
Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors


Abstract
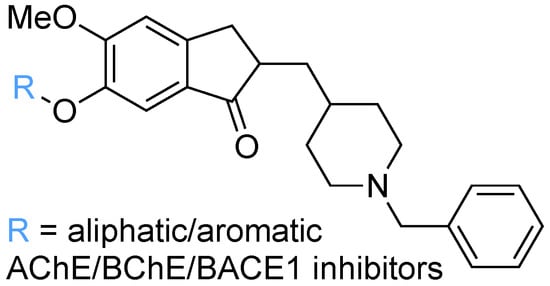
:
1. Introduction
2. Results and Discussion
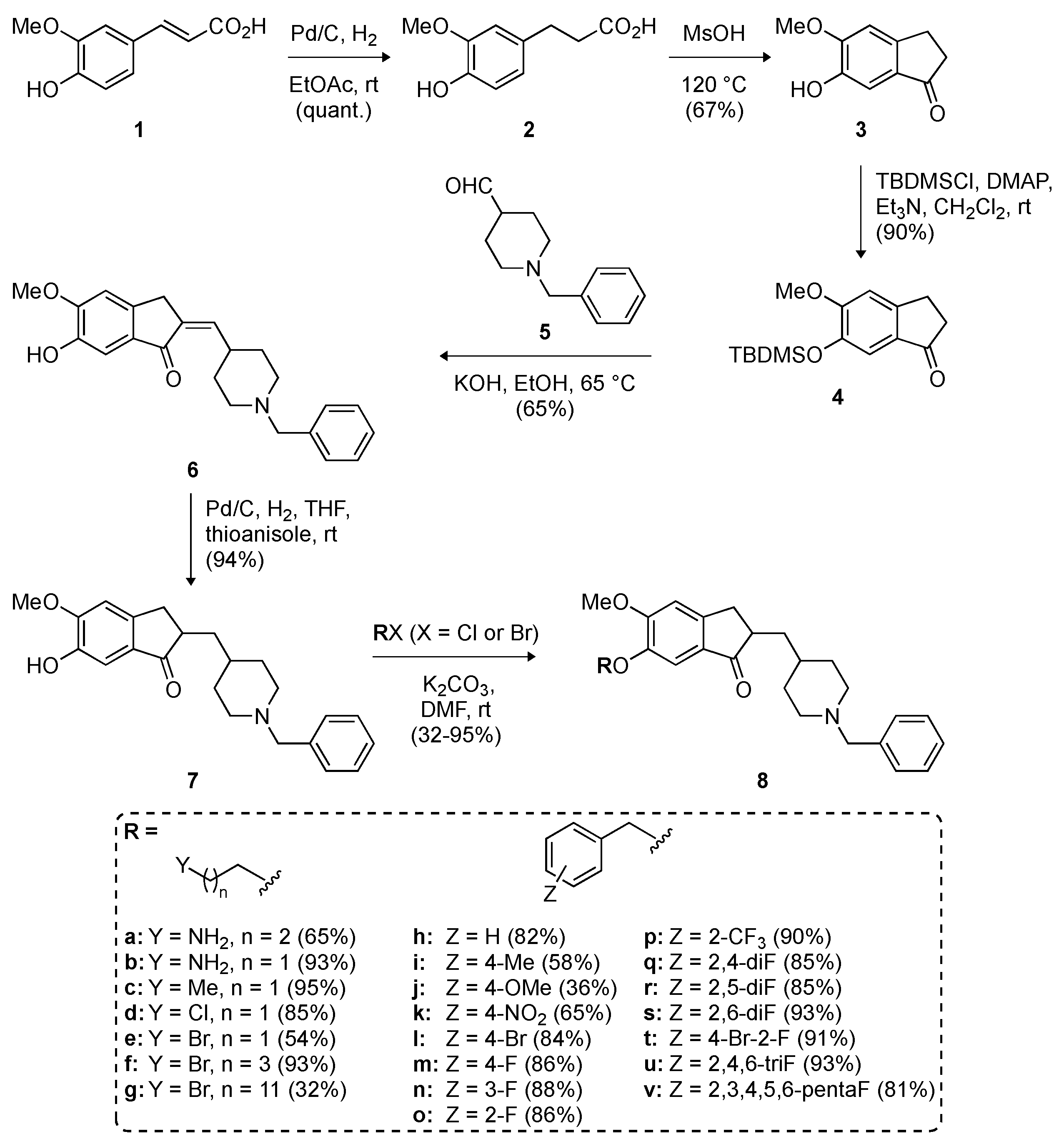
2.1. Chemistry
2.2. Cholinesterase Inhibition
2.2.1. AChE Inhibition
2.2.2. EfBChE Inhibition
2.3. BACE1 Inhibition
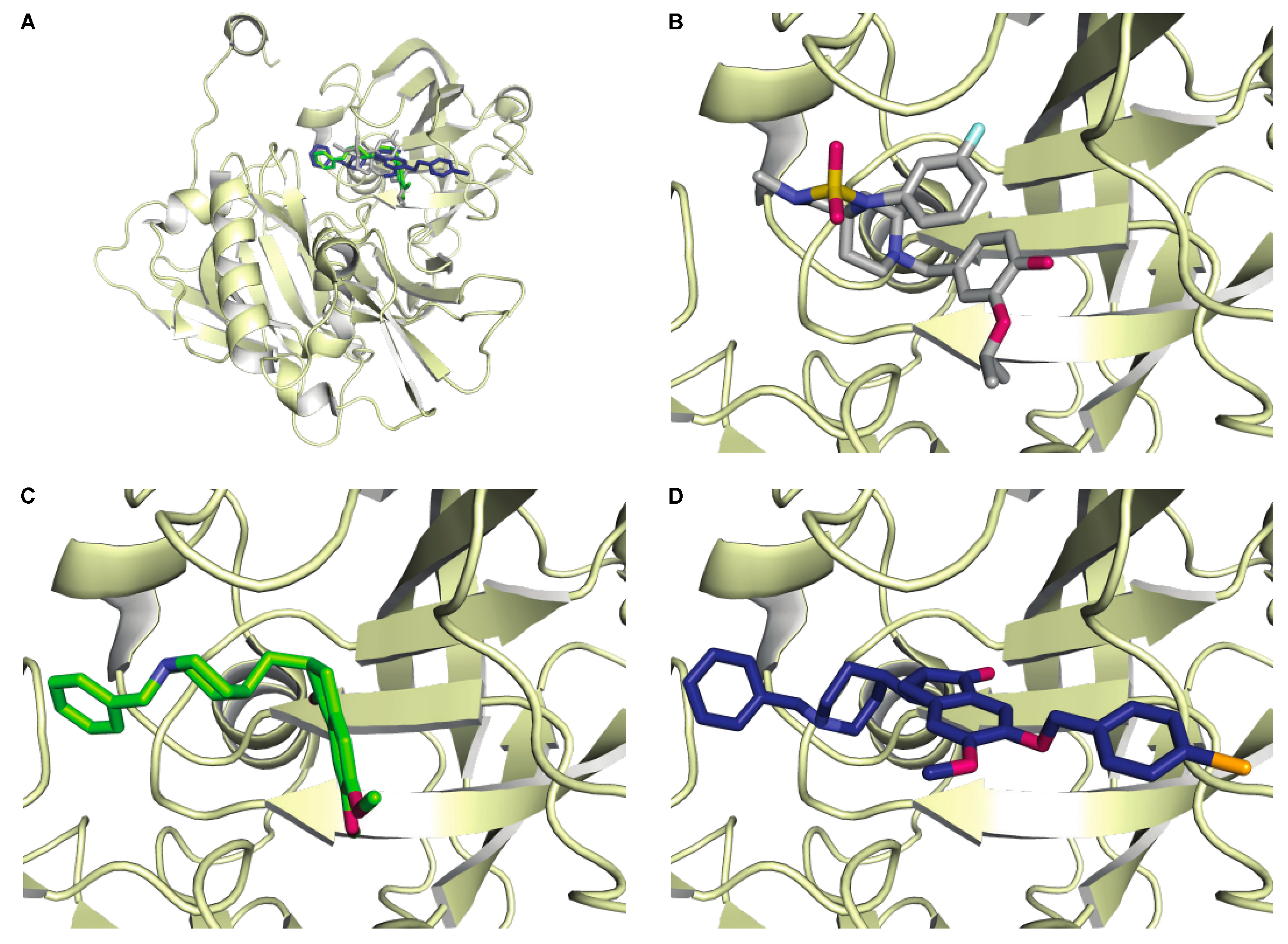
2.4. BACE1 Modeling
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Compounds 2–8v
3.2.1. 3-(4-Hydroxy-3-methoxyphenyl)propanoic acid (2)
3.2.2. 6-Hydroxy-5-methoxy-2,3-dihydroinden-1-one (3)
3.2.3. 6-[tert-Butyl(dimethyl)silyl]oxy-5-methoxy-2,3-dihydroinden-1-one (4)
3.2.4. (E)-2-[(1-Benzylpiperidin-4-yl)methylene]-6-hydroxy-5-methoxy-2,3-dihydroinden-1-one (6)
3.2.5. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-hydroxy-5-methoxy-2,3-dihydroinden-1-one (7)
