
Radical Smiles Rearrangement: An Update

Abstract
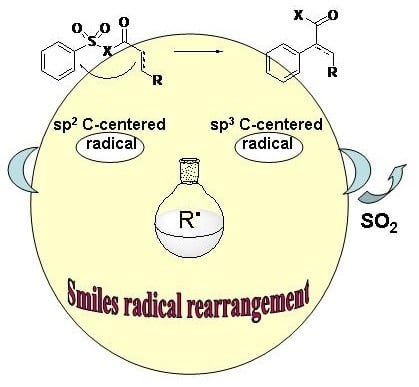
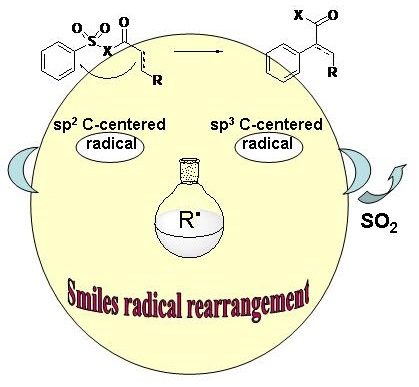
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
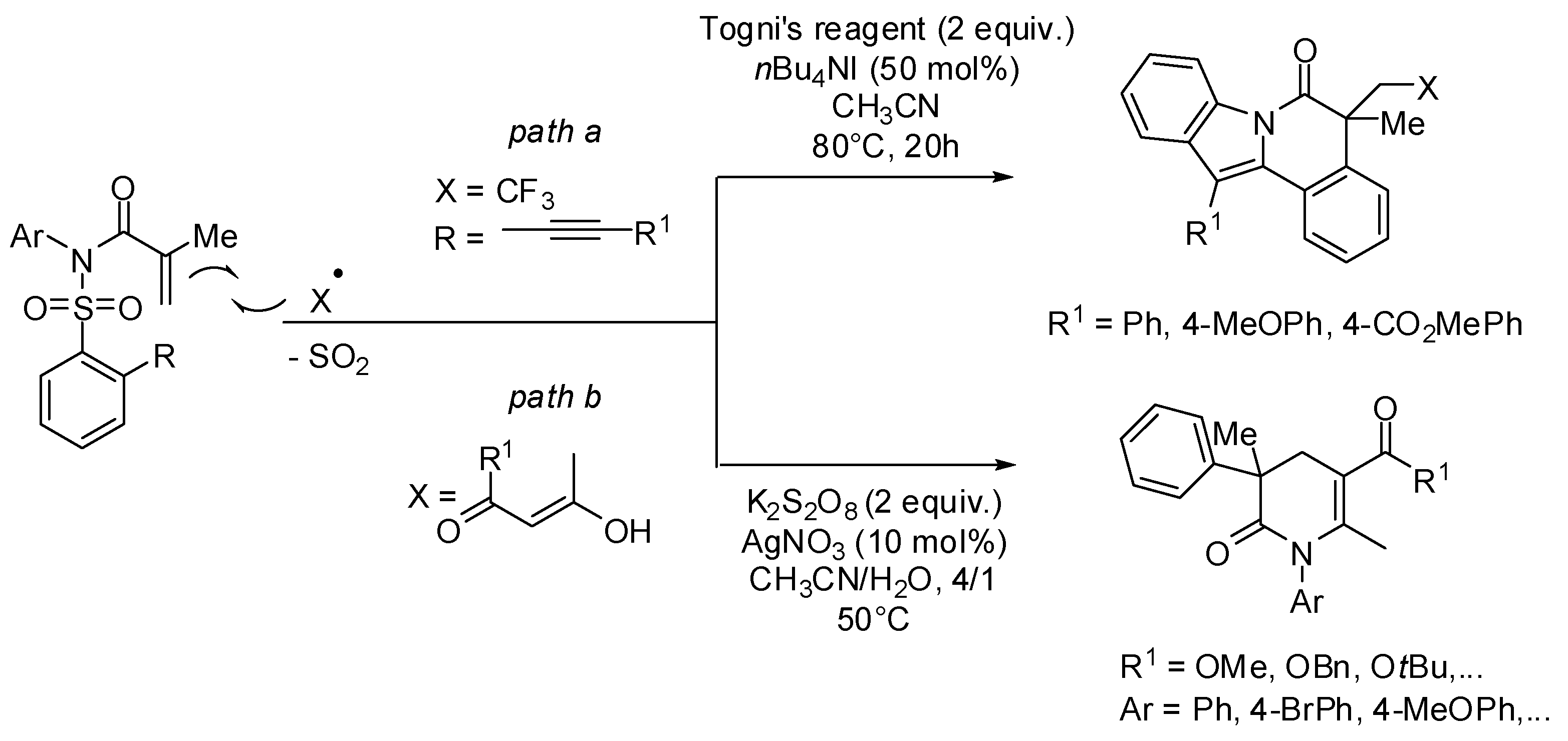{kind=link}
{kind=link}
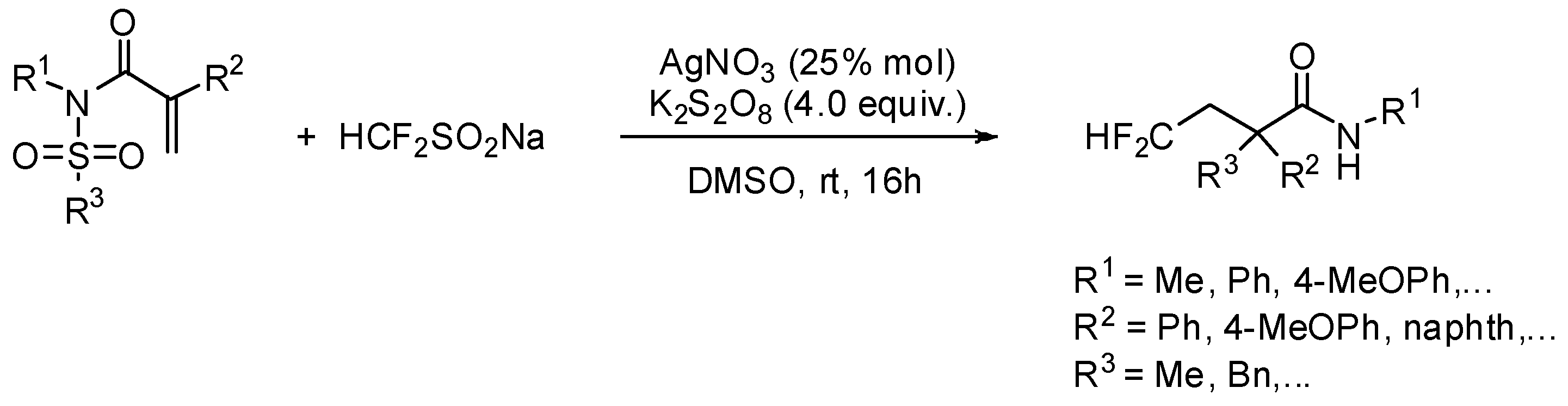{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
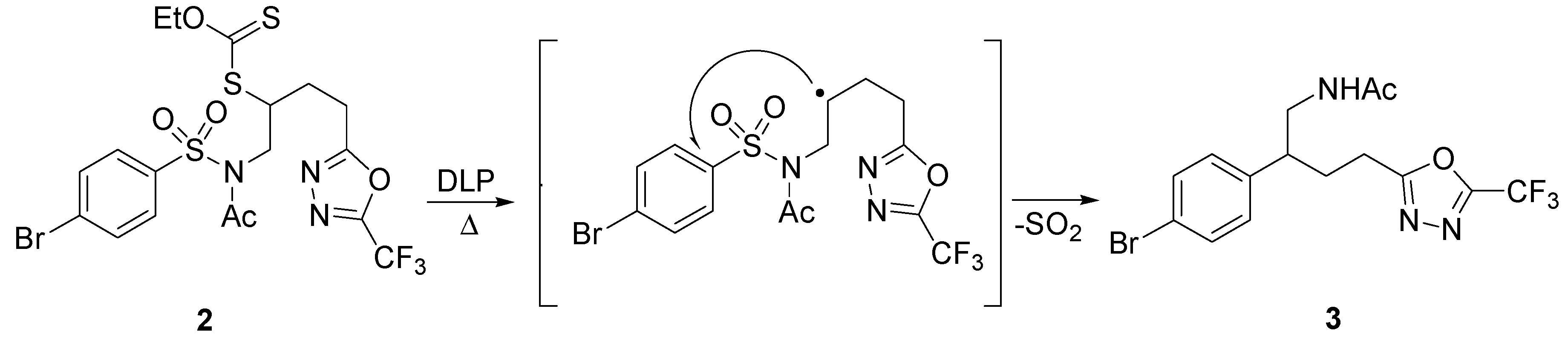
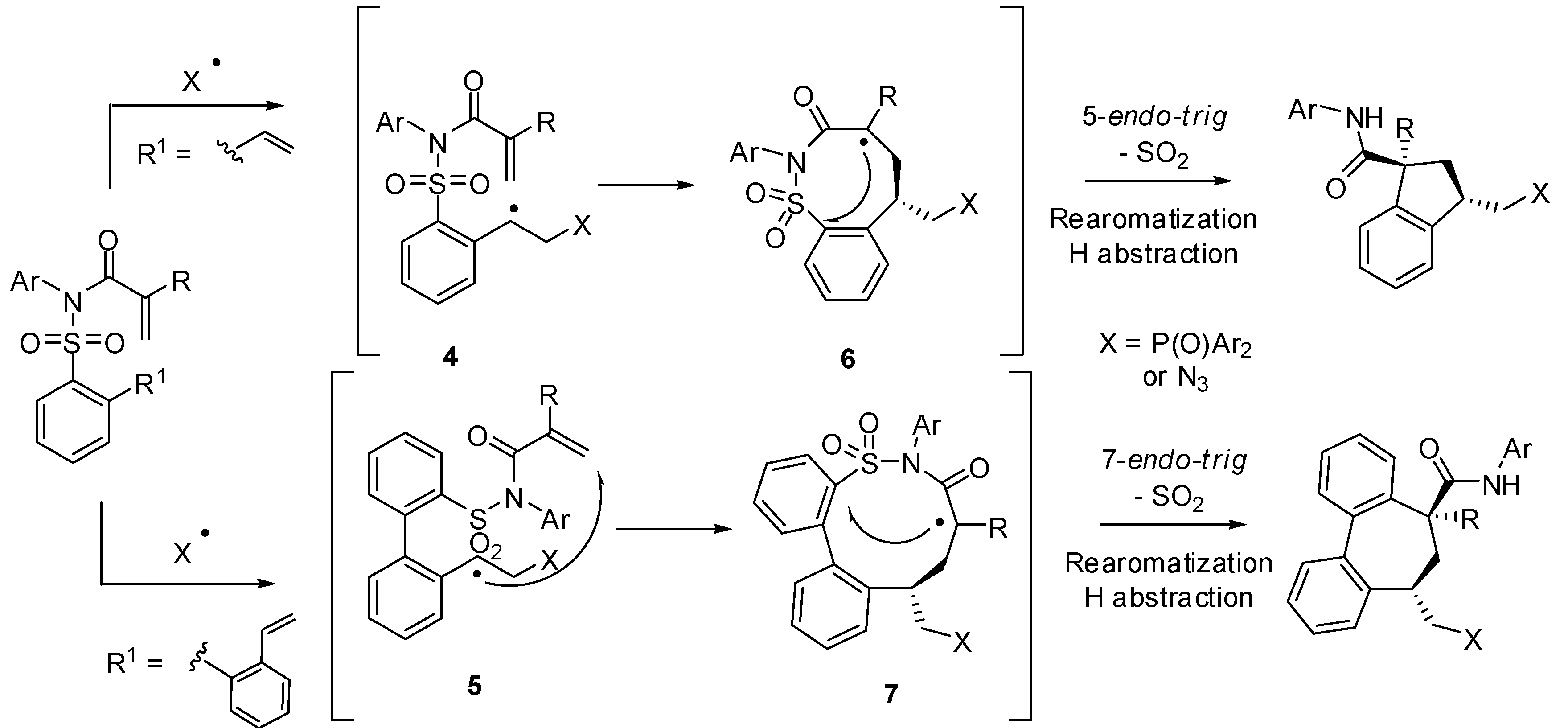
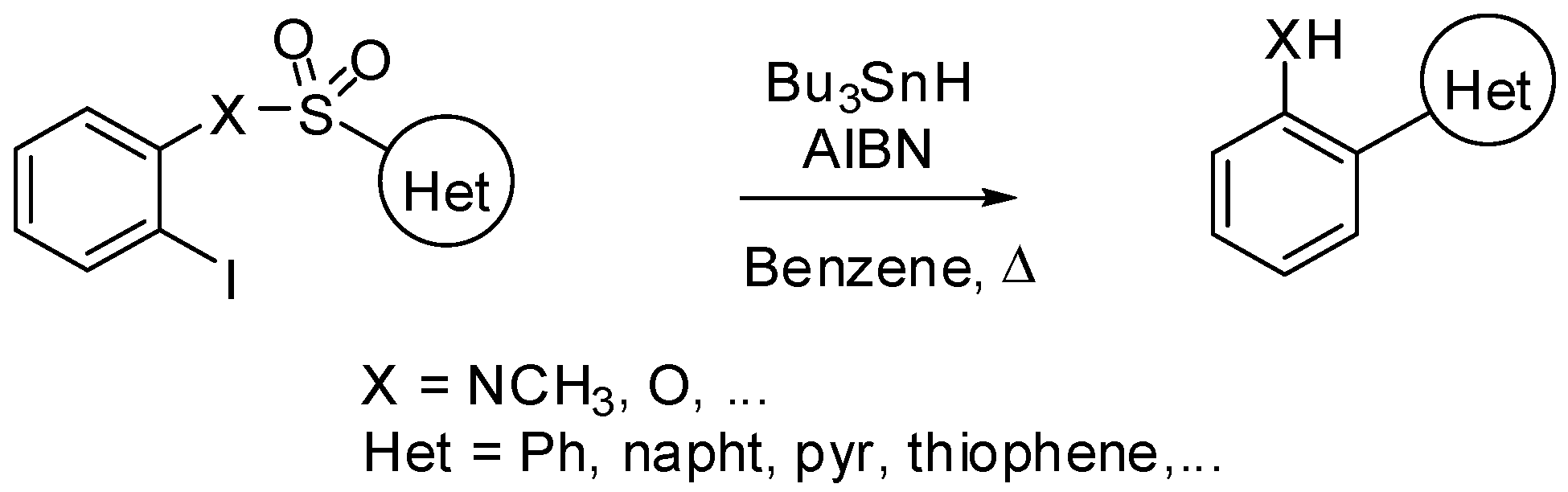
2. sp3 C-Centered Radical-Promoted Rearrangements
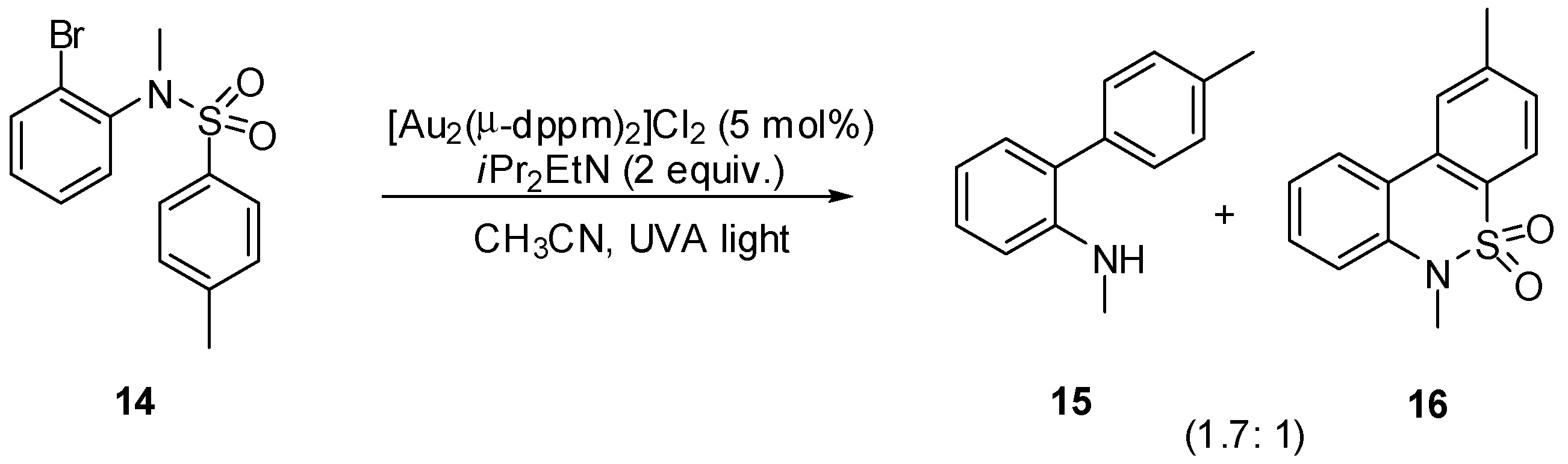
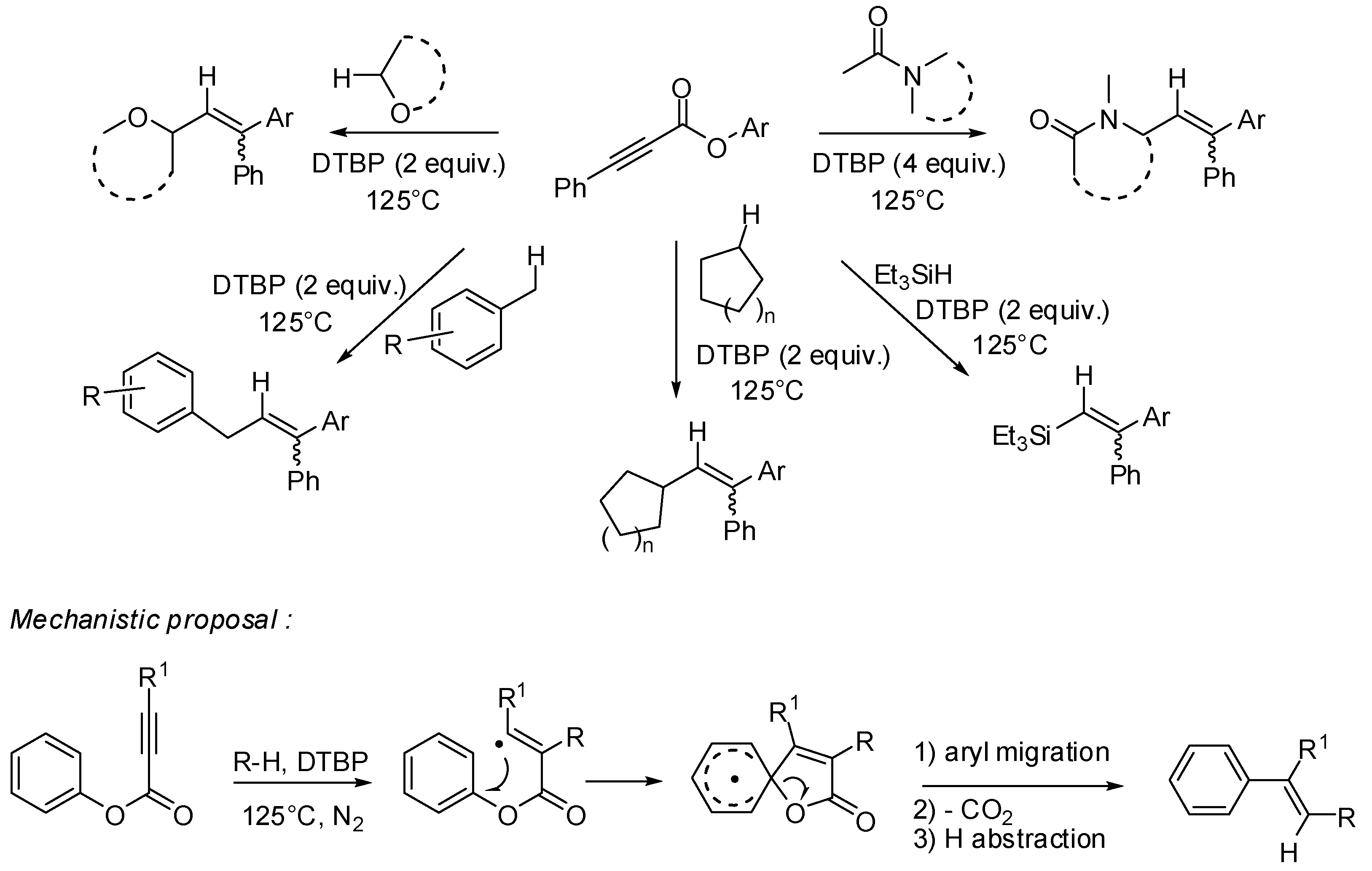
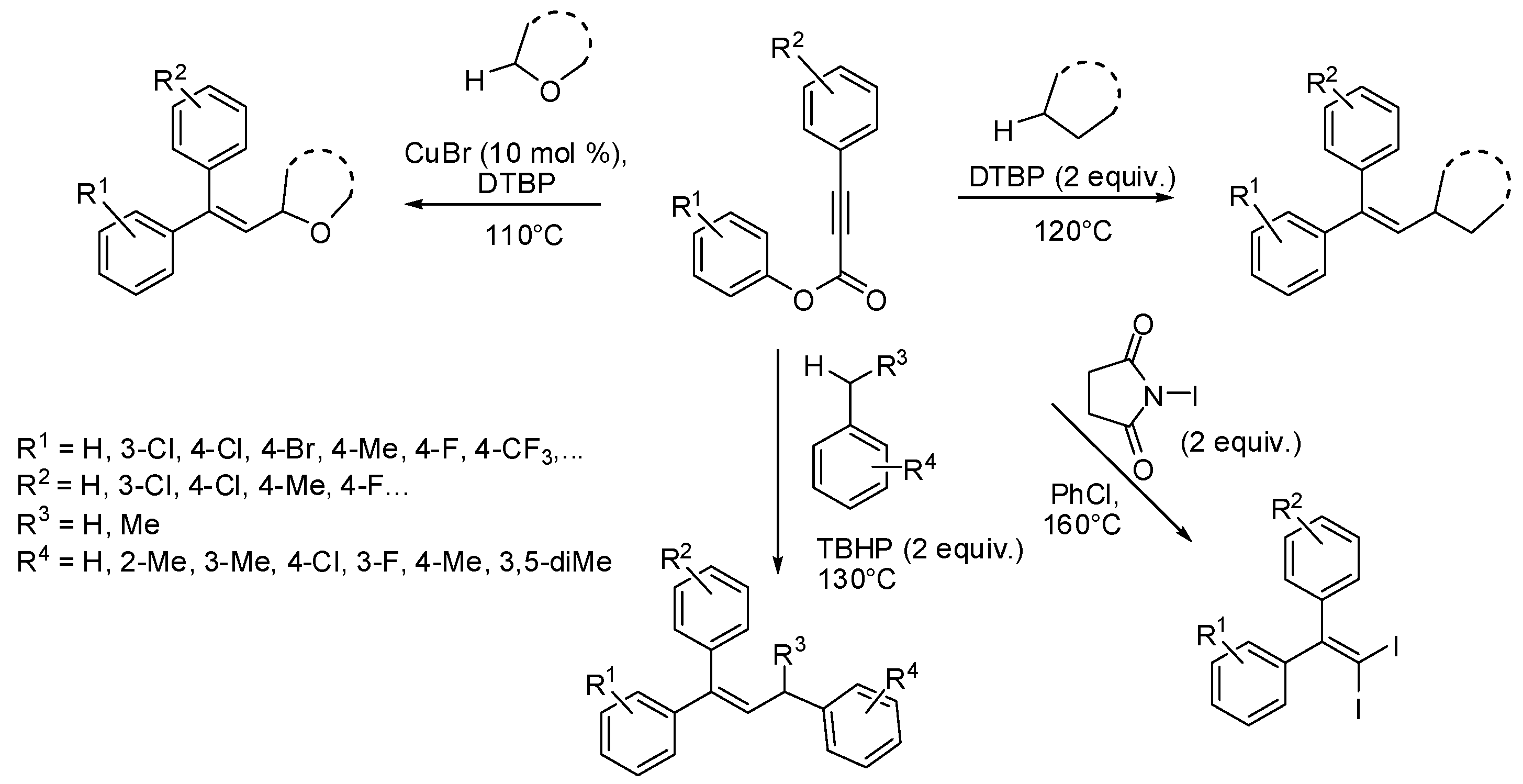
3. sp2 C-Centered Radical-Promoted Rearrangements
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Li, J.J. Smiles Rearrangement. In Name Reactions: A Collection on Detailed Reaction Mechanisms; Springer: Berlin, Heidelberg, Germany, 2006; pp. 549–554. [Google Scholar]
- Levy, A.A.; Rains, H.C.; Smiles, S. The rearrangement of hydroxy-sulphones. J. Chem. Soc. 1931, 3264–3269. [Google Scholar] [CrossRef]
