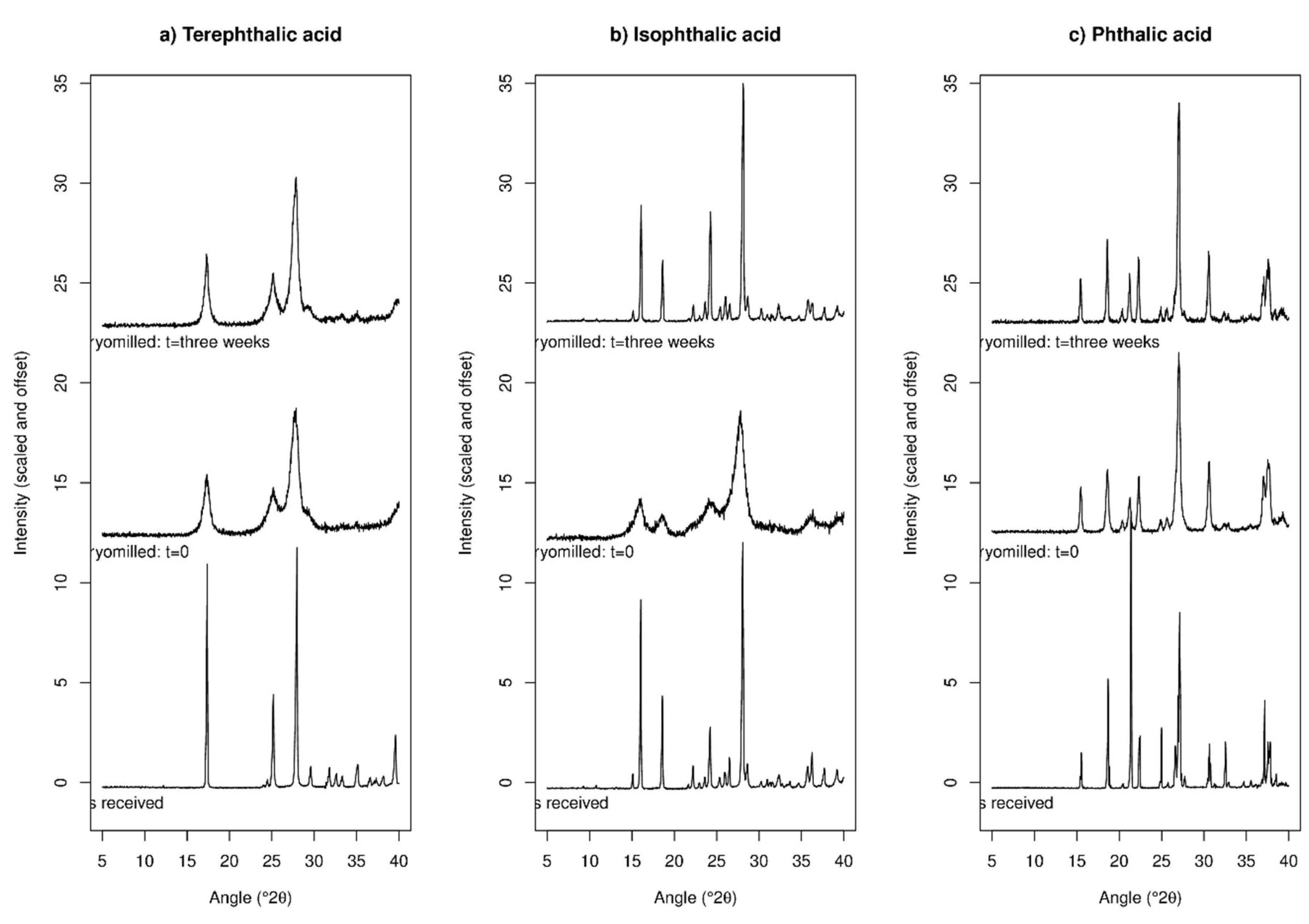
2.1.2. XRPD Experiments
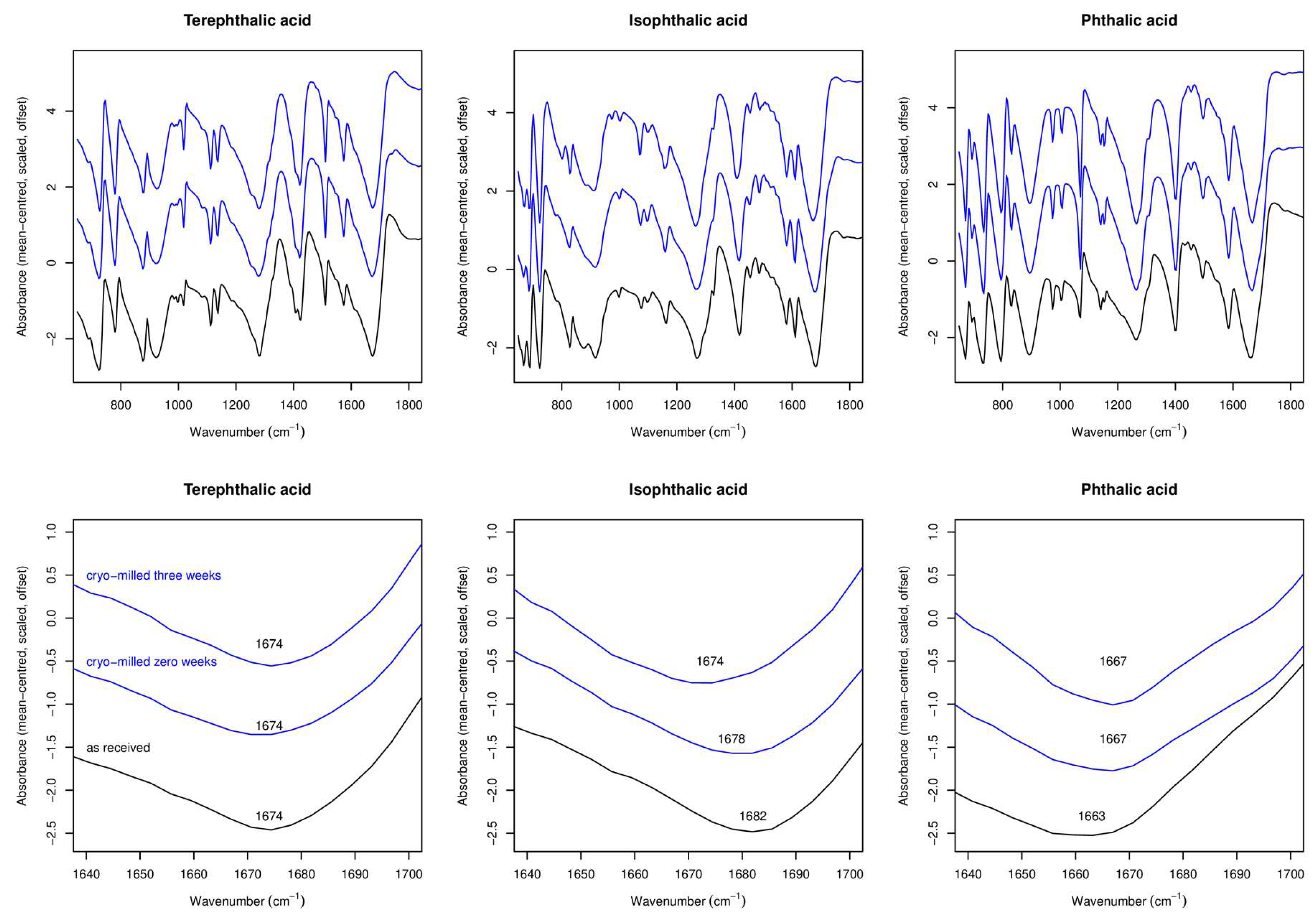
2.1.3. ATR-FTIR Experiments
2.1.4. Summary of Experimental Results for Single-Component Systems
2.2. Multi-Component Samples
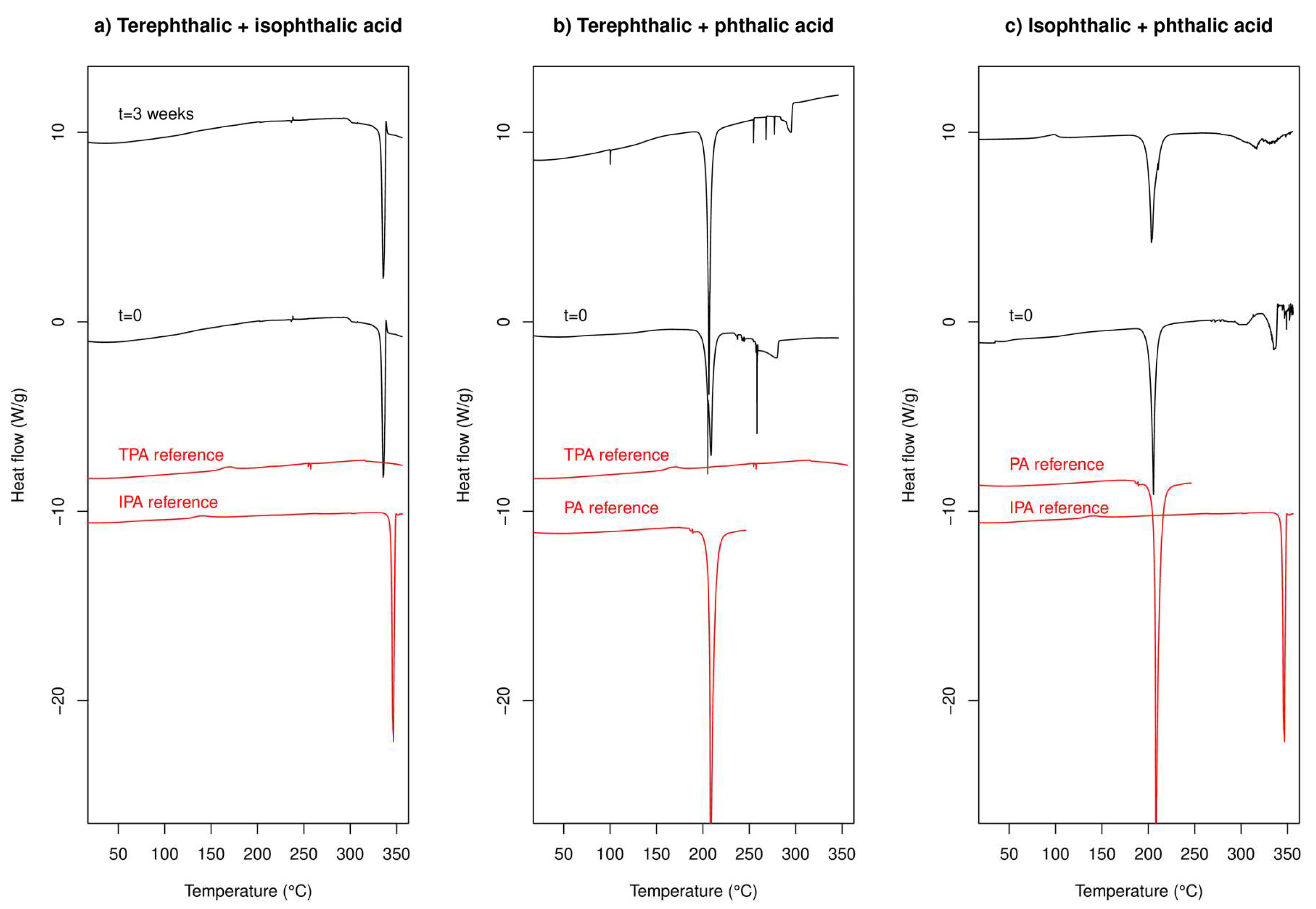
2.2.1. DSC Experiments
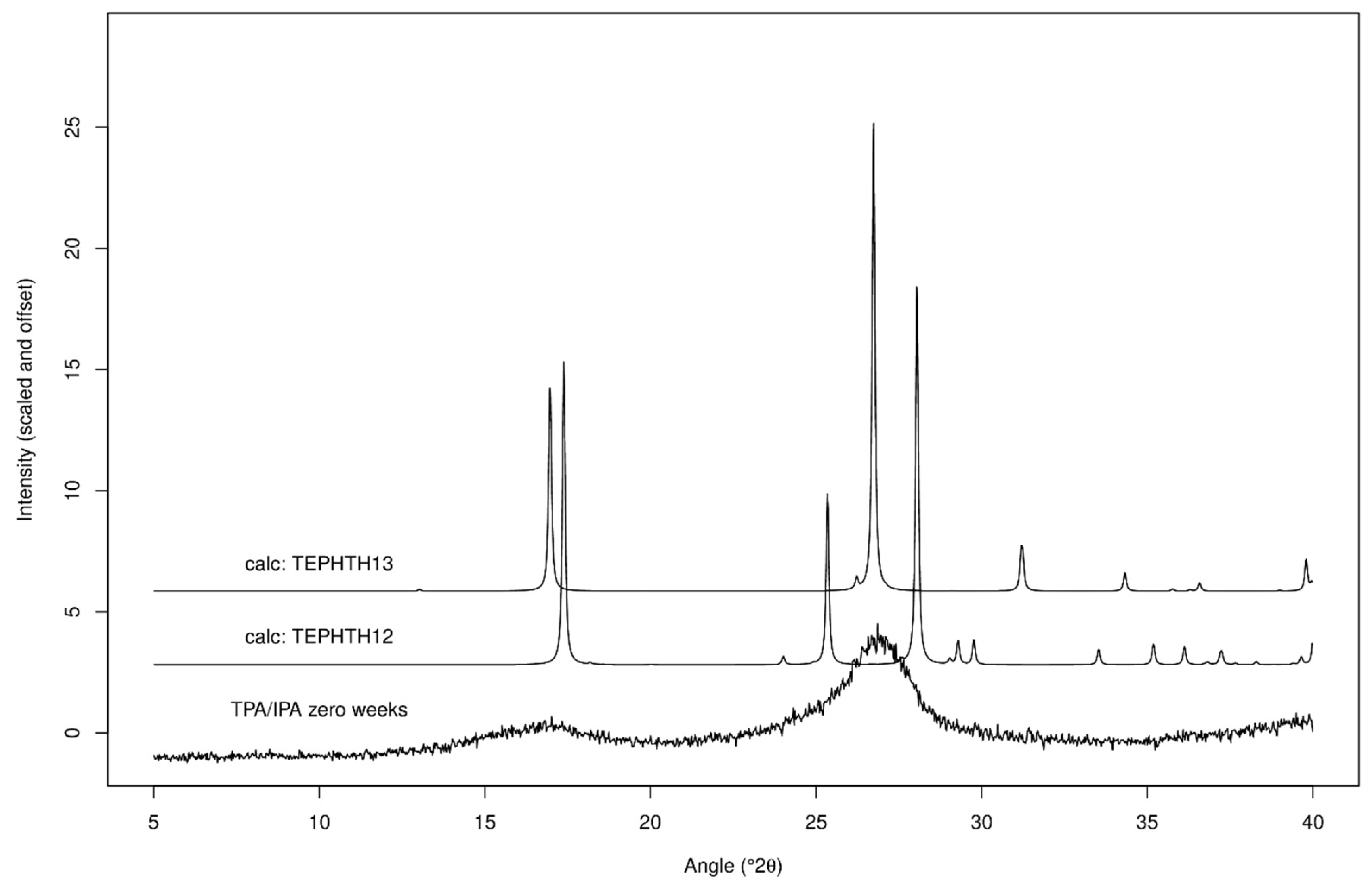
2.2.2. XRPD Experiments
2.2.3. ATR-FTIR Experiments
3. Discussion
3.1. Single Components
3.2. Multiple Components
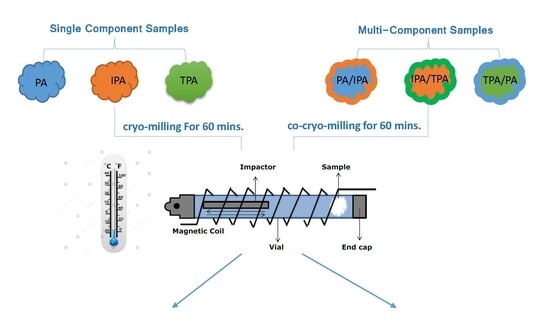
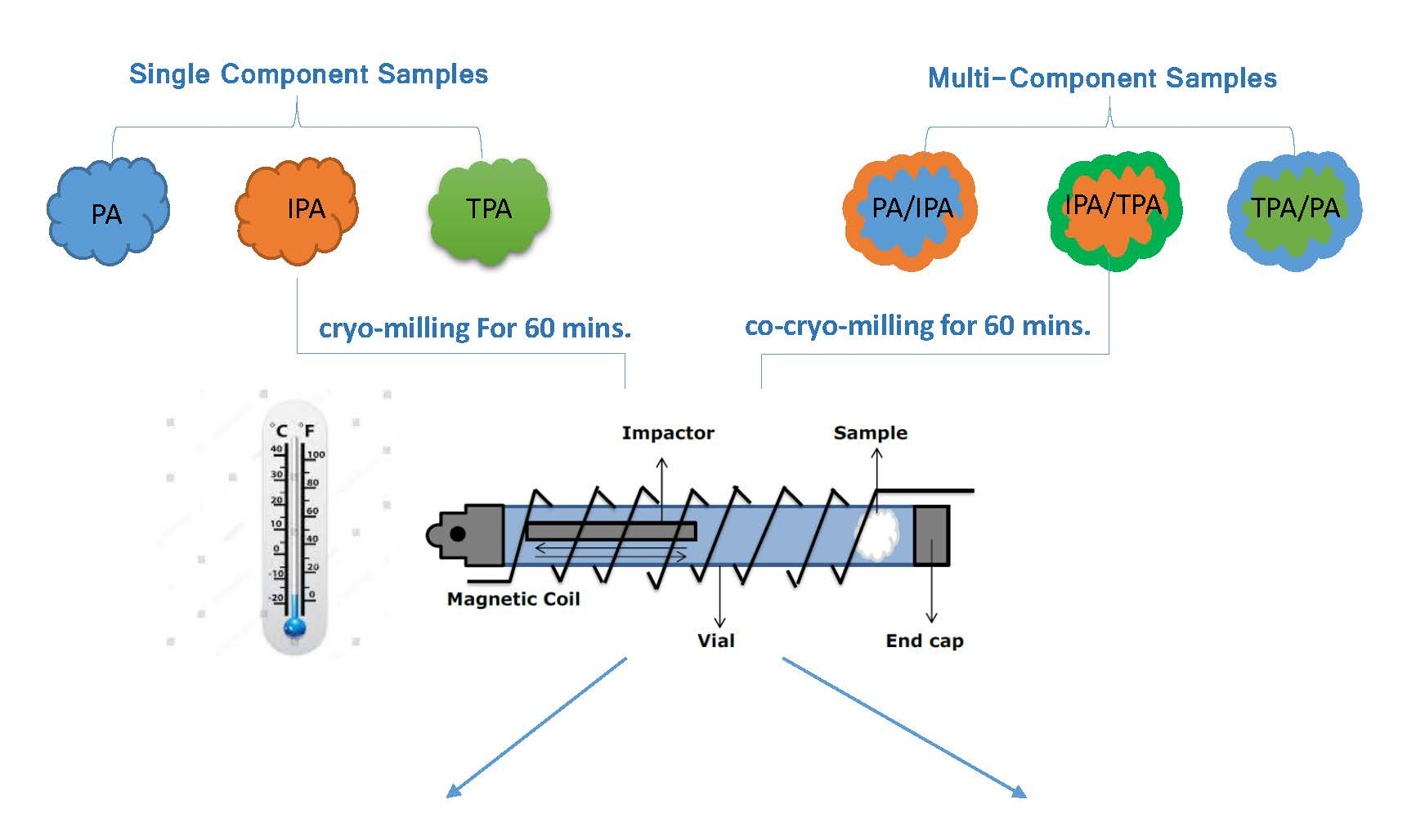
4. Materials and Methods
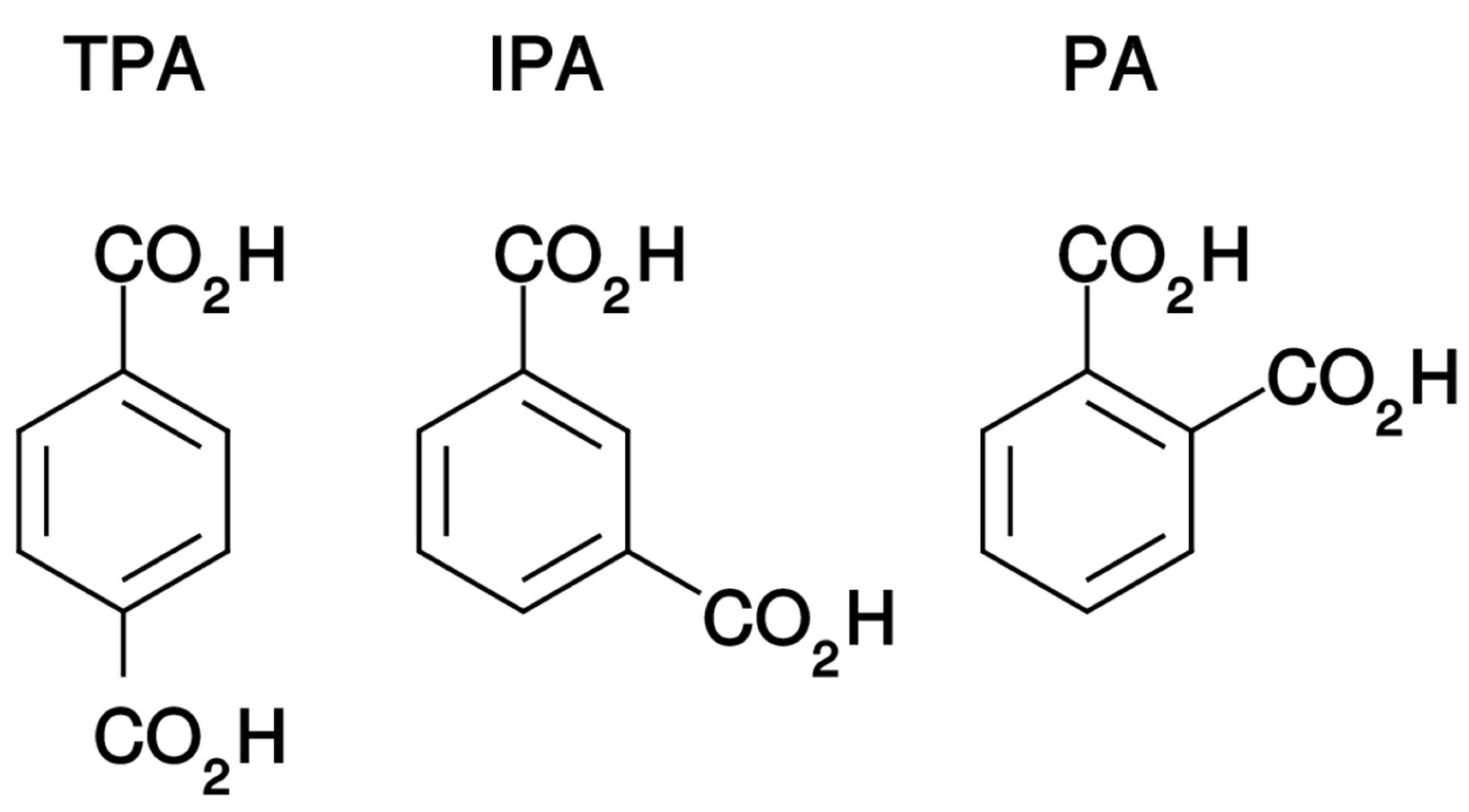
4.1. Materials
4.2. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 11, 1607–1621. [Google Scholar] [CrossRef]
- Laitinen, R.; Lbmann, K.; Grohganz, H.; Priemel, P.; Strachan, C.J.; Rades, T. Supersaturating drug delivery systems: The potential of co-amorphous drug formulations. Int. J. Pharm. 2017, 532, 1–12. [Google Scholar] [CrossRef]
- Bastin, R.J.; Bowker, M.J.; Slater, B.J. Salt Selection and Optimisation Procedures for Pharmaceutical New Chemical Entities. Org. Process Res. Dev. 2000, 4, 427–435. [Google Scholar] [CrossRef]