3.2.6. tert-Butyl N-(3-chloropropyl)carbamate (Boc-protected 3-chloropropylamine).
3.2.7. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(3-tert-butyl-N-propylcarbamate)oxy]-5-methoxy-2,3-dihydroinden-1-one (Boc-protected compound 8a)
3.2.8. 6-[(3-Aminopropyl)oxy]-2-[(1-benzylpiperidin-4-yl)methyl]-5-methoxy-2,3-dihydroinden-1-one (8a)
3.2.9. tert-Butyl N-(2-chloroethyl)carbamate.
3.2.10. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(3-tert-butyl-N-ethylcarbamate)oxy]-5-methoxy-2,3-dihydroinden-1-one (Boc-protected compound 8b).
3.2.11. 6-[(3-Aminoethyl)oxy]-2-[(1-benzylpiperidin-4-yl)methyl]-5-methoxy-2,3-dihydroinden-1-one (8b)
3.2.12. 2-[(1-Benzylpiperidin-4-yl)methyl]-5-methoxy-6-propoxy-2,3-dihydroinden-1-one (8c).
3.2.13. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(chloroethyl)oxy]-5-methoxy-2,3-dihydroinden-1-one (8d)
3.2.14. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(bromoethyl)oxy]-5-methoxy-2,3-dihydroinden-1-one (8e)
3.2.15. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(bromobutyl)oxy]-5-methoxy-2,3-dihydroinden-1-one (8f)
3.2.16. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(bromododecyl)oxy]-5-methoxy-2,3-dihydroinden-1-one (8g)
3.2.17. 6-[(Benzyl)oxy-2-[(1-benzylpiperidin-4-yl)methyl]-5-methoxy-2,3-dihydroinden-1-one (8h)
3.2.18. 2-[(1-Benzylpiperidin-4-yl)methyl]-5-methoxy-6-[(4-methylbenzyl)oxy-2,3-dihydroinden-1-one (8i)
3.2.19. 2-[(1-Benzylpiperidin-4-yl)methyl]-5-methoxy-6-[(4-methoxybenzyl)oxy-2,3-dihydroinden-1-one (8j)
3.2.20. 2-[(1-Benzylpiperidin-4-yl)methyl]-5-methoxy-6-[(4-nitrobenzyl)oxy-2,3-dihydroinden-1-one (8k).