- Truce, W.E.; Kreider, E.M.; Brand, W.W. The Smiles and related rearrangements of aromatic systems. In Organic Reactions; Dauben, W.G., Fried, J., House, H.O., Kende, A.S., Marshall, J.A., McKusick, B.C., Meinwald, J., Eds.; John Wiley: New York, NY, USA, 1970; Volume 18, pp. 99–215. [Google Scholar]
- For an update on Truce-Smiles rearrangement, see: Snape, T.J. A truce on the Smiles rearrangement: Revisiting an old reaction—The Truce–Smiles rearrangement. Chem. Soc. Rev. 2008, 37, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Loven, R.; Speckamp, W.N. A novel 1,4-aryl radical rearrangement. Tetrahedron Lett. 1972, 13, 1567–1570. [Google Scholar] [CrossRef]
- Koehler, J.J.; Speckamp, W.N. Intramolecular radical reactions in α-halomethyl substituted piperidine sulfonamides. Tetrahedron Lett. 1977, 18, 631–634. [Google Scholar] [CrossRef]
- Motherwell, W.B.; Pennell, A.M.K. A novel route to biaryls via intramolecular free radical ipso-substitution reactions. J. Chem. Soc. Chem. Commun. 1991, 877–879. [Google Scholar] [CrossRef]
- Da Mata, M.L.E.N.; Motherwell, W.B.; Ujjainwalla, F. Steric and electronic effects in the synthesis of biaryls and their heterocyclic congeners using intramolecular free radical [1,5]ipso substitution reactions. Tetrahedron Lett. 1997, 38, 137–140. [Google Scholar] [CrossRef]
