
Crystal Structures and Cytotoxicity of ent-Kaurane-Type Diterpenoids from Two Aspilia Species

 , , , , , and
, , , , , and
Abstract
:1. Introduction
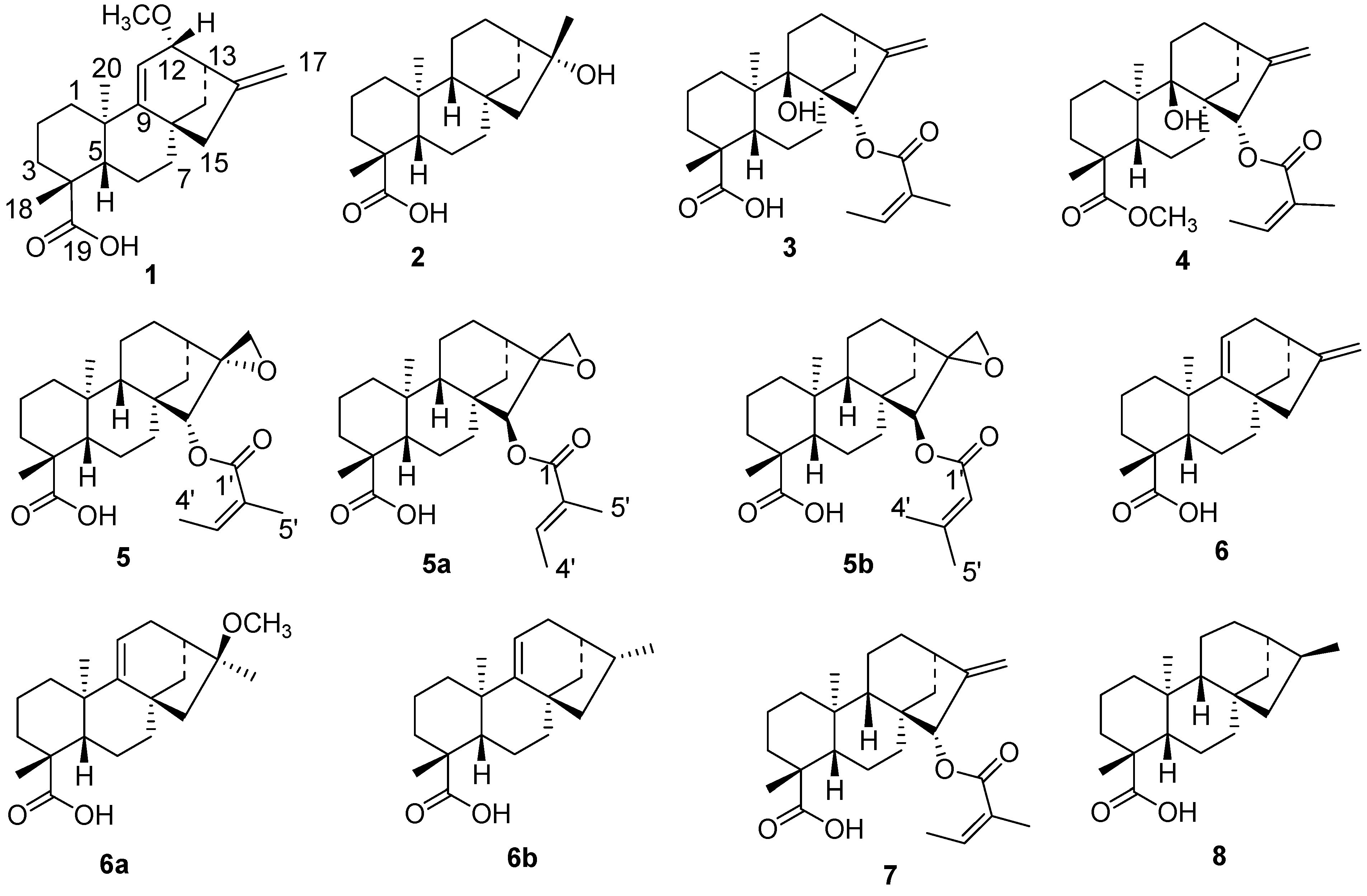
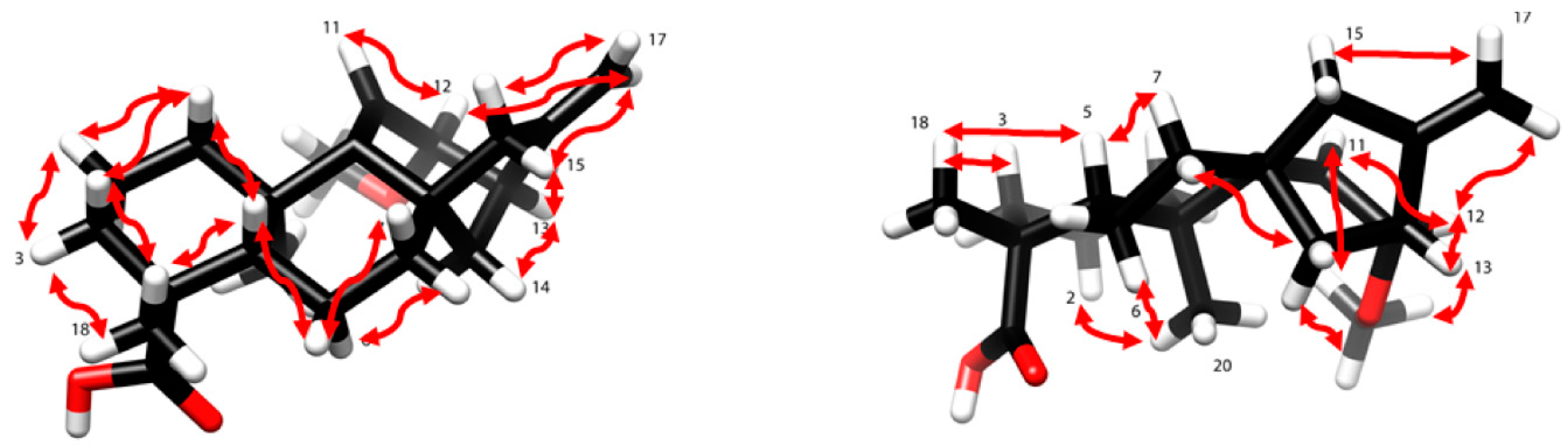
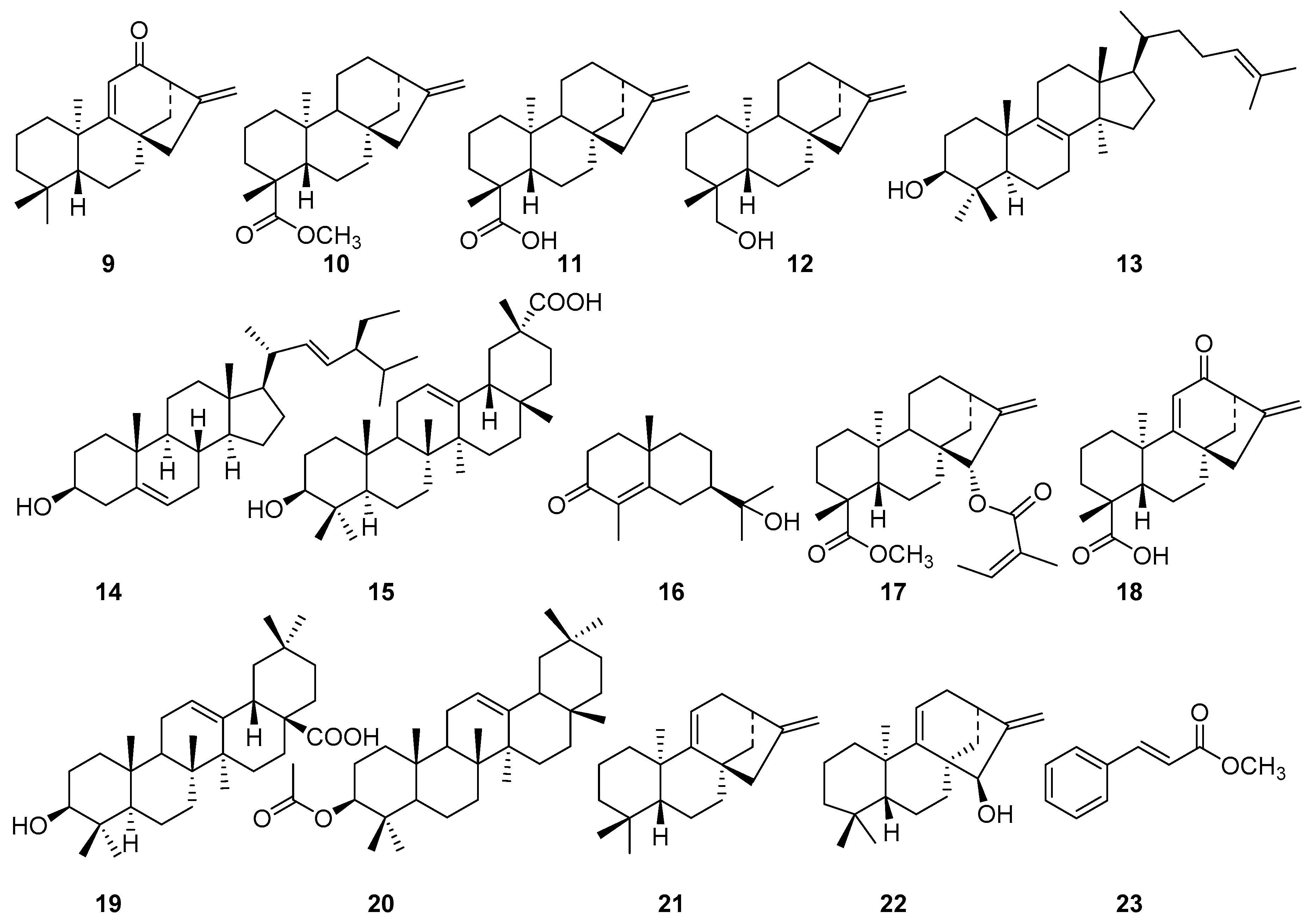
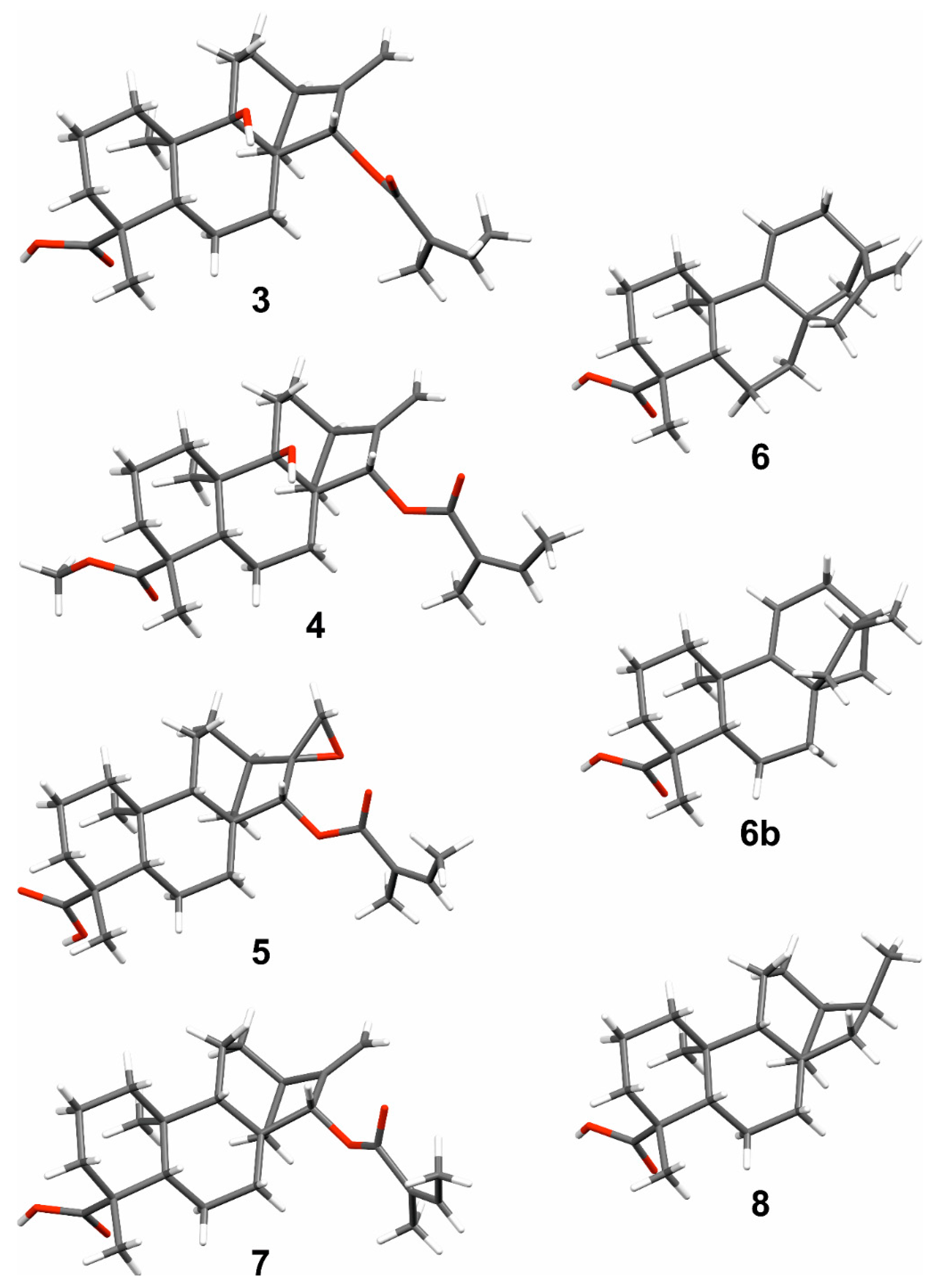
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. X-ray Diffraction Analyses
3.3. Plant Materials
3.4. Extraction, Isolation and Derivatization
3.5. Cell Culture
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faleye, F.J.; Ogundaini, O.A. Evaluation of antioxidant and antimicrobial activities of two isolates from Aspilia africana (Pers) C.D. Adams. Int. Res. J. Pharm. 2012, 655, 135–138. [Google Scholar]
- Bohm, B.A.; Stuessy, T.F. Flavonoids of the Sunflower Family (Asteraceae); Springer Science & Business Media: Wien, Austria, 2001; pp. 116–119. ISBN 3-211-83479-6. [Google Scholar]
- Alvarenga, S.A.; Ferreira, M.J.; Rodrigues, G.V.; Emerenciano, V.P. A general survey and some taxonomic implications of diterpenes in the Asteraceae. Bot. J. Linnean. Soc. 2005, 147, 291–308. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Muñoz, A.; Muñoz-Schick, M. Classification, diversity, and distribution of Chilean Asteraceae: Implications for biogeography and conservation. Divers. Distrib. 2007, 13, 818–828. [Google Scholar] [CrossRef]
- Souza, J.M.; Chang, M.R.; Brito, D.Z.; Farias, K.S.; Damasceno-Junior, G.A.; Turatti, I.C.; Lopes, N.P.; Santos, E.A.; Carollo, C.A. Antimicrobial activity of Aspilia latissima (Asteraceae). Braz. J. Microbiol. 2015, 46, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Seaman, F.; Bohlmann, F.; Zdero, C.; Mabry, T.J. Diterpenes of Flowering Plants—Compositae (Asteraceae); Springer: New York, NY, USA, 2012. [Google Scholar]
