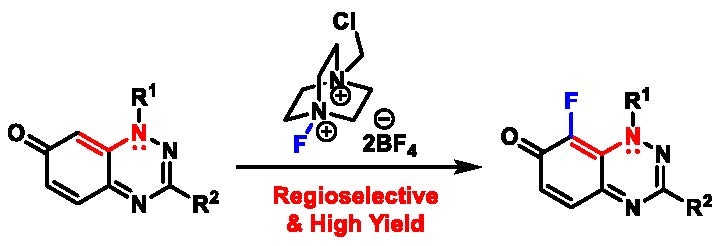
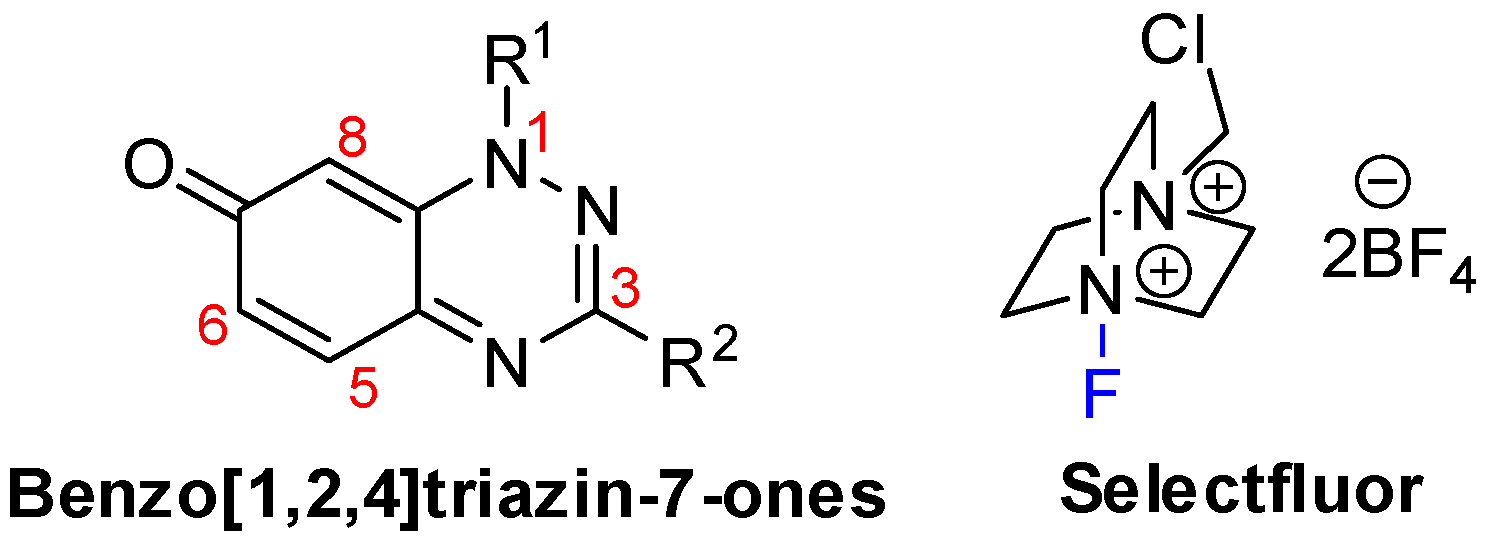
Regioselective Fluorination of 7-Oxo-1,2,4-benzotriazines Using Selectfluor



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Fluorinations
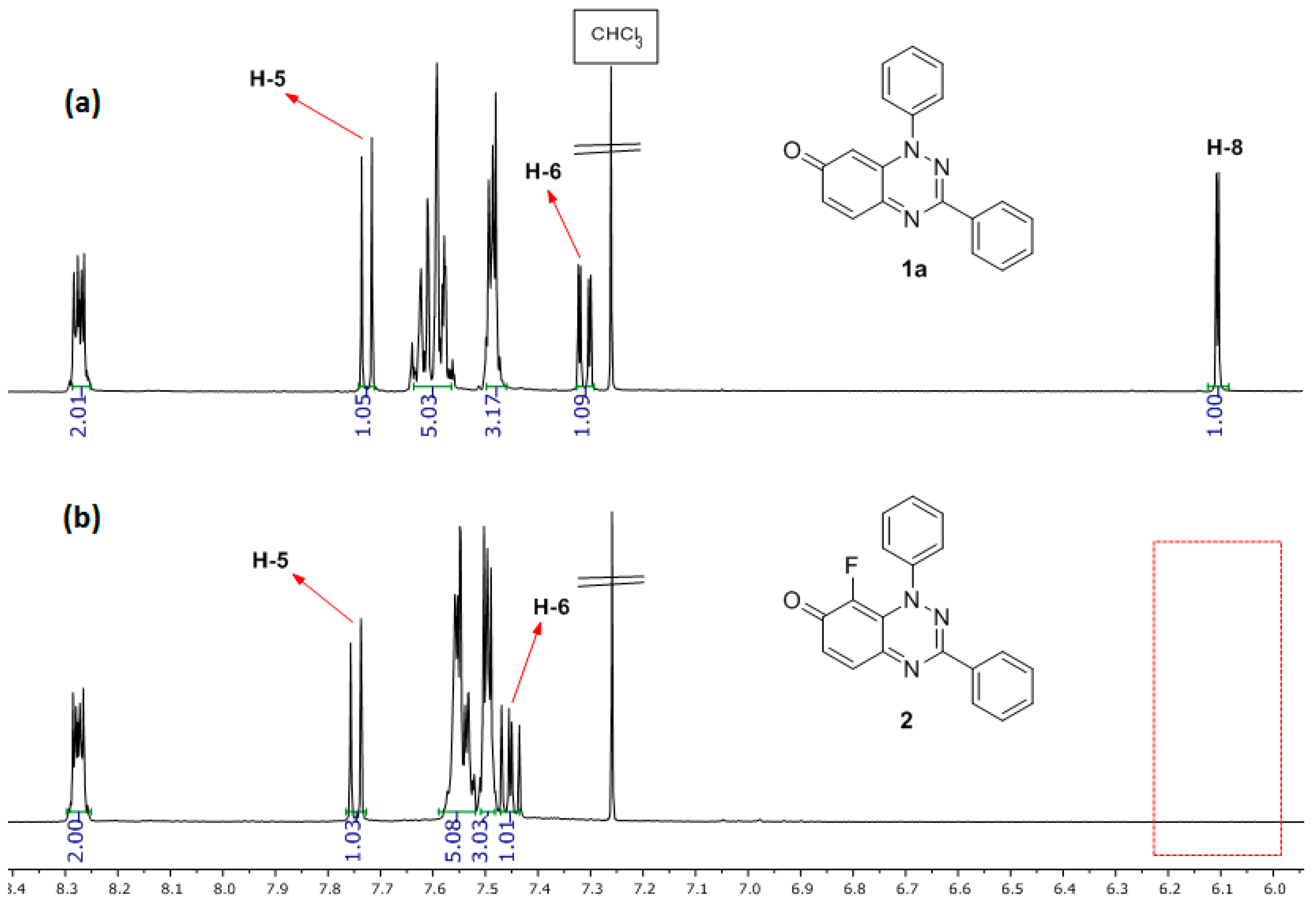
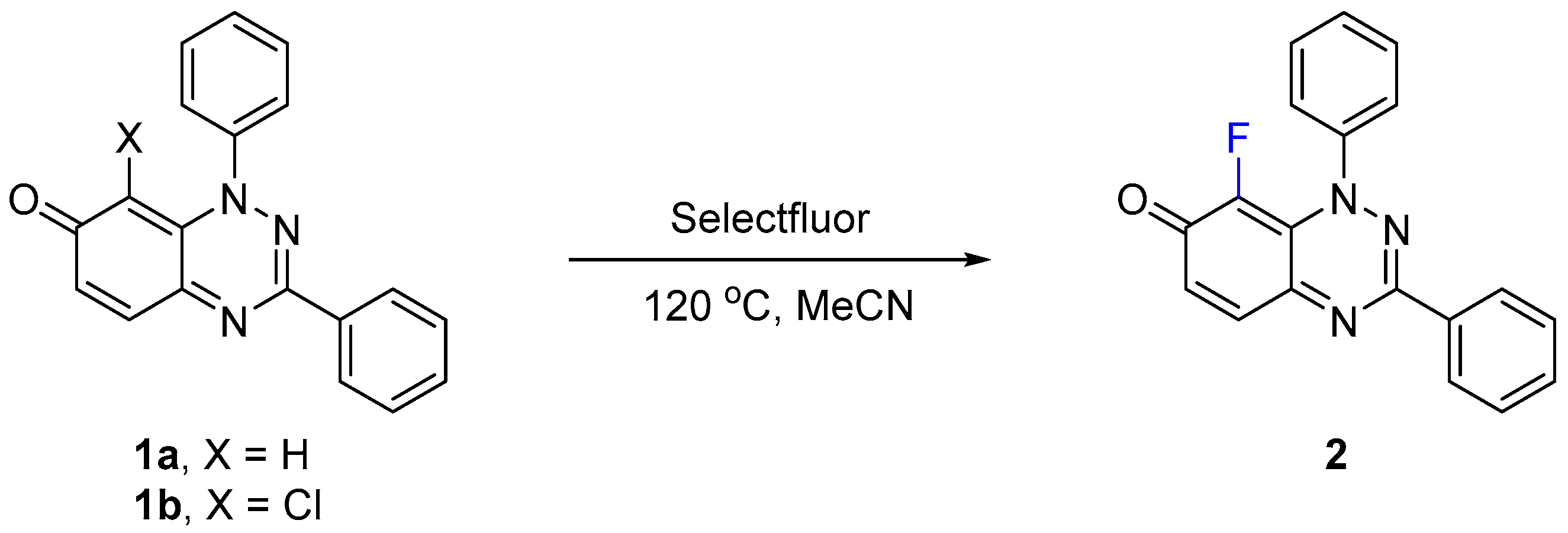
2.1.1. Optimizing the Fluorination and Confirming Selectivity
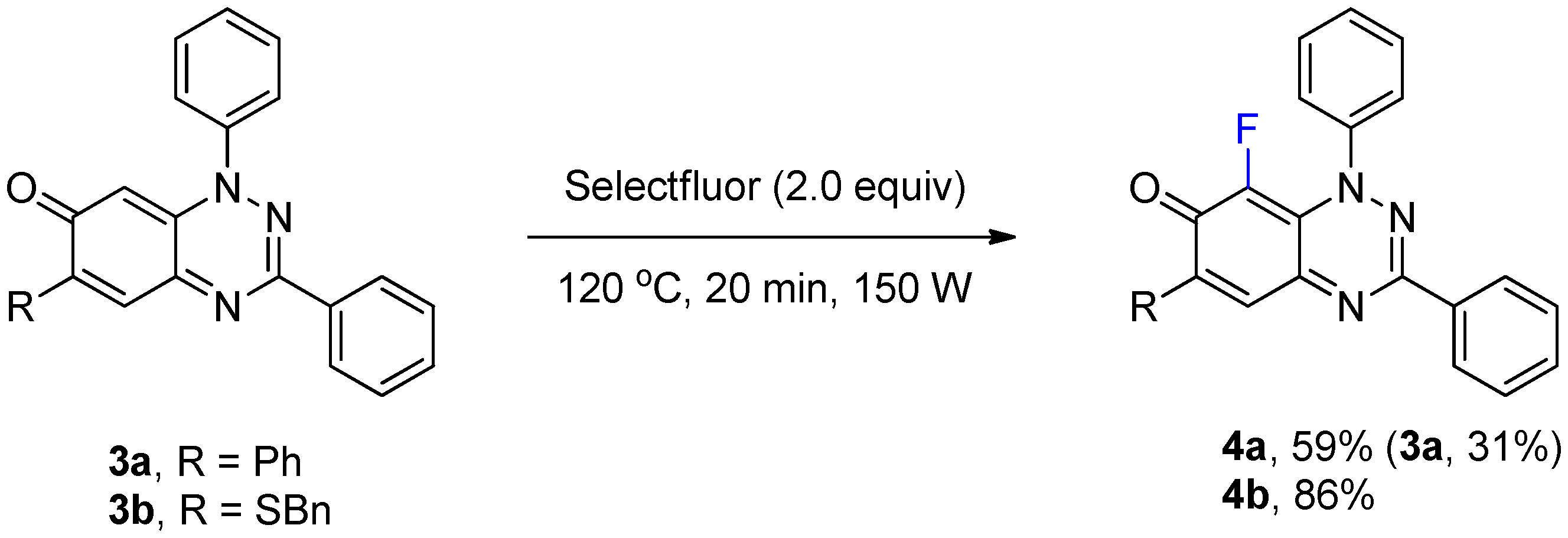
2.1.2. Fluorination of C-6-Substituted Benzo[1,2,4]triazin-7-ones
2.1.3. Fluorination of 1,2,5-Thiadiazolo-Fused Benzotriazinones
2.2. Cytotoxicity against MCF-7 using the MTT Assay
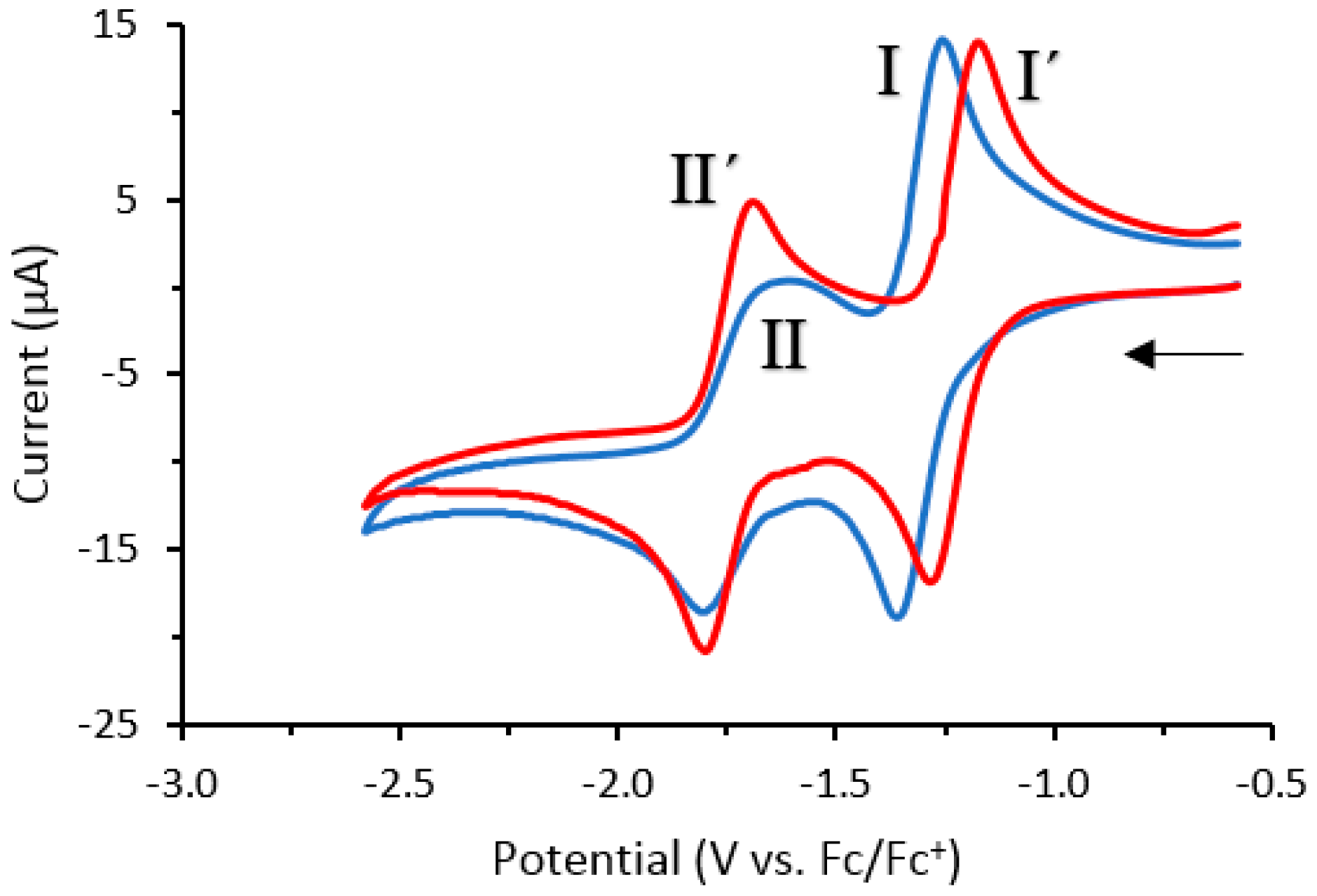
2.3. Cyclic Voltammetry
3. Experimental Section
3.1. General Materials and Methods
3.2. Synthetic Procedures and Characterization





3.3. Cell Culture and Cytotoxicity Evaluation
3.3.1. Materials and Cell Lines
3.3.2. Cytotoxicity Measurements Using the MTT Assay