- Da Mata, M.L.E.N.; Motherwell, W.B.; Ujjainwalla, F. Observation on the nature of the tethering chain in the synthesis of biaryls via intramolecular free radical ipso substitution reactions. Tetrahedron Lett. 1997, 38, 141–144. [Google Scholar] [CrossRef]
- El Kaïm, L.; Gizolme, M.; Grimaud, L. O-Arylative Passerini reactions. Org. Lett. 2006, 8, 5021–5023. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-M.; Li, H. Lewis acid-catalyzed formation of Ugi four-component reaction product from Passerini three-component reaction system without an added amine. Tetrahedron 2007, 63, 12866–12876. [Google Scholar] [CrossRef]
- El Kaïm, L.; Grimaud, L.; Oble, J.; Wagschal, S. Three-component Ugi-Smiles couplings of cyclic imines. Tetrahedron Lett. 2009, 50, 1741–1743. [Google Scholar] [CrossRef]
- Xiang, J.; Zheng, L.; Chen, F.; Dang, Q.; Bai, X. A cascade reaction consisting of Pictet-Spengler-type cyclization and Smiles rearrangement: Application to the synthesis of novel pyrrol-fused dihydropteridines. Org. Lett. 2007, 9, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Xie, H.; Wen, D.; Dang, Q.; Bai, X. Synthesis of pyrido[2,3-e]pyrrolo[1,2-a]pyrazine derivatives via tandem iminium cyclization and Smiles rearrangement. J. Org. Chem. 2008, 73, 3281–3283. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Zheng, L.; Xie, H.; Hu, X.; Dang, Q.; Bai, X. Pyrrolo-dihydropteridines via a cascade reaction consisting of iminium cyclization and O-N Smiles rearrangement. Tetrahedron 2008, 64, 9101–9107. [Google Scholar] [CrossRef]
- Pudlo, M.; Allart-Simon, I.; Tinant, B.; Gérard, S.; Sapi, J. First domino radical cyclisation/Smiles rearrangement combination. Chem. Commun. 2012, 48, 2442–2444. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 2015, 44, 5220–5245. [Google Scholar] [CrossRef] [PubMed]
- For a recent review on light-induced radical reactions, see Gurry, M.; Aldabbagh, F. A new era for homolytic aromatic substitution: Replacing Bu3SnH with efficient light-induced chain reactions. Org. Biomol. Chem. 2016, 14, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Gurry, M.; Allart-Simon, I.; McArdle, P.; Gérard, S.; Sapi, J.; Aldabbagh, F. Photochemical aryl radical cyclizations to give (E)-3-ylidenoxindoles. Molecules 2014, 19, 15891–15899. [Google Scholar] [CrossRef] [PubMed]
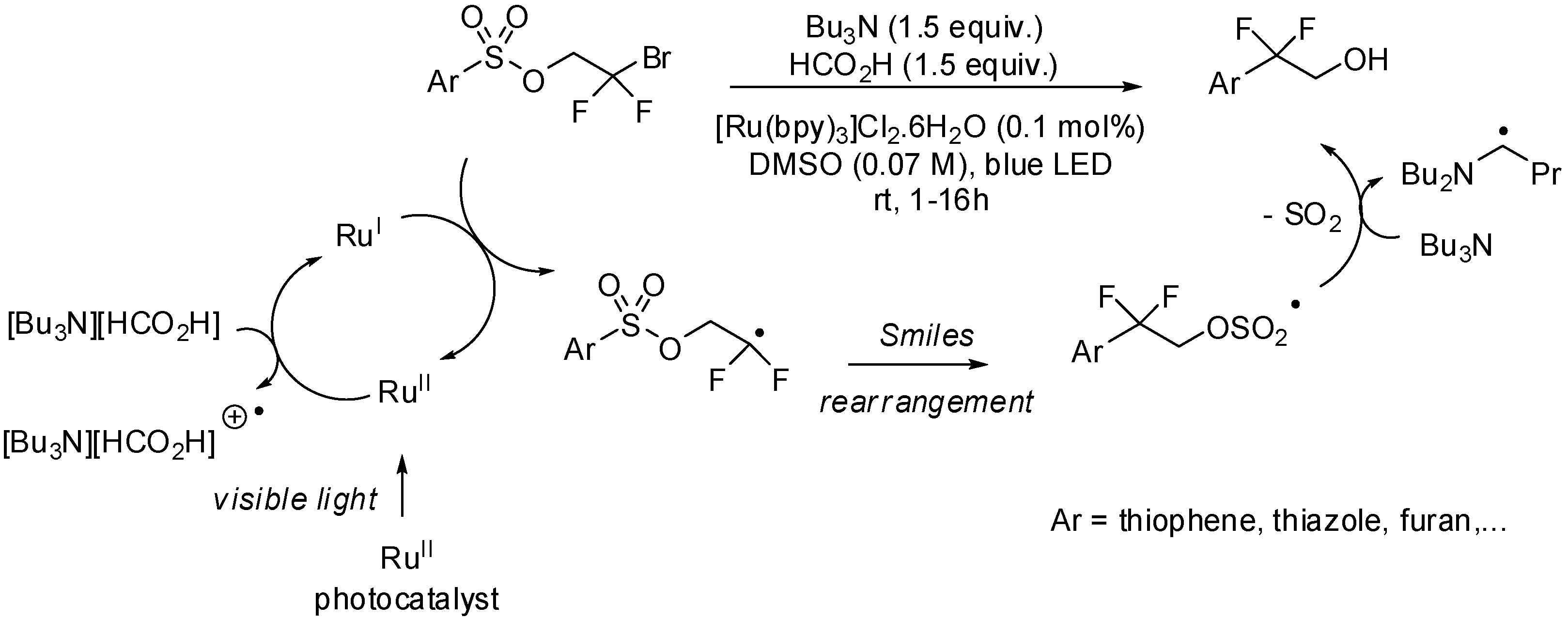
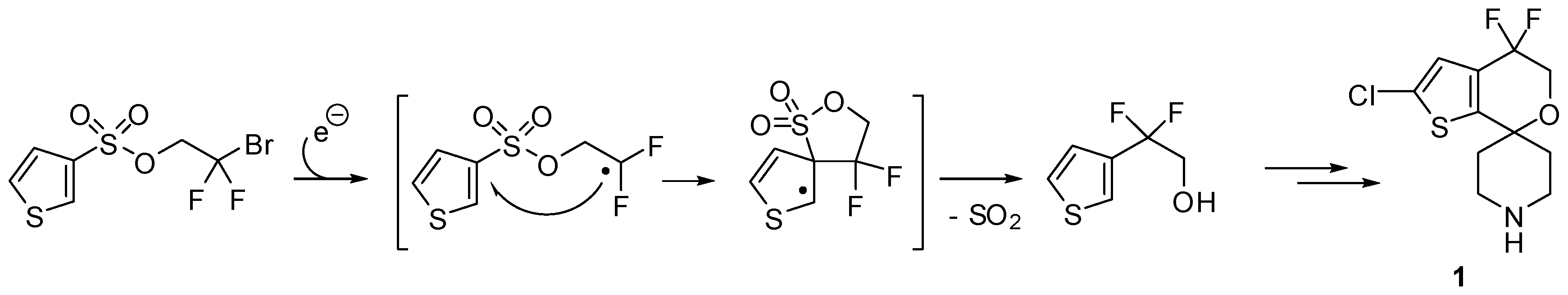
- Douglas, J.; Albright, H.; Sevrin, M.; Cole, K.; Stephenson, C.R.J. A visible-light-mediated radical Smiles rearrangement and its application to the synthesis of a difluoro-substituted spirocyclic ORL-1 antagonist. Angew. Chem. Int. Ed. 2015, 54, 14898–14902. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zard, S.Z. Radical-based route to 2-(trifluoromethyl)-1,3,4-oxadiazoles and trifluoromethyl-substituted polycyclic 1,2,4-triazoles and dihydrofurans. Org. Lett. 2015, 17, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Kong, W.; Fernandez-Sanchez, L.; Merino, E.; Nevado, C. Cyclization cascades via N-amidyl radicals toward highly functionalized heterocyclic scaffolds. J. Am. Chem. Soc. 2015, 137, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Fuentes, N.; Garcia-Dominguez, A.; Merino, E.; Nevado, C. Stereoselective synthesis of highly functionalized indanes and dibenzocycloheptadienes through complex radical cascade reactions. Angew. Chem. Int. Ed. 2015, 54, 2487–2491. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Tan, P.; Ni, C.; Hu, J. Fluoroalkylative aryl migration of conjugated N-arylsulfonylated amides using easily accessible sodium di- and monofluoroalkanesulfinates. Org. Lett. 2015, 17, 1838–1841. [Google Scholar] [CrossRef] [PubMed]
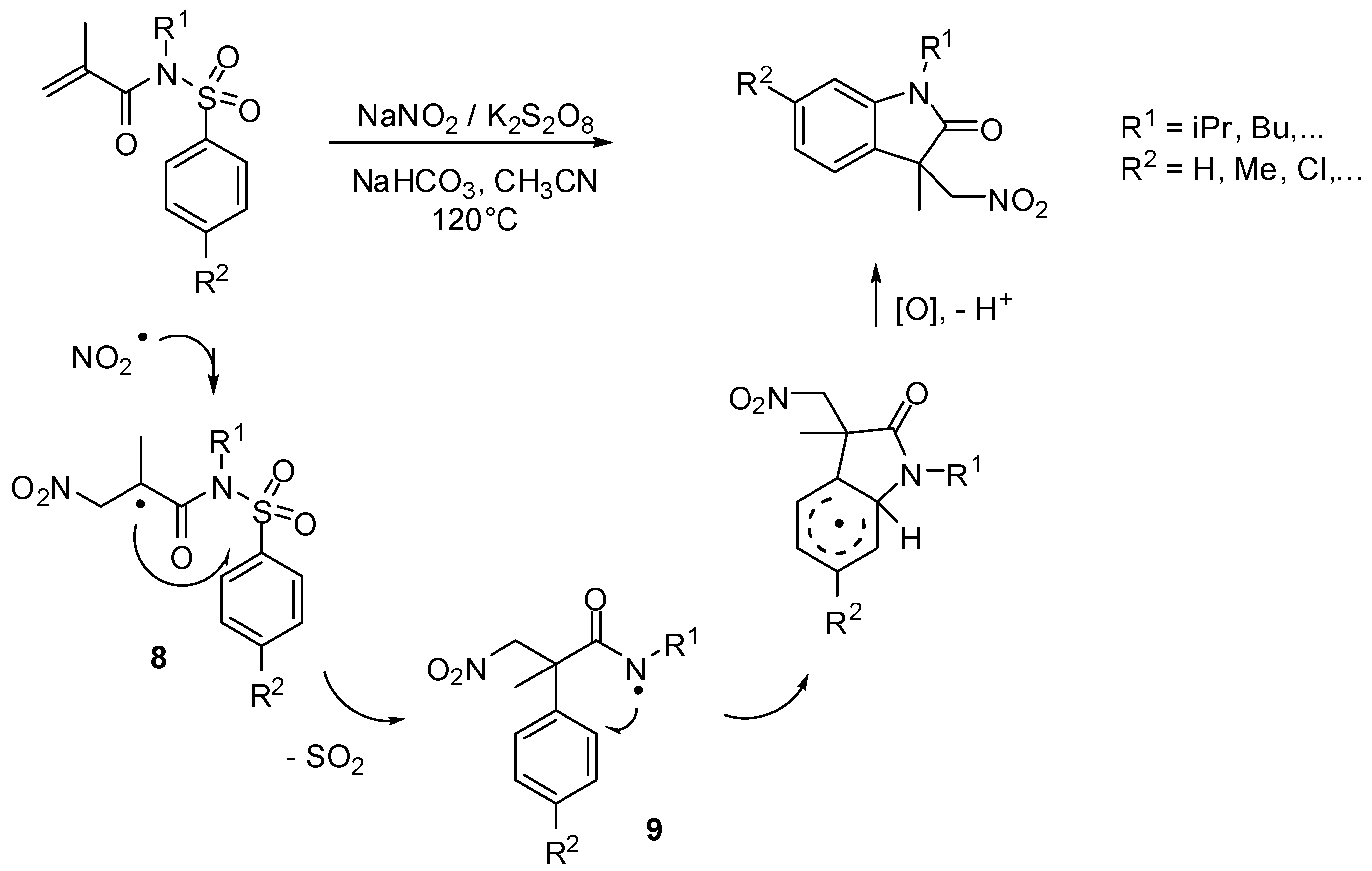
- Niu, B.; Xie, P.; Bian, Z.; Zhao, W.; Zhang, M.; Zhou, Y.; Feng, L.; Pittman, C.U., Jr.; Zhou, A. Synthesis of nitromethyl-substituted oxindole derivatives via a desulfonylation cascade. Synlett 2015, 26, 635–638. [Google Scholar]
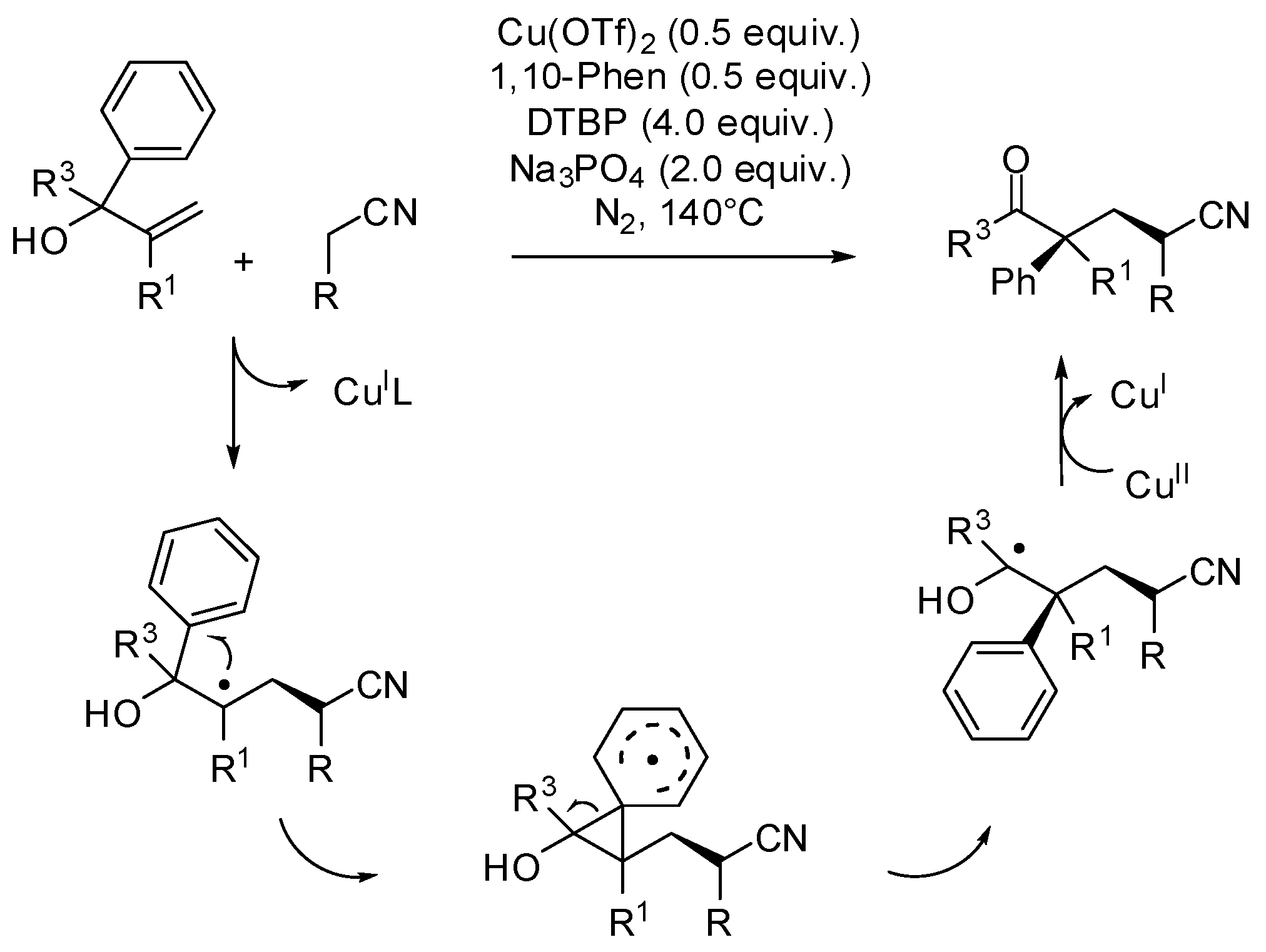
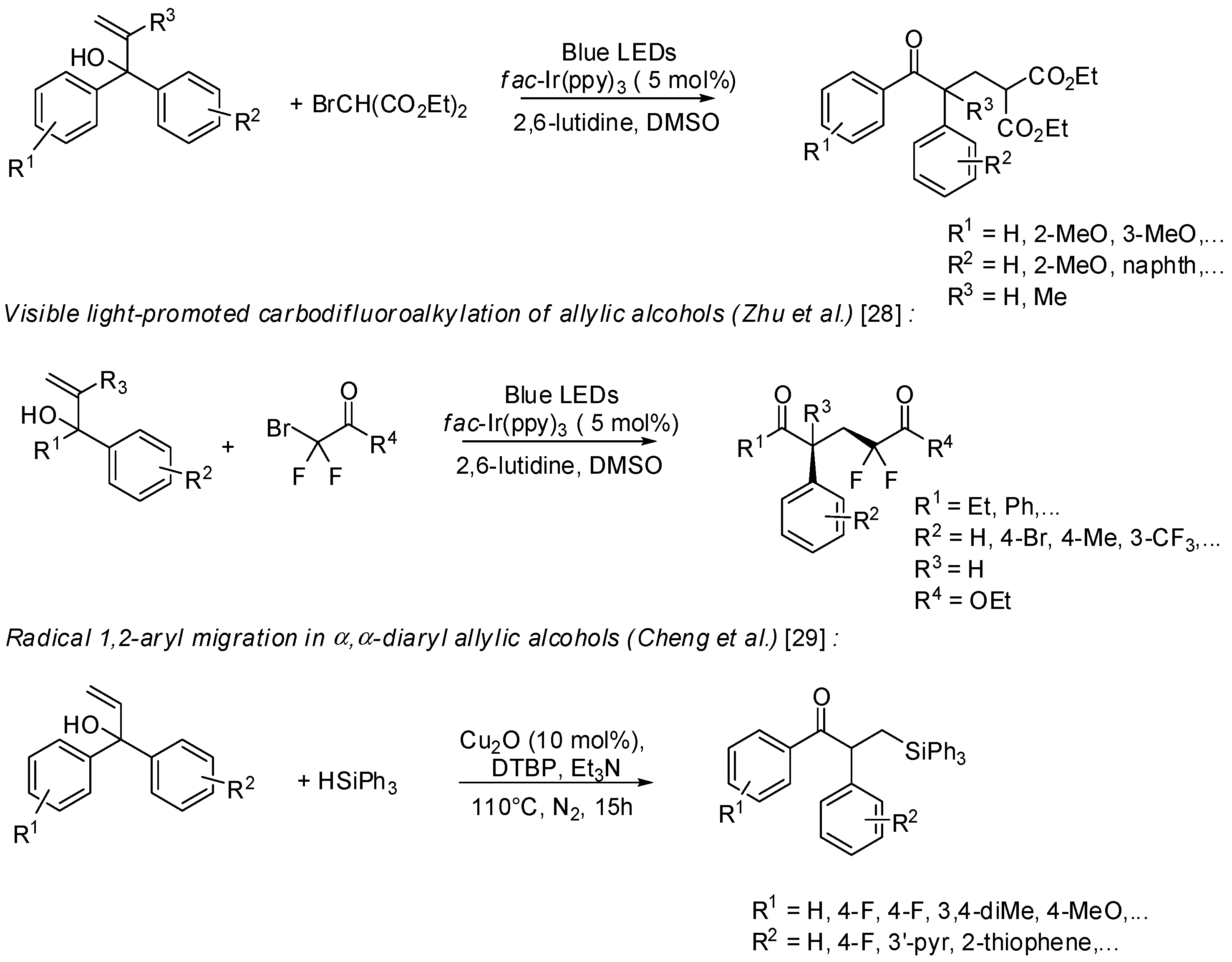
- Bunescu, A.; Wang, Q.; Zhu, J. Copper-catalyzed cyanomethylation of allylic alcohols with concomitant 1,2-aryl migration: Efficient synthesis of functionalized ketones containing an α-quaternary center. Angew. Chem. Int. Ed. 2015, 54, 3132–3135. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-L.; Yan, H.; Yang, C.; Xia, W. Visible light-mediated arylalkylation of allylic alcohols through concomitant 1,2-aryl migration. Chem. Commun. 2015, 51, 4910–4913. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hu, K.; Gu, Z.; Cheng, Y.; Zhu, C. Visible light promoted carbodifluoroalkylation of allylic alcohols via concomitant 1,2-aryl migration. Chem. Commun. 2015, 51, 7222–7225. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yu, J.-T.; Jiang, Y.; Cheng, J. Radical 1,2-aryl migration in α,α-diarylallylic alcohols toward β-silyl ketones. Org. Biomol. Chem. 2015, 13, 10299–10302. [Google Scholar] [CrossRef] [PubMed]
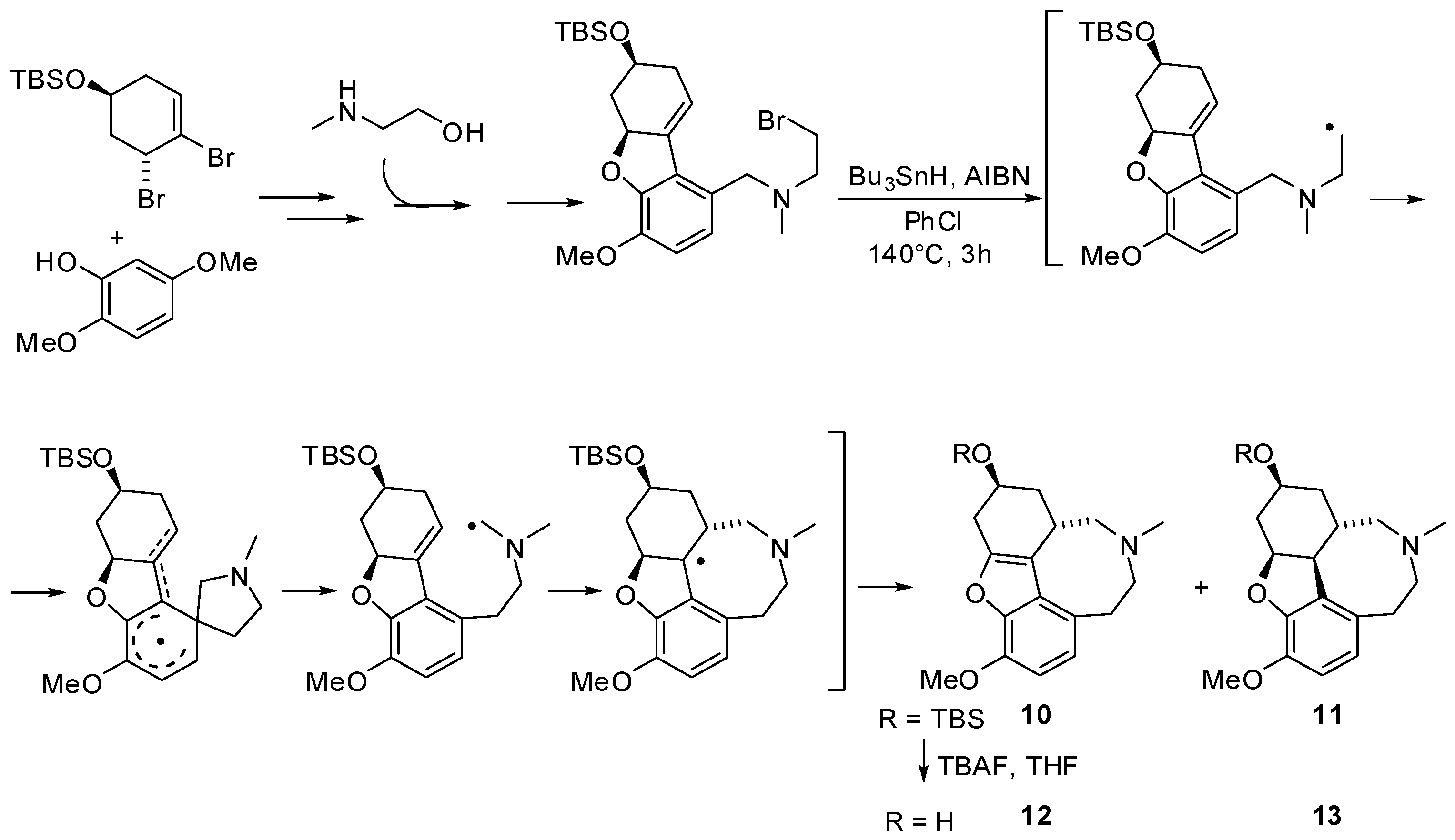
- Lan, P.; Jackson, C.J.; Banwell, M.G.; Willis, A.C. Synthesis of a D-ring isomer of galanthamine via a radical-based Smiles rearrangement reaction. J. Org. Chem. 2014, 79, 6759–6764. [Google Scholar] [CrossRef] [PubMed]
- Ujjainwalla, F.; Da Mata, M.L.; Pennell, A.M.K.; Escolano, C.; Motherwell, W.B.; Vasquez, S. Synthesis of biaryls via intramolecular free radical/ipso-substitution reactions. Tetrahedron 2015, 71, 6701–6719, and references cited therein. [Google Scholar] [CrossRef]
- Revol, G.; McCallum, T.; Morin, M.; Gagosz, F.; Barriault, L. Photoredox transformations with dimeric gold complexes. Angew. Chem. Int. Ed. 2013, 52, 13342–13345. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-L.; Cheng, L.; Wu, H.-R.; Li, Y.