
Oxygen Evolution Reaction at IrO2/Ir(Ni) Film Electrodes Prepared by Galvanic Replacement and Anodization: Effect of Precursor Ni Film Thickness

 , ,
, ,
Abstract
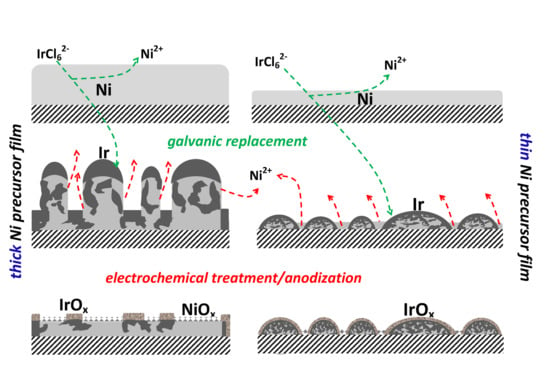
:
1. Introduction
2. Results and Discussion
2.1. Microscopic and Spectroscopic Characterization of IrO2/Ir(Ni)/GC Catalyst Film Electrodes
2.2. Surface Electrochemistry of the Electrodes
2.3. Oxygen Evolution Reaction (OER)
3. Materials and Methods
3.1. Preparation of IrO2/Ir(Ni)/GC Electrodes
3.2. Electrochemical Setup and Procedures
3.3. Microscopic and Spectroscopic Characterization
4. Conclusions
4.1. IrO2/Ir(Ni) Catalyst Morphological, Compositional and Electrochemical Characterization; Mechanism of Catalyst Preparation
4.2. Catalyst Performance as an OER Anode
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guerrini, E.; Trasatti, S. Electrocatalysis in water electrolysis. In Catalysis for Sustainable Energy Production; Barbaro, P., Bianchini, C., Eds.; WILEY-VCH Verlag GmbH&Co.: Weinheim, Germany, 2009; Chapter 7; pp. 235–269. [Google Scholar] [CrossRef]
- Trasatti, S. Physical electrochemistry of ceramic oxides. Electrochim. Acta 1991, 36, 225–241. [Google Scholar] [CrossRef]
- Zhang, W.; Ghali, E.; Houlachi, G. Review of oxide coated catalytic titanium anodes performance for metal electrowinning. Hydrometallurgy 2017, 169, 456–467. [Google Scholar] [CrossRef]
- Shan, R.; Zhang, Z.; Kan, M.; Zhang, T.; Zan, Q.; Zhao, Y. A novel highly active nanostructured IrO2/Ti anode for water oxidation. Int. J. Hydrogen Energy 2015, 40, 14279–14283. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Zafar, M.S.; Tausif, M.; Ashrafq, M.; Hussain, S. New development of anodic electro-catalyst for chlor-alkali industry. Port. Electrochim. Acta 2016, 34, 257–266. [Google Scholar] [CrossRef]
- Papaderakis, A.; Pliatsikas, N.; Prochaska, C.; Vourlias, G.; Patsalas, P.; Tsiplakides, D.; Balomenou, S.; Sotiropoulos, S. Oxygen evolution at IrO2 shell-ir-ni core electrodes prepared by galvanic replacement. J. Phys. Chem. C 2016, 120, 19995–20005. [Google Scholar] [CrossRef]
- Forgie, R.; Bugosh, G.; Neyerlin, K.C.; Liu, Z.; Strasser, P. Bimetallic ru electrocatalysts for the OER and electrolytic water splitting in acidic media. Electrochem. Solid-State Lett. 2010, 13, B36–B39. [Google Scholar] [CrossRef]
- Zequine, C.; Bhoyate, S.; Siam, K.; Kahol, P.; Kostoglou, N.; Mitterer, C.; Hinder, S.J.; Baker, M.A.; Constantinides, G.; Rebholz, C.; et al. Needle grass array of nanostructured nickel cobalt sulfide electrode for clean energy generation. Surf. Coat. Technol. 2018, 354, 306–312. [Google Scholar] [CrossRef]
- Zhang, C.; Bhoyate, S.; Zhao, C.; Kahol, P.; Kostoglou, N.; Mitterer, C.; Hinder, S.; Baker, M.; Constantinides, G.; Polychronopoulou, K.; et al. Electrodeposited nanostructured CoFe2O4 for overall water splitting and supercapacitor applications. Catalysts 2019, 9, 176. [Google Scholar] [CrossRef]
- Liu, J.; Nai, J.; You, T.; An, P.; Zhang, J.; Ma, G.; Niu, X.; Liang, C.; Yang, S.; Guo, L. The flexibility of an amorphous cobalt hydroxide nanomaterial promotes the electrocatalysis of oxygen evolution reaction. Small 2018, 14, 1703514. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Armyanov, S.; Valova, E.; Hubin, A.; Steenhaut, O.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Methanol oxidation at Pt-Cu, Pt-Ni, and Pt-Co electrode coatings prepared by a galvanic replacement process. J. Phys. Chem. C 2010, 114, 5217–5223. [Google Scholar] [CrossRef]