3.2.21. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(4-bromobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8l)
3.2.22. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(4-fluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8m)
3.2.23. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(3-fluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8n)
3.2.24. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2-fluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8o)
3.2.25. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2-trifluoromethylbenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8p)
3.2.26. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2,4-difluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8q)
3.2.27. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2,5-difluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8r)
3.2.28. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2,6-difluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8s)
3.2.29. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(4-bromo-2-fluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8t)
3.2.30. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2,4,6-trifluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8u)
3.2.31. 2-[(1-Benzylpiperidin-4-yl)methyl]-6-[(2,3,4,5,6-pentafluorobenzyl)oxy-5-methoxy-2,3-dihydroinden-1-one (8v)
3.3. In Vitro Cholinesterase (ChE) Inhibition Assays
3.4. BACE1 Inhibition
3.5. Molecular Docking of Donepezil and Compound 8l with BACE1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| APP | amyloid precursor protein |
| BACE | β-secretase |
| ChE | cholinesterase |
| EeAChE | acetylcholinesterase (from Electrophorus electricus) |
| EfBChE | butyrylcholinesterase (from Equus ferus) |
| HsAChE | acetylcholinesterase (from Homo sapiens) |
| IC50 | half maximal inhibitory concentration |
| KOH | potassium hydroxide |
| MsOH | methanesulfonic acid |
| TBDMS | tert-butyldimethylsilyl |
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- World Health Organization. Global health estimates 2016: Deaths by cause, age, sex, by country, and by region, Geneva, Switzerland. 2018. [Google Scholar]
- Schmidt, C.; Wolff, M.; Weitz, M.; Bartlau, T.; Korth, C.; Zerr, I. Rapidly progressive Alzheimer disease. Arch. Neurol. 2011, 68, 1124–1130. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martinez-Murillo, R.; Carreiras, M.C.; Ismaili, L. Ass234, as a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L., 3rd; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Shah, A.A.; Dar, T.A.; Dar, P.A.; Ganie, S.A.; Kamal, M.A. A current perspective on the inhibition of cholinesterase by natural and synthetic inhibitors. Curr. Drug Metab. 2017, 18, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Unzeta, M.; Esteban, G.; Bolea, I.; Fogel, W.A.; Ramsay, R.R.; Youdim, M.B.; Tipton, K.F.; Marco-Contelles, J. Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front. Neurosci. 2016, 10, 205. [Google Scholar] [CrossRef]
- Eckroat, T.J.; Mayhoub, A.S.; Garneau-Tsodikova, S. Amyloid-β probes: Review of structure-activity and brain-kinetics relationships. Beilstein J. Org. Chem. 2013, 9, 1012–1044. [Google Scholar] [CrossRef] [PubMed]
- Coulson, E.J.; Paliga, K.; Beyreuther, K.; Masters, C.L. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem. Int. 2000, 36, 175–184. [Google Scholar] [CrossRef]
- Dobrowolska Zakaria, J.A.; Vassar, R.J. A promising, novel, and unique BACE1 inhibitor emerges in the quest to prevent Alzheimer’s disease. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Eckroat, T.J.; Green, K.D.; Reed, R.A.; Bornstein, J.J.; Garneau-Tsodikova, S. Investigation of the role of linker moieties in bifunctional tacrine hybrids. Bioorg. Med. Chem. 2013, 21, 3614–3623. [Google Scholar] [CrossRef] [PubMed]