-X.; Wang, D.; Liu, L. A metal-free yne-addition/1,4-aryl migration/decarboxylation cascade reaction of alkynoates with Csp3-H centers. Org. Biomol. Chem. 2016, 14, 2210–2217. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Zhang, H.; Zhu, C. Oxidative difunctionalization of alkynoates via cascade radical addition, aryl migration, and decarboxylation. Tetrahedron Lett. 2016, 57, 595–598. [Google Scholar] [CrossRef]
- Miao, T.; Xia, D.; Li, Y.; Li, P.; Wang, L. Direct difunctionalization of activated alkynes via domino oxidative benzylation/1,4-aryl migration/decarboxylation reactions under metal-free conditions. Chem. Commun. 2016, 52, 3175–3178. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Zhang, Y.; Xie, C.; Mei, H.; Han, J.; Pan, Y. Oxidative difunctionalization of alkynoates through alkylation and migration decarboxylative arylation. Org. Lett. 2015, 17, 5524–5527. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Sha, W.; Zhang, L.; Xie, C.; Mei, H.; Han, J.; Pan, Y. N-Iodosuccinimide-promoted cascade trifunctionalization of alkynoates: Access to 1,1-diiodoalkenes. Org. Lett. 2016, 18, 712–715. [Google Scholar] [CrossRef] [PubMed]