3.4. Electrochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; DurÓn, S.G.; Burkart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic insight and applications. Angew. Chem. Int. Ed. 2005, 44, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Stavber, S. Recent Advances in the Application of SelectfluorTM F-TEDA-BF4 as a Versatile Mediator or Catalyst in Organic Synthesis. Molecules 2011, 6, 6432–6464. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Li, Y.; Gao, S.; Li, R.; Li, X.; Wang, B.; Yang, H. Amide-assisted radical strategy: Metal-free direct fluorination of arenes in aqueous media. Green Chem. 2017, 19, 3344–3349. [Google Scholar] [CrossRef]
- Heravi, M.R.P. Fluorination of activated aromatic systems with Selectfluor™ F-TEDA-BF4 in ionic liquids. J. Fluor. Chem. 2008, 129, 217–221. [Google Scholar] [CrossRef]
- Keinan, S.; Paquette, W.D.; Skoko, J.J.; Beratan, D.N.; Yang, W.; Shinde, S.; Johnston, P.A.; Lazo, J.S.; Wipf, P. Computational design, synthesis and biological evaluation of para-quinone-based inhibitors for redox regulation of the dual-specificity phosphatase Cdc25B. Org. Biomol. Chem. 2008, 6, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-T.; Zhou, D.-C.; Mai, Y.-W.; Huo, L.; Yao, P.-F.; Huang, S.-L.; Wang, H.-G.; Huang, Z.-S.; Gu, L.-Q. Construction of the oxaphenalene skeletons of mansonone F derivatives through C-H bond functionalization and their evaluation for anti-proliferative activities. RSC Adv. 2017, 7, 20919–20928. [Google Scholar] [CrossRef]
- Cameron, D.W.; Feutrill, G.I.; Griffiths, P.G.; Richards, K.R. Synthesis of fluoronaphthoquinones: Halide displacement by naked fluoride. Aust. J. Chem. 1982, 35, 1509–1512. [Google Scholar] [CrossRef]
- Cameron, D.W.; Chalmers, P.J.; Feutrill, G.I. Regiochemistry of nucleophilic displacements in chloroquinones. Tetrahedron Lett. 1984, 25, 6031–6032. [Google Scholar] [CrossRef]
- Kim, B.G.; Chun, T.G.; Lee, H.-Y.; Snapper, M.L. A new structural class of S-adenosylhomocysteine hydrolase inhibitors. Bioorg. Med. Chem. 2009, 17, 6707–6714. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, F.; Yang, J.; Yu, P.; Yi, P.; Sun, Y.; Wang, Y. Fluorination-oxidation of 2-hydroxymethylindole using Selectfluor. Adv. Synth. Catal. 2017, 359, 853–858. [Google Scholar] [CrossRef]