- Kochi, A.; Eckroat, T.J.; Green, K.D.; Mayhoub, A.S.; Lim, M.H.; Garneau-Tsodikova, S. A novel hybrid of 6-chlorotacrine and metal-amyloid-β modulator for inhibition of acetylcholine and metal-induced amyloid-β aggregation. Chem. Sci. 2013, 4, 4137–4145. [Google Scholar] [CrossRef]
- Bornstein, J.J.; Eckroat, T.J.; Houghton, J.L.; Jones, C.K.; Green, K.D.; Garneau-Tsodikova, S. Tacrine-mefenamic acid hybrids for inhibition of acetycholinesterase. Med. Chem. Comm. 2011, 2, 406–412. [Google Scholar] [CrossRef]
- Fosso, M.Y.; McCarty, K.; Head, E.; Garneau-Tsodikova, S.; LeVine, H., 3rd. Differential effects of structural modifications on the competition of chalcones for the PIB amyloid imaging ligand-binding site in Alzheimer’s disease brain and synthetic abeta fibrils. ACS Chem. Neurosci. 2016, 7, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Fosso, M.Y.; LeVine, H., 3rd; Green, K.D.; Tsodikov, O.V.; Garneau-Tsodikova, S. Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones. Org. Biomol. Chem. 2015, 13, 9418–9426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xiao, K.; Ma, L.; Xiong, B.; Fu, Y.; Yu, H.; Wang, W.; Wang, X.; Hu, D.; Peng, H.; et al. Design, synthesis and biological evaluation of novel dual inhibitors of acetylcholinesterase and β-secretase. Bioorg. Med. Chem. 2009, 17, 1600–1613. [Google Scholar] [CrossRef] [PubMed]
- Panek, D.; Wieckowska, A.; Pasieka, A.; Godyn, J.; Jonczyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, synthesis, and biological evaluation of 2-(benzylamino-2-hydroxyalkyl)isoindoline-1,3-diones derivatives as potential disease-modifying multifunctional anti-Alzheimer’s agents. Molecules 2018, 23, 347. [Google Scholar] [CrossRef]
- Zhao, X.J.; Gong, D.M.; Jiang, Y.R.; Guo, D.; Zhu, Y.; Deng, Y.C. Multipotent AChE and BACE-1 inhibitors for the treatment of Alzheimer’s disease: Design, synthesis and bio-analysis of 7-amino-1,4-dihydro-2h-isoquilin-3-one derivates. Eur. J. Med. Chem. 2017, 138, 738–747. [Google Scholar] [CrossRef]
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, synthesis, and evaluation of donepezil-like compounds as AChE and BACE-1 inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475. [Google Scholar] [CrossRef]
- Meng, F.C.; Mao, F.; Shan, W.J.; Qin, F.; Huang, L.; Li, X.S. Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents. Bioorg. Med. Chem. Lett. 2012, 22, 4462–4466. [Google Scholar] [CrossRef]
- Hu, J.; Yan, J.; Chen, J.; Pang, Y.; Huang, L.; Li, X. Synthesis, biological evaluation and mechanism study of a class of benzylideneindanone derivatives as novel anticancer agents. MedChemComm 2015, 6, 1318–1327. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Nochi, S.; Asakawa, N.; Sato, T. Kinetic study on the inhibition of acetylcholinesterase by 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl]methylpiperidine hydrochloride (e2020). Biol. Pharm. Bull. 1995, 18, 1145–1147. [Google Scholar] [CrossRef]
- Saxena, A.; Redman, A.M.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Biochemistry 1997, 36, 14642–14651. [Google Scholar] [CrossRef]
- Brodney, M.A.; Barreiro, G.; Ogilvie, K.; Hajos-Korcsok, E.; Murray, J.; Vajdos, F.; Ambroise, C.; Christoffersen, C.; Fisher, K.; Lanyon, L.; et al. Spirocyclic sulfamides as β-secretase 1 (BACE-1) inhibitors for the treatment of Alzheimer’s disease: Utilization of structure based drug design, watermap, and CNS penetration studies to identify centrally efficacious inhibitors. J. Med. Chem. 2012, 55, 9224–9239. [Google Scholar] [CrossRef] [PubMed]