- Kuria, J.M. Efficacy of Aspilia pluriseta Schweinf in Cutaneous Wound Healing in a Mouse Model. Ph.D. Thesis, University of Nairobi, Nairobi, Kenya, 2014. [Google Scholar]
- Sebisubi, F.M.; Odyek, O.; Anokbonggo, W.W.; Ogwal-Okeng, J.; Carcache-Blanco, E.J.; Ma, C.; Orjala, J.; Tan, G.T. Antimalarial activity of Aspilia pluriseta, a medicinal plant from Uganda. Planta Med. 2010, 76, 1870–1873. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.A.; Huang, D.Q.; Rodriguez, E. Aspilia mossambicensis: In Vitro Propagation and Production of Antibiotic Polyacetylenes by Root Cultures. In Medicinal and Aromatic Plants V; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 54–63. [Google Scholar]
- Musyimi, D.; Ogur, J.; Muema, P. Phytochemical compounds and antimicrobial activity of extracts of Aspilia plant (Aspilia mossambicensis) Oliv. wild. Int. J. Botany 2008, 4, 56–61. [Google Scholar] [CrossRef]
- Page, J.E.; Balza, F.; Nishida, T.; Towers, G.N. Biologically active diterpenes from Aspilia mossambicensis, a Chimpanzee medicinal plant. Phytochemistry 1992, 31, 3437–3439. [Google Scholar] [CrossRef]
- Page, J.E.; Huffman, M.; Smith, V.; Towers, G. Chemical basis for Aspilia leaf-swallowing by Chimpanzees: A reanalysis. J. Chem. Ecol. 1997, 23, 2211–2226. [Google Scholar] [CrossRef]
- Ahmed, M.; Jakupovic, J.; Castro, V. Kaurene derivatives from Lasianthea fruticosa, revision of stereochemistry of related compounds. Phytochemistry 1991, 30, 1712–1714. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, Y.; Yang, D.; Hao, X.; Li, S. Two new kaurane-type diterpenoids from Wedelia chinensis (Osbeck.) Merr. Nat. Prod. Res. 2017, 31, 2531–2536. [Google Scholar] [CrossRef]