- Sweeney, M.; Coyle, R.; Kavanagh, P.; Berezin, A.A.; Lo Re, D.; Zissimou, G.A.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Discovery of anti-cancer for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorg. Med. Chem. 2016, 24, 3565–3570. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Berezin, A.A.; Lo Re, D.; Loizou, G.; Demetriades, M.; De Stradis, A.; Campagna, F.; Koutentis, P.A.; Carotti, A. Design, synthesis and biological evaluation of benzo[e][1,2,4]triazin-7(1H)-one and [1,2,4]-triazino[5,6,1-jk]carbazol-6-one derivatives as dual inhibitors of beta-amyloid aggregation and acetyl/butyryl cholinesterase. Eur. J. Med. Chem. 2012, 58, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Keane, L.-A.J.; Mirallai, S.I.; Sweeney, M.; Carty, M.P.; Zissimou, G.A.; Berezin, A.A.; Koutentis, P.A.; Aldabbagh, F. Anti-cancer activity of phenyl and pyrid-2-yl 1,3-substituted benzo[1,2,4]triazin-7-ones and stable free radical precursors. Molecules 2018, 23, 574. [Google Scholar] [CrossRef] [PubMed]
- Zissimou, G.A.; Kourtellaris, A.; Manoli, M.; Koutentis, P.A. Redox active quinoidal 1,2,4-Benzotriazines. J. Org. Chem. 2018, 83, 9391–9402. [Google Scholar] [CrossRef] [PubMed]
- Koutentis, P.A.; Lo Re, D. Catalytic oxidation of N-phenylamidrazones to 1,3-diphenyl-1,4-dihydro-1,2,4-benzotriazin-4-yls: An improved synthesis of Blatter’s radical. Synthesis 2010, 2075–2079. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Krassos, H.; Lo Re, D. 1,3-Diphenylbenzo[e][1,2,4]-7(1H)-one: Selected chemistry at the C-6, C-7 and C-8 positions. Org. Biomol. Chem. 2011, 9, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Harwood, L.M. Dry-column flash chromatography. Aldrichim. Acta 1985, 18, 25. [Google Scholar]
Sample Availability: Samples of the compounds 2, 4a–4b, 6a–6b are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Iminoquinone | Selectfluor (equiv) | Time (h) | Isolated Yield 2 (%) |
|---|---|---|---|---|