- Allais, F.; Pla, T.J.L.; Ducrot, P.-H. An access to chiral b-benzyl-γ-butyrolactones and its application to the synthesis of enantiopure (+)-secoisolariciresinol, (−)-secoisolariciresinol, and (−)-enterolactone. Synthesis 2011, 9, 1456–1464. [Google Scholar] [CrossRef]
- Percec, V.; Wilson, D.A.; Leowanawat, P.; Wilson, C.J.; Hughes, A.D.; Kaucher, M.S.; Hammer, D.A.; Levine, D.H.; Kim, A.J.; Bates, F.S.; et al. Self-assembly of Janus dendrimers into uniform dendrimersomes and other complex architectures. Science 2010, 328, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, C.L.; Dobri, N.; Freeman, E.E.; Conlon, M.P.; Chen, P.; Stafford, D.G.; Schwarz, D.M.; Golden, K.C.; Zhu, L.; Kitchen, D.B.; et al. Design, synthesis, and evaluation of nonretinoid retinol binding protein 4 antagonists for the potential treatment of atrophic age-related macular degeneration and stargardt disease. J. Med. Chem. 2014, 57, 7731–7757. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Swissdock, a protein-small molecule docking web service based on eadock dss. Nucl. Acids Res. 2011, 39, 270–277. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the charmm force field with eadock dss. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds synthesized are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Cpd | EeAChE | EfBChE | SI |
|---|---|---|---|
| Donepezil HCl | 0.12 ± 0.01 | 2.0 ± 0.1 | 17 |
| 7 | 0.41 ± 0.05 | 4.3 ± 0.4 | 11 |
| 8a | 0.054 ± 0.003 | 0.57 ± 0.04 | 11 |
| 8b | 0.021 ± 0.003 | 0.48 ± 0.03 | 23 |
| 8c | 0.14 ± 0.02 | 2.1 ± 0.3 | 15 |
| 8d | 0.059 ± 0.004 | 1.3 ± 0.1 | 22 |
| 8e | 0.044 ± 0.003 | 1.3 ± 0.2 | 30 |
| 8f | 0.061 ± 0.007 | 1.3 ± 0.2 | 21 |
| 8g | 0.79 ± 0.28 | 5.2 ± 1.6 | 6.6 |
| 8h | 0.13 ± 0.01 | 0.70 ± 0.05 | 5.4 |
| 8i | 0.23 ± 0.03 | 1.0 ± 0.2 | 4.3 |
| 8j | 0.13 ± 0.01 | 0.67 ± 0.17 | 5.2 |
| 8k | 0.13 ± 0.02 | 0.46 ± 0.06 | 3.5 |
| 8l | 0.071 ± 0.015 | 0.72 ± 0.10 | 10 |
| 8m | 0.081 ± 0.005 | 0.57 ± 0.10 | 7.0 |
| 8n | 0.16 ± 0.02 | 0.96 ± 0.15 | 6.0 |
| 8o | 0.12 ± 0.02 | 0.76 ± 0.12 | 6.3 |
| 8p | 0.032 ± 0.010 | 0.25 ± 0.08 | 7.8 |
| 8q | 0.11 ± 0.01 | 0.48 ± 0.08 | 4.4 |
| 8r | 0.090 ± 0.009 | 0.60 ± 0.15 | 6.7 |
| 8s | 0.016 ± 0.001 | 0.44 ± 0.05 | 28 |
| 8t | 0.054 ± 0.007 | 0.37 ± 0.05 | 6.9 |
| 8u | 0.027 ± 0.004 | 0.20 ± 0.03 | 7.4 |
| 8v | 0.17 ± 0.02 | 0.11 ± 0.01 | 0.69 |
| Cpd | IC50 (μM) | SI a |
|---|---|---|
| Donepezil HCl | 0.032 ± 0.011 | 3.8 |
| 8t | 0.0018 ± 0.0006 | 30 |
| Cpd | IC50 (μM) |
|---|---|
| Donepezil HCl | 1.5 ± 0.3 |
| 7 | -- |
| 8a | 95 ± 12 |
| 8b | ~100 |
| 8c | 6.1 ± 0.1 |
| 8d | ~100 |
| 8e | 7.9 ± 0.9 |
| 8f | 7.9 ± 2.4 |
| 8g | -- |
| 8h | 58 ± 1 |
| 8i | 58 ± 2 |
| 8j | -- |
| 8k | -- |
| 8l | 3.4 ± 0.1 |
| 8m | ~100 |
| 8n | 12 ± 3 |
| 8o | 21 ± 4 |
| 8p | 34 ± 9 |
| 8q | 37 ± 5 |
| 8r | ~100 |
| 8s | 30 ± 8 |
| 8t | 169 ± 2 |
| 8u | 91 ± 14 |
| 8v | 29 ± 6 |
| BACE1 inhibitor IV | 0.63 ± 0.18 nM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, K.D.; Fosso, M.Y.; Garneau-Tsodikova, S. Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors. Molecules 2018, 23, 3252. https://doi.org/10.3390/molecules23123252
Green KD, Fosso MY, Garneau-Tsodikova S. Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors. Molecules. 2018; 23(12):3252. https://doi.org/10.3390/molecules23123252
Chicago/Turabian StyleGreen, Keith D., Marina Y. Fosso, and Sylvie Garneau-Tsodikova. 2018. "Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors" Molecules 23, no. 12: 3252. https://doi.org/10.3390/molecules23123252