- Li, S.F.; Ding, J.Y.; Li, Y.T.; Hao, X.J.; Li, S.L. Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc. Molecules 2016, 21, 457. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Takaishi, Y.; Momota, H.; Ohmoto, Y.; Taki, T.; Jia, Y.; Li, D. Immunodepressive diterpenoids from Tripterygium wilfordii. J. Nat. Prod. 1999, 62, 1522–1525. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Jin, W.Y.; Zhang, X.; Hung, T.M.; Song, K.S.; Seong, Y.H.; Bae, K. Cytotoxic and COX-2 inhibitory constituents from the aerial parts of Aralia cordata. Arch. Pharm. Res. 2006, 29, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Tirapelli, C.R.; de Oliveira, A.M.; Murillo, R.; Castro, V.; Merfort, I. Studies of ent-kaurane diterpenes from Oyedaea verbesinoides for their inhibitory activity on vascular smooth muscle contraction. Phytochemistry 2003, 63, 391–396. [Google Scholar] [CrossRef]
- Delgado, G.; Vivar, A.R.D. Ent-kaurenoid methyl esters from Viguiera stenoloba, structural revision of stenolobin and its biomimetic conversion to zoapatlin. Chem. Lett. 1984, 13, 1237–1240. [Google Scholar] [CrossRef]
- Wafo, P.; Kamdem, R.S.; Ali, Z.; Anjum, S.; Begum, A.; Oluyemisi, O.O.; Khan, S.N.; Ngadjui, B.T.; Etoa, X.F.; Choudhary, M.I. Kaurane-type diterpenoids from Chromoleana odorata, their X-ray diffraction studies and potent α-glucosidase inhibition of 16-kauren-19-oic acid. Fitoterapia 2011, 82, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, A.; Pérez-Castorena, A.L.; Meléndez-Aguirre, M.; Ávila, J.G.; García-Bores, A.M.; Villaseñor, J.L.; Romod, V.A. Chemical composition and antimicrobial activity of Ageratina deltoidea. Chem. Biodivers. 