- Kumar, L.; Amin, A.; Bansal, A.K. An overview of automated systems relevant in pharmaceutical salt screening. Drug Discov. Today 2007, 12, 1046–1053. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Morissette, S. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Deliv. Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Brittain, H.G. Cocrystal Systems of Pharmaceutical Interest: 2010. Cryst. Growth Des. 2011, 12, 1046–1054. [Google Scholar] [CrossRef]
- Chadha, R.; Bala, M.; Rani, D.; Arora, P. Retrospection on Polymorphism and Cocrystallization of Anti-Retrovirals. Curr. Top. Med. Chem. 2013, 13, 1933–1962. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBoyace, K.; Wildfong, P.L.D. The Application of Modeling and Prediction to the Formation and Stability of Amorphous Solid Dispersions. J. Pharm. Sci. 2018, 107, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Haser, A.; Zhang, F. New Strategies for Improving the Development and Performance of Amorphous Solid Dispersions. AAPS PharmSciTech 2018, 19, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Pillai, J. Solid lipid matrix mediated nanoarchitectonics for improved oral bioavailability of drugs. Expert Opin. Drug Metab. Toxicol. 2019, 15, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Faham, A.; Clas, S.D.; Boyd, B.J.; Jannin, V.; Bernkop-Schnürch, A.; Merkel, O.M. Lipids and polymers in pharmaceutical technology: Lifelong companions. Int. J. Pharm. 2019, 558, 128–142. [Google Scholar] [CrossRef]
- Vishwakarma, N.; Jain, A.; Sharma, R.; Mody, N.; Vyas, S.; Vyas, S.P. Lipid-Based Nanocarriers for Lymphatic Transportation. AAPS PharmSciTech 2019, 20, 83. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Shi, Q.; Moinuddin, S.M.; Cai, T. Advances in coamorphous drug delivery systems. Acta Pharm. Sin. B 2019, 9, 19–35. [Google Scholar] [CrossRef]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous Drug Systems: Enhanced Physical Stability and Dissolution Rate of Indomethacin and Naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Larsen, F.H.