- Papaderakis, A.; Pliatsikas, N.; Prochaska, C.; Papazisi, K.M.; Balomenou, S.P.; Tsiplakides, D.; Patsalas, P.; Sotiropoulos, S. Ternary Pt-Ru-Ni catalytic layers for methanol electrooxidation prepared by electrodeposition and galvanic replacement. Front. Chem. 2014, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mintsouli, I.; Georgieva, J.; Valova, E.; Armyanov, S.; Kakaroglou, A.; Hubin, A.; Steenhaut, O.; Dille, J.; Papaderakis, A.; Kokkinidis, G.; et al. Pt-Ni carbon-supported catalysts for methanol oxidation prepared by Ni electroless deposition and its galvanic replacement by Pt. J. Solid State Electrochem. 2013, 17, 435–443. [Google Scholar] [CrossRef]
- Papaderakis, A.; Pliatsikas, N.; Patsalas, P.; Tsiplakides, D.; Balomenou, S.; Touni, A.; Sotiropoulos, S. Hydrogen evolution at Ir-Ni bimetallic deposits prepared by galvanic replacement. J. Electroanal. Chem. 2017, 808, 21–27. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; M. Dekker: New York, NY, USA, 1985; pp. 1–834. [Google Scholar]
- Reier, T.; Pawolek, Z.; Cherevko, S.; Bruns, M.; Jones, T.; Teschner, D.; Selve, S.; Bergmann, A.; Nong, H.N.; Schlögl, R.; et al. Molecular insight in structure and activity of highly efficient, low-Ir Ir-Ni oxide catalysts for electrochemical water splitting (OER). J. Am. Chem. Soc. 2015, 137, 13031–13040. [Google Scholar] [CrossRef] [PubMed]
- Pickup, P.G.; Birss, V.I. A model for anodic hydrous oxide growth at iridium. J. Electroanal. Chem. 1987, 220, 83–100. [Google Scholar] [CrossRef] [Green Version]
- Burke, L.D.; Sullivan, E.J.M.O. Oxygen gas evolution on hydrous oxides - an example of three-dimensional electrocatalysis? J. Electroanal. Chem. 1981, 117, 155–160. [Google Scholar] [CrossRef]
- Mozota, J.; Conway, B.E. Surface and bulk processes at oxidized iridium electrodes—I. monolayer stage and transition to reversible multilayern oxide film behaviour. Electrochim. Acta 1983, 28, 1–8. [Google Scholar] [CrossRef]
- McIntyre, N.S.; Chan, T.C.; Chen, C. Characterization of oxide structures formed on nickel-chromium alloy during low pressure oxidation at 500–600 °C. Oxid. Met. 1990, 33, 457–479. [Google Scholar] [CrossRef]
- Liu, Y.X.; Masumoto, H.; Goto, T. Preparation of IrO2 thin films by oxidating laser-ablated Ir. Mater. Trans. 2004, 45, 900–903. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, W.; Zhou, Z.; Zaman, W.Q.; Tariq, M.; Cao, L.; Yang, J. Highly efficient oxygen evolution activity of Ca2IrO4 in an acidic environment due to its crystal configuration. ACS Omega 2018, 3, 2902–2908. [Google Scholar] [CrossRef]
- Lee, Y.W.; Hwang, E.T.; Kwak, D.H.; Park, K.W. Preparation and characterization of PtIr alloy dendritic nanostructures with superior electrochemical activity and stability in oxygen reduction and ethanol oxidation reactions. Catal. Sci. Technol. 2016, 6, 569–576. [Google Scholar] [CrossRef]
- Buckley, D.; Burke, L. The oxygen electrode. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1975, 71, 1447–1459. [Google Scholar] [CrossRef]