2018, 15, e1700529. [Google Scholar] [CrossRef]
- El Marsni, Z.; Torres, A.; Varela, R.M.; Molinillo, J.M.; Casas, L.; Mantell, C.; Martinez, E.J.D.L.O.; Macias, F.A. Isolation of bioactive compounds from sunflower leaves (Helianthus annuus L.) extracted with supercritical carbon dioxide. J. Agric. Food Chem. 2015, 63, 6410–6421. [Google Scholar] [CrossRef]
- Pinto, A.C.; Prado, S.K.D.; Pinchin, R. Two kaurenes from Vellozia caput-ardeae. Phytochemistry 1981, 20, 520–521. [Google Scholar] [CrossRef]
- Aráoz, M.V.C.; Mercado, M.I.; Grau, A.; Catalán, C.A. Ent-kaurane derivatives from the root cortex of Yacon and other three Smallanthus species (Heliantheae, Asteraceae). Biochem. Syst. Ecol. 2010, 38, 1042–1048. [Google Scholar] [CrossRef]
- Chen, Q.; Lin, H.; Wu, X.; Song, H.; Zhu, X. Preparative separation of six terpenoids from Wedelia prostrata Hemsl. By two-step high-speed counter-current chromatography. J. Liq. Chromatogr. Relat. Technol. 2018, 41, 408–414. [Google Scholar] [CrossRef]
- Peña, A.; Alarcón, L.; Baptista, J.G.; Aparicio, R.; Villasmil, T.; Usubillaga, A. A phytochemical analysis of Espeletia nana Cuatrec. A midget Espeletiinae from Paramo Ortiz, Venezuela. Av. Quim. 2012, 7, 187–192. [Google Scholar]