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of variation in molar ratio on co-amorphous drug-amino acid systems. Eur. J. Pharm. Biopharm. 2016, 107, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Lobmann, K.; Grohganz, H.; Strachan, C.; Rades, T. Amino Acids as Co-amorphous Excipients for Simvastatin and Glibenclamide: Physical Properties and Stability. Mol. Pharm. 2014, 11, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Ojarinta, R.; Heikkinen, A.T.; Sievanen, E.; Laitinen, R. Dissolution behavior of co-amorphous amino acid-indomethacin mixtures: The ability of amino acids to stabilize the supersaturated state of indomethacin. Eur. J. Pharm. Biopharm. 2017, 112, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, Y.; Sue, K.; Yoda, S.; Hakuta, Y.; Furuya, T. Solubility of Terephthalic Acid in Subcritical Water. J. Chem. Eng. Data 2012, 57, 1810–1816. [Google Scholar] [CrossRef]
- Long, B.-W.; Wang, L.-S.; Wu, J.-S. Solubilities of 1,3-Benzenedicarboxylic Acid in Water + Acetic Acid Solutions. J. Chem. Eng. Data 2005, 50, 136–137. [Google Scholar] [CrossRef]
- Han, N.; Zhu, L.; Wang, L.; Fu, R. Aqueous solubility of m-phthalic acid, o-phthalic acid and p-phthalic acid from 298 to 483 K. Sep. Purif. Technol. 1999, 16, 175–180. [Google Scholar] [CrossRef]
- Fajardo, C.; Guyot, J.P.; Macarie, H.; Monroy, O. Inhibition of anaerobic digestion by terephthalic acid and its aromatic by products. Water Sci. Technol. 1997, 36, 83–90. [Google Scholar] [CrossRef]
- Bailey, M.; Brown, C.J. The crystal structure of terephthalic acid. Acta Crystallogr. 1967, 22, 387–391. [Google Scholar] [CrossRef]
- Alcala, R.; Martínez-Carrera, S. The crystal structure of isophthalic acid. Acta Crystallogr. Sect. B 1972, 28, 1671–1677. [Google Scholar] [CrossRef]
- Schalkwyk, T.G.D.V. The crystal structure of phthalic acid. Acta Crystallogr. 1954, 7, 775. [Google Scholar] [CrossRef]
- Fukuoka, E.; Makita, M.; Yamamura, S. Glassy State of Pharmaceuticals. III: Thermal Properties and Stability of Glassy Pharmaceuticals and Their Binary Glass Systems. Chem. Pharm. Bull. 1989, 37, 1047–1050. [Google Scholar] [CrossRef]