- Rubel, M.; Haasch, R.; Mrozek, P.; Wieckowski, A.; De Pauli, C.; Trasatti, S. Characterization of IrO2SnO2 thin layers by electron and ion spectroscopies. Vacuum 1994, 45, 423–427. [Google Scholar] [CrossRef]
- Hara, M.; Asami, K.; Hashimoto, K.; Masumoto, T. An X-Ray photoelectron spectroscopic study of electrocatalytic activity of platinum group-metals for chlorine evolution. Electrochim. Acta 1983, 28, 1073–1081. [Google Scholar] [CrossRef]
- Geiger, S.; Kasian, O.; Shrestha, B.R.; Mingers, A.M.; Mayrhofer, K.J.J.; Cherevko, S. Activity and Stability of Electrochemically and Thermally Treated Iridium for the Oxygen Evolution Reaction. J. Electrochem. Soc. 2016, 163, F3132–F3138. [Google Scholar] [CrossRef] [Green Version]
- Saveleva, V.A.; Wang, L.; Teschner, D.; Jones, T.; Gago, A.S.; Friedrich, K.A.; Zafeiratos, S.; Schlögl, R.; Savinova, E.R. Operando evidence for a universal oxygen evolution mechanism on thermal and electrochemical iridium oxides. J. Phys. Chem. Lett. 2018, 9, 3154–3160. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Velasco Vélez, J.J.; Massué, C.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Greiner, M.T.; Allan, J.; et al. The electronic structure of iridium and its oxides. Surf. Interface Anal. 2016, 48, 261–273. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Velasco Vélez, J.J.; Massué, C.; Greiner, M.T.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Allan, J.; et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 2016, 18, 2292–2296. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Wrabetz, S.; Massué, C.; Velasco Vélez, J.J.; Arrigo, R.; Scherzer, M.; Piccinin, S.; Hävecker, M.; Knop-Gericke, A.; et al. Reactive oxygen species in iridium-based OER catalysts. Chem. Sci. 2016, 7, 6791–6795. [Google Scholar] [CrossRef] [Green Version]
- Kasian, O.; Grote, J.P.; Geiger, S.; Cherevko, S.; Mayrhofer, K.J.J. The common intermediates of oxygen evolution and dissolution reactions during water electrolysis on iridium. Angew. Chem. Int. Ed. 2018, 57, 2488–2491. [Google Scholar] [CrossRef]
- Atanasoska, L.; Atanasoski, R.; Trasatti, S. XPS and AES study of mixed layers of RuO2 and IrO2. Vacuum 1990, 40, 91–94. [Google Scholar] [CrossRef]
- Angelinetta, C.; Trasatti, S.; Atanasoska, L.D.; Minevski, Z.S.; Atanasoski, R.T. Effect of preparation on the surface and electrocatalytic properties of RuO2 + IrO2 mixed oxide electrodes. Mater. Chem. Phys. 1989, 22, 231–247. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In Situ observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. Int. Ed. 2014, 53, 7169–7172. [Google Scholar] [CrossRef]
- Peuckert, M. XPS study on thermally and electrochemically prepared oxidic adlayers on iridium. Surf. Sci. 1984, 144, 451–464. [Google Scholar] [CrossRef]
- Conway, B.E.; Augustynski, J.; Koudelka, M.; Sanchez, J. ESCA study of the state of iridium and oxygen in electrochemically and thermally formed iridium oxide. J. Electroanal. Chem. Interfacial Electrochem. 1984, 160, 233–248. [Google Scholar] [CrossRef]
- Powell, C.J. Summary Abstract: Accurate determination of the energies of Auger electrons and photoelectrons from nickel, copper, and gold. J. Vac. Sci. Technol. 1982, 20, 625. [Google Scholar] [CrossRef]