- Dias, J.R.; Gao, H. 13C nuclear magnetic resonance data of lanosterol derivatives—profiling the steric topology of the steroid skeleton via substituent effects on its 13C-NMR. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2009, 74, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedula, V.S.P.; Prakash, I. Isolation of stigmasterol and β-sitosterol from the dichloromethane extract of Rubus suavissimus. Int. Curr. Pharm. J. 2012, 1, 239–242. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, B.; Liu, Z.; Jiang, Y.; Liu, Y.; Fu, L.; Wang, X.; Kuang, H. Cytotoxicity of triterpenes from green walnut husks of Juglans mandshurica maxim in HepG-2 cancer cells. Molecules 2015, 20, 19252–19262. [Google Scholar] [CrossRef] [PubMed]
- Reisch, J.; Hussain, R.A.; Krebs, B.; Dartmann, M. Natural product chemistry, part 100: The structure of carissone, C15H24O2. Monatsh. Chem. 1990, 121, 941–944. [Google Scholar] [CrossRef]
- Ohno, N.; Mabry, T.J.; Zabelt, V.; Watson, W.H. Tetrachyrin, a new rearranged kaurenoid lactone, and diterpene acids from Tetrachyron orizabaensis and Helianthus debilis. Phytochemistry 1979, 18, 1687–1689. [Google Scholar] [CrossRef]
- Huang, W.; Liang, Y.; Wang, J.; Li, G.; Wang, G.; Li, Y.; Chung, H.Y. Anti-angiogenic activity and mechanism of kaurane diterpenoids from Wedelia chinensis. Phytomedicine 2016, 23, 283–292. [Google Scholar] [CrossRef]
- Tincusi, B.M.; Jiménez, I.A.; Bazzocchi, I.L.; Moujir, L.M.; Mamani, Z.A.; Barroso, J.P.; Ravelo, A.G.; Hernandez, B.V. Antimicrobial terpenoids from the oleoresin of the peruvian medicinal plant Copaifera paupera. Planta Med. 2002, 68, 808–812. [Google Scholar] [CrossRef]
- Seebacher, W.; Simic, N.; Weis, R.; Saf, R.; Kunert, O. Complete assignments of 1H and 13C-NMR resonances of oleanolic acid, 18α-oleanolic acid, ursolic acid and their 11-oxo derivatives. Magn. Reson. Chem. 2003, 41, 636–638. [Google Scholar] [CrossRef]