- Shayanfar, A.; Jouyban, A. Drug–Drug Coamorphous Systems: Characterization and Physicochemical Properties of Coamorphous Atorvastatin with Carvedilol and Glibenclamide. J. Pharm. Innov. 2013, 8, 218–228. [Google Scholar] [CrossRef]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Loebmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef]
- Lucchesi, C.A.; Lewis, W.T. Latent heat of sublimation of terephthalic acid from differential thermal analysis data. J. Chem. Eng. Data 1968, 13, 389–391. [Google Scholar] [CrossRef]
- Marsac, P.J.; Konno, H.; Rumondor, A.C.F.; Taylor, L.S. Recrystallization of nifedipine and felodipine from amorphous molecular level solid dispersions containing poly(vinylpyrrolidone) and sorbed water. Pharm. Res. 2008, 25, 647–656. [Google Scholar] [CrossRef]
- Monroe, K.P. Phthalic Anhydride. II—The Melting Point of Pure Phthalic Anhydride. The System: Phthalic Anhydride–Phthalic Acid. J. Ind. Eng. Chem. 1919, 11, 1116–1119. [Google Scholar] [CrossRef]
- Tellez, C.A.; Hollauer, E.; Mondragon, M.A.; Castano, V.M. Fourier transform infrared and Raman spectra, vibrational assignment and ab initio calculations of terephthalic acid and related compounds. Spectrochim. Acta Part -Mol. Biomol. Spectrosc. 2001, 57, 993–1007. [Google Scholar] [CrossRef]
- Bardak, F.; Karaca, C.; Bilgili, S.; Atac, A.; Mavis, T.; Asiri, A.M.; Kose, E. Conformational, electronic, and spectroscopic characterization of isophthalic acid (monomer and dimer structures) experimentally and by DFT. Spectrochim. Acta Part -Mol. Biomol. Spectrosc. 2016, 165, 33–46. [Google Scholar] [CrossRef]
- Loring, J.S.; Karlsson, M.; Fawcett, W.R.; Casey, W.H. Infrared spectra of phthalic acid, the hydrogen phthalate ion, and the phthalate ion in aqueous solution. Spectrochim. Acta Part -Mol. Biomol. Spectrosc. 2001, 57, 1635–1642. [Google Scholar] [CrossRef]
- Peisheng, M.; Qing, X. Determination and Correlation for Solubility of Aromatic Acids in Solvents. Chin. J. Chem. Eng. 2001, 9, 39–44. [Google Scholar]
- Śledź, M.; Janczak, J.; Kubiak, R. New crystalline modification of terephthalic acid. J. Mol. Struct. 2001, 595, 77–82. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity Fixed-Points of Binary Saturated Aqueous-Solutions. J. Res. Natl. Bur. Stand. Sect. -Phys. Chem. 1977, 81, 89–96. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2012. [Google Scholar]
Sample Availability: Not available. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elfakhri, R.; Burley, J.C. Investigation of Potential Amorphisation and Co-Amorphisation Behaviour of the Benzene Di-Carboxylic Acids upon Cryo-Milling. Molecules 2019, 24, 3990. https://doi.org/10.3390/molecules24213990
Elfakhri R, Burley JC. Investigation of Potential Amorphisation and Co-Amorphisation Behaviour of the Benzene Di-Carboxylic Acids upon Cryo-Milling. Molecules. 2019; 24(21):3990. https://doi.org/10.3390/molecules24213990
Chicago/Turabian StyleElfakhri, Rehab, and Jonathan C. Burley. 2019. "Investigation of Potential Amorphisation and Co-Amorphisation Behaviour of the Benzene Di-Carboxylic Acids upon Cryo-Milling" Molecules 24, no. 21: 3990. https://doi.org/10.3390/molecules24213990