- Lebugle, A.; Axelsson, U.; Nyholm, R.; Maertensson, N. Experimental L and M Core Level Binding Energies for the Metals 22 Ti to 30 Zn. Phys. Scr. 1981, 23, 825–827. [Google Scholar] [CrossRef]
- Ulrich, F.; Fuggle, J.C.; Bennett, P.A.; Zolnierek, Z. Electronic structure of Ni and Pd alloys. II. X-ray photoelectron core-level spectra. Phys. Rev. B 1983, 27, 2179–2193. [Google Scholar] [CrossRef]
- Ertl, G.; Hierl, R.; Knozinger, H.; Thiele, N.; Urbach, H.P. XPS Study of Copper Aluminate Catalysts. Appl. Surf. Sci. 1980, 5, 49–64. [Google Scholar] [CrossRef]
- Mansour, A.N. Characterization of NiO by XPS. Surf. Sci. Spectra 1994, 3, 231–238. [Google Scholar] [CrossRef]
- Mansour, A.N. Nickel Monochromated Al K α XPS Spectra from the Physical Electronics Model 5400 Spectrometer. Surf. Sci. Spectra 1994, 3, 221–230. [Google Scholar] [CrossRef]
- Fan, L.; Liu, P.F.; Yan, X.; Gu, L.; Yang, Z.Z.; Yang, H.G.; Qiu, S.; Yao, X. Atomically isolated nickel species anchored on graphitized carbon for efficient hydrogen evolution electrocatalysis. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Pu, Z.; Zhang, C.; Amiinu, I.S.; Li, W.; Wu, L.; Mu, S. General strategy for the synthesis of transition-metal phosphide/N-Doped carbon frameworks for hydrogen and oxygen evolution. ACS Appl. Mater. Interfaces 2017, 9, 16187–16193. [Google Scholar] [CrossRef]
- Narayanan, S. X.p.s. studies on the reduction of nickel mordenite. Zeolites 1984, 4, 231–234. [Google Scholar] [CrossRef]
- Roberts, M.W.; Smart, S.T.C. The defect structure of nickel oxide surfaces as revealed by photoelectron spectroscopy. J. Chem. Soc. Faraday Trans. 1 1984, 80, 2957–2968. [Google Scholar] [CrossRef]
- Petrović, S.; Peruško, D.; Kovač, J.; Siketić, Z.; Radović-Bogdanović, I.; Gaković, B.; Radak, B.; Trtica, M. Laser-induced surface oxidation of (Ni/Ti)/Si system with picosecond laser pulses. Mater. Chem. Phys. 2014, 143, 530–535. [Google Scholar] [CrossRef]
- Mayer, B.; Uhlenbrock, S.; Neumann, M. XPS satellites in transition metal oxides. J. Electron. Spectrosc. 1996, 81, 63–67. [Google Scholar] [CrossRef]
- Zucchi, F.; Fonsati, M.; Trabanelli, G. Corrosion and corrosion inhibition of nickel in HCIO4 solutions using the EQCM technique. J. Appl. Electrochem. 1999, 29, 347–353. [Google Scholar] [CrossRef]
- Vanrenterghem, B.; Papaderakis, A.; Sotiropoulos, S.; Tsiplakides, D.; Balomenou, S.; Bals, S.; Breugelmans, T. The reduction of benzylbromide at Ag-Ni deposits prepared by galvanic replacement. Electrochim. Acta 2016, 196, 756–768. [Google Scholar] [CrossRef]
- Fierro, S.; Kapałka, A.; Comninellis, C. Electrochemical comparison between IrO2 prepared by thermal treatment of iridium metal and IrO2 prepared by thermal decomposition of H2IrCl6 solution. Electrochem. Commun. 2010, 12, 172–174. [Google Scholar] [CrossRef]
- Ouattara, L.; Fierro, S.; Frey, O.; Koudelka, M.; Comninellis, C. Electrochemical comparison of IrO2 prepared by anodic oxidation of pure iridium and IrO2 prepared by thermal decomposition of H2IrCl6 precursor solution. J. Appl. Electrochem. 2009, 39, 1361–1367. [Google Scholar] [CrossRef]