- Faleye, F.J. Terpenoid constituents of Aspilia africana [Pers.] C.D. Adams leaves. Int. J. Pharm. Sci. Rev. Res. 2012, 13, 138–142. [Google Scholar]
- Ogihara, K.; Iraha, R.; Higa, M.; Yogi, S. Studies on constituents from the twigs of Messerschmidia argentea II. Bull. Coll. Sci., Univ. Ryukyus 1997, 64, 53–59. [Google Scholar]
- Zhang, H.; Wynne, G.; Mander, L.N. Synthesis of ent-9α, 15α-cyclokaurene from grandiflorenic acid. ARKIVOC 2001, 8, 40–58. [Google Scholar]
- Nagashima, F.; Kondoh, M.; Fujii, M.; Takaoka, S.; Watanabe, Y.; Asakawa, Y. Novel cytotoxic kaurane-type diterpenoids from the New Zealand liverwort Jungermannia species. Tetrahedron 2005, 61, 4531–4544. [Google Scholar] [CrossRef]
- Gao, L.; Xu, X.; Nan, H.; Yang, J.; Sun, G.; Wu, H.; Zhong, M. Isolation of cinnamic acid derivatives from the root of Rheum tanguticum Maxim. Ex balf. And its significance. J. Med. Plants Res. 2012, 6, 929–931. [Google Scholar]
- Reynolds, W.F.; Lough, A.J.; Sawyer, J.F.; Enriquez, R.G.; Ortiz, B.; Walls, F. Structure of (4α)-kaura-9(11),16-dien-18-oic acid (grandiflorenic acid), an active ingredient of the Mexican medicinal plant zoapatle. Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 1991, 47, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Banfi, D.; Patiny, L. www.nmrdb.org: Resurrecting and processing NMR spectra on-line. Chimia 2008, 62, 280–281. [Google Scholar] [CrossRef]
- CrysAlisPro, version 1.171.38.41q; Rigaku Oxford Diffraction: Yarnton, UK, 2015; Available online: https://www.rigaku.com/en/products/smc/crysalis (accessed on 11 November 2018).