| 1 | 1a | 1.2 | 24 | 32 b |
| 2 | 1a | 1.5 | 24 | 55 c |
| 3 | 1a | 2 | 1 | 94 d |
| 4 | 1a | 2 | 0.3 | 97 e |
| 5 | 1b | 2 | 1 | 96 e |
| Compound | IC50 MCF-7 (μΜ) a |
|---|---|
| 1a | 0.810 ± 0.080 b |
| 2 | 0.157 ± 0.001 |
| Compound | E0′ [V] versus Fc/Fc+ | |
|---|---|---|
| E0′ (I) | E0′ (II) | |
| 1a | −1.31 | −1.72 |
| 2 | −1.21 | −1.73 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirallai, S.I.; Koutentis, P.A.; Aldabbagh, F. Regioselective Fluorination of 7-Oxo-1,2,4-benzotriazines Using Selectfluor. Molecules 2019, 24, 282. https://doi.org/10.3390/molecules24020282
Mirallai SI, Koutentis PA, Aldabbagh F. Regioselective Fluorination of 7-Oxo-1,2,4-benzotriazines Using Selectfluor. Molecules. 2019; 24(2):282. https://doi.org/10.3390/molecules24020282
Chicago/Turabian StyleMirallai, Styliana I., Panayiotis A. Koutentis, and Fawaz Aldabbagh. 2019. "Regioselective Fluorination of 7-Oxo-1,2,4-benzotriazines Using Selectfluor" Molecules 24, no. 2: 282. https://doi.org/10.3390/molecules24020282