- Pavlovic, Z.; Ranjan, C.; Gao, Q.; Van Gastel, M.; Schlögl, R. Probing the structure of a water-Oxidizing anodic iridium oxide catalyst using raman spectroscopy. ACS Catal. 2016, 6, 8098–8105. [Google Scholar] [CrossRef]
- Papaderakis, A.; Tsiplakides, D.; Balomenou, S.; Sotiropoulos, S. Electrochemical impedance studies of IrO2 catalysts for oxygen evolution. J. Electroanal. Chem. 2015, 757, 216–224. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2016, 4, 3800–3821. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Tazawa, T.; Muroi, N.; Sato, E.-I. New types of anodes for the oxygen evolution reaction in acidic solution IrO2/TiO2/Ti. J. Electrochem. Soc. 1986, 133, 2257–2262. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Kokkinidis, G.; Sotiropoulos, S. A rotating disc electrode study of oxygen reduction at platinised nickel and cobalt coatings. J. Solid State Electr. 2010, 14, 175–184. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrodes | Ir/% at | Ni/% at |
|---|---|---|
| “27 mC” as prepared | 77.1 (±1.3) | 22.9 (±1.3) |
| “27 mC” anodized | 87.8 (±1.0) | 12.2 (±1.0) |
| “75 mC” as prepared | 5.3 (±1.8) | 94.7 (±1.8) |
| “75 mC” anodized | 32.9 (±7.2) | 67.1 (±7.2) |
| Sputter-Etch Depth/nm | Ir(total)/% at | Ni(total)/% at | Ir(0)/Ir(total) | Ni(0)/Ni(total) |
|---|---|---|---|---|
| 0 | 93.18 | 6.82 | 19.7 | n/a (very poor Ni signal) |
| 1 | 92.68 | 7.32 | 61.9 | 43.36 |
| 3 | 93.35 | 6.65 | 68.6 | 58.14 |
| Sputter-Etch Depth/nm | Ir(total)/% at | Ni(total)/% at | Ir(0)/Ir(total) | Ni(0)/Ni(total) |
|---|---|---|---|---|
| 0 | 48.34 | 51.66 | 13.6 | 0 |
| 1 | 70.67 | 29.33 | 54.4 | 75.7 |
| 3 | 62.4 | 37.6 | 65.3 | 60.3 |
| Electrode | qIrO2/mC cm−2 (before OER) | qIrO2/mC cm−2 (after OER) |
|---|---|---|
| “27 mC” | 2.82 | 5.17 |
| “50 mC” | 4.77 | 5.40 |
| “75 mC” | 4.18 | 6.76 |
| Catalyst | j = 10 mA·cm−2 | j = 0.5 mA mC−1 | η = 300 mV | |
|---|---|---|---|---|
| η/mV | η/mV | j/mA cm−2 | j/mA mC−1 | |
| Electrochemically anodized IrO2-Ni film via galvanic deposition (707 mC cm−2 electrodeposited Ni) [7] | 360 | 354 | 1 | 0.05 |
| Thermally prepared mixed Ir-Ni oxides (varied Ni content) [17] | 200–370 | 290–315 | 1–7 | 0.33–0.85 |
| Electrochemically anodized bulk Ir (300 cycles) [56] | 390 | 301 | 9.7 | 0.47 |
| This work (varied Ni content) | 299–307 | 253–270 | 9–11 | 1.8–3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touni, A.; Papaderakis, A.; Karfaridis, D.; Vourlias, G.; Sotiropoulos, S. Oxygen Evolution Reaction at IrO2/Ir(Ni) Film Electrodes Prepared by Galvanic Replacement and Anodization: Effect of Precursor Ni Film Thickness. Molecules 2019, 24, 2095. https://doi.org/10.3390/molecules24112095
Touni A, Papaderakis A, Karfaridis D, Vourlias G, Sotiropoulos S. Oxygen Evolution Reaction at IrO2/Ir(Ni) Film Electrodes Prepared by Galvanic Replacement and Anodization: Effect of Precursor Ni Film Thickness. Molecules. 2019; 24(11):2095. https://doi.org/10.3390/molecules24112095
Chicago/Turabian StyleTouni, Aikaterini, Athanasios Papaderakis, Dimitrios Karfaridis, Georgios Vourlias, and Sotiris Sotiropoulos. 2019. "Oxygen Evolution Reaction at IrO2/Ir(Ni) Film Electrodes Prepared by Galvanic Replacement and Anodization: Effect of Precursor Ni Film Thickness" Molecules 24, no. 11: 2095. https://doi.org/10.3390/molecules24112095