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Boeck, P.; Sá, M.M.; Souza, B.S.D.; Cercená, R.; Escalante, A.M.; Zachino, S.A.; Cechinel, F.V.; Yunes, R.A. A simple synthesis of kaurenoic esters and other derivatives and evaluation of their antifungal activity. J. Brazil. Chem. Soc. 2005, 16, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.K.W.; Zhang, M.M.; Zhou, H.; Lam, K.Y.C.; Chan, P.L.; Law, C.K.M.; Yue, P.Y.K.; Liu, L. Saikosaponin-D enhances the anticancer potency of TNF-via overcoming its undesirable response of activating NF-kappa B signalling in cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pupier, M.; Nuzillard, J.M.; Wist, J.; Schlorer, N.E.; Kuhn, S.; Erdelyi, M.; Steinbeck, C.; Williams, A.J.; Butts, C.; Claridge, T.D.W.; et al. NMReDATA, a standard to report the NMR assignment and parameters of organic compounds. Magn. Reson. Chem. 2018, 56, 703–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC Lit. [13] * | δC | δH, mult. (J in Hz) | HMBC (2J, 3J) |
|---|---|---|---|---|
| 1 | 38.17 | 40.6 | 1.14 ddd (13.5, 9.5, 4.2) | C-2, C-3, C-10, C-20 |
| 1.90 ddd (13.5, 3.5, 1.4) | C-2, C-3, C-10, C-20 | |||
| 2 | 18.35 | 20.0 | 1.43 dddd (14.2, 9.5, 3.9, 3.5) | C-1, C-3, C-4, C-5, C-10 |
| 1.79 ddddd (14.2, 11.1, 4.2, 3.5, 1.4) | C-1, C-4, C-5 | |||
| 3 | 29.03 | 38.1 | 0.93 ddd (13.4, 11.1, 3.9) | C-1, C-2, C-4, C-18, C-19 |
| 2.08 ddd (13.4, 3.5, 3.5) | C-1, C-4, C-5, C-7 | |||
| 4 | 43.43 | 44.6 | ||
| 5 | 43.81 | 46.1 | 1.56 dd (11.1, 8.5) | C-4, C-7, C-9, C-10, C-18, C-19, C-20 |
| 6 | 20.07 | 18.3 | 1.82 dddd (14.2, 10.0, 8.5, 2.5) | C-3, C-4, C-5, C-7, C-10 |
| 2.43 dddd (14.2, 11.1, 9.5, 3.5) | C-4, C-5, C-8 | |||
| 7 | 40.60 | 28.9 | 1.42 ddd (13.8, 3.5, 2.5) | C-5, C-6, C-8, C-9, C-15 |
| 1.95 ddd (13.8, 10.0, 9.5) | C-6, C-8, C-9, C-14, C-15 | |||
| 8 | 44.66 | 43.4 | ||
| 9 | 160.28 | 160.2 | ||
| 10 | 38.94 | 38.9 | ||
| 11 | 115.42 | 115.3 | 5.30 dd (4.3, 1.4) | C-8, C-9, C-10, C-12, C-13, C-15, C-20 |
| 12 | 81.79 | 81.7 | 3.38 dd (4.3, 2.9) | C-9, C-11, C-13, C-16, C-20, OCH3-12 |
| 13 | 46.17 | 43.7 | 2.89 dd (2.9, 1.4) | C-10, C-11, C-12, C-15, C-16 |
| 14 | 40.60 | 40.5 | 1.31 dd (10.8, 4.3) | C-7, C-8, C-9, C-12, C-13, C-15 |
| 1.58 dd (10.8, 2.5) | C-9, C-12, C-13, C-15, C-16 | |||
| 15 | 47.17 | 47.1 | 2.08 dd (15.4, 4.3) | C-7, C-8, C-9, C-16, C-17 |
| 2.35 dd (15.4, 2.5) | C-7, C-9, C-13, C-14, C-16, C-17 | |||
| 16 | 153.00 | 152.9 | ||
| 17 | 108.12 | 108.1 | 4.84 dd (3.0, 1.6) | C-12, C-13, C-15, C-16 |
| 4.94 dd (3.0, 1.6) | C-12, C-13, C-15, C-16 | |||
| 18 | 28.22 | 28.2 | 1.17 s | C-3, C-4, C-5, C-8, C-19 |
| 19 | 182.98 | 183.2 | ||
| 20 | 23.41 | 23.4 | 1.01 s | C-1, C-5, C-9, C-10 |
| OCH3-12 | 56.53 | 56.5 | 3.34 s | C-12 |
| Position | 5 [20] | 5a [8] | 5b [8] | 5 | 5 |
|---|---|---|---|---|---|
| δC | δC | δC | δC | δH, mult. (J in Hz) | |
| 1 | 41.2 | 40.6 | 40.6 | 40.6 | 0.80 ddd (7.2, 7.1, 1.3) |
| 1.86* dd (2.9, 1.4) | |||||
| 2 | 28.9 | 19.8 | 19.0 | 19.7 | 1.55 ddd (7.3, 3.6, 2.4) |
| 1.75 dd (3.7, 3.6) | |||||
| 3 | 37.7 | 36.7 | 36.4 | 37.6 | 0.96 ddd (13.7, 13.6, 4.3) |
| 2.11 dd (13.7, 3.1) | |||||
| 4 | 43.6 | 46.9 | 47.8 | 43.5 | |
| 5 | 56.7 | 20.3 | 56.6 | 56.5 | 1.16 dd (9.1, 7.1) |
| 6 | 19.0 | 41.2 | 20.3 | 20.8 | 1.76 ddd (5.7, 3.4, 2.1) |
| 1.86* ddd (3.4, 3.4, 2.7) | |||||
| 7 | 35.4 | 47.8 | 41.2 | 35.3 | 1.25 ddd (14.4, 13.9, 4.4) |
| 1.79 ddd (13.8, 13.2, 4.3) | |||||
| 8 | 47.9 | 52.9 | 43.6 | 47.8 | |
| 9 | 52.9 | 43.6 | 53.0 | 52.8 | 1.28 dd (13.8, 3.8) |
| 10 | 39.8 | 56.6 | 39.8 | 39.7 | |
| 11 | 19.8 | 20.8 | 19.8 | 18.9 | 1.40 ddd (13.8, 3.4, 3.4, 3.1) |
| 1.81 dd (13.8, 4.3) | |||||
| 12 | 20.8 | 28.9 | 28.9 | 28.8 | 1.50 ddd (13.5, 7.8, 7.2) |
| 13 | 41.2 | 36.4 | 35.1 | 41.1 | 1.82 dd (13.8, 4.4) |
| 14 | 36.5 | 37.7 | 37.7 | 36.4 | 1.68 dd (14.5, 3.3) |
| 1.97 dd (13.1, 3.4) | |||||
| 15 | 81.9 | 81.2 | 81.2 | 81.9 | 4.73 br s |
| 16 | 66.3 | 66.4 | 66.4 | 66.3 | |
| 17 | 49.6 | 49.6 | 49.6 | 49.6 | 2.78 dd (5.6, 1.3) |
| 3.09 dd (5.8, 1.3) | |||||
| 18 | 28.8 | 28.9 | 28.9 | 28.7 | 1.28 s |
| 19 | 182.3 | 182.6 | 182.6 | 182.7 | |
| 20 | 15.7 | 15.8 | 16.0 | 15.9 | 1.03 s |
| 1’ | 167.9 | 166.5 | 166.5 | 167.8 | |
| 2’ | 128.1 | 129.0 | 115.9 | 128.0 | |
| 3’ | 137.3 | 137.1 | 156.8 | 137.3 | 5.96 q (7.1) |
| 4’ | 15.9 | 27.4 | 20.8 | 15.7 | 1.96 d (1.9) |
| 5’ | 20.6 | 20.8 | 27.4 | 20.6 | s |
| Compound | Normal Cell Lines | Cancer Cell Lines | ||
|---|---|---|---|---|
| BEAS-2B | LO2 | A549 | Hep-G2 | |
| 1 | >100 | >100 | >100 | 27.3 ± 1.9 |
| 2 | >100 | >100 | >100 | >100 |
| 3 | 89.9 ± 2.0 | 57.2 ± 1.2 | >100 | 24.7 ± 2.8 |
| 4 | >100 | >100 | >100 | >100 |
| 5 | >100 | >100 | 30.7 ± 1.7 | >100 |
| 6 | >100 | >100 | >100 | >100 |
| 6a | >100 | >100 | >100 | >100 |
| 6b | >100 | >100 | >100 | >100 |
| 7 | >100 | >100 | >100 | >100 |
| 9 | >100 | 75.3 ± 2.8 | >100 | >100 |
| 10 | >100 | >100 | >100 | >100 |
| 11 | >100 | >100 | >100 | >100 |
| 14 | >100 | >100 | >100 | >100 |
| 17 | >100 | >100 | >100 | >100 |
| 18 | 38.6 ± 2.5 | 30.0 ± 1.7 | 80.5 ± 1.8 | 81.3 ± 0.3 |
| Paclitaxel | <0.1 | <0.1 | 0.0033 | 0.19 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaouba, S.; Valkonen, A.; Coghi, P.; Gao, J.; Guantai, E.M.; Derese, S.; Wong, V.K.W.; Erdélyi, M.; Yenesew, A. Crystal Structures and Cytotoxicity of ent-Kaurane-Type Diterpenoids from Two Aspilia Species. Molecules 2018, 23, 3199. https://doi.org/10.3390/molecules23123199
Yaouba S, Valkonen A, Coghi P, Gao J, Guantai EM, Derese S, Wong VKW, Erdélyi M, Yenesew A. Crystal Structures and Cytotoxicity of ent-Kaurane-Type Diterpenoids from Two Aspilia Species. Molecules. 2018; 23(12):3199. https://doi.org/10.3390/molecules23123199
Chicago/Turabian StyleYaouba, Souaibou, Arto Valkonen, Paolo Coghi, Jiaying Gao, Eric M. Guantai, Solomon Derese, Vincent K. W. Wong, Máté Erdélyi, and Abiy Yenesew. 2018. "Crystal Structures and Cytotoxicity of ent-Kaurane-Type Diterpenoids from Two Aspilia Species" Molecules 23, no. 12: 3199. https://doi.org/10.3390/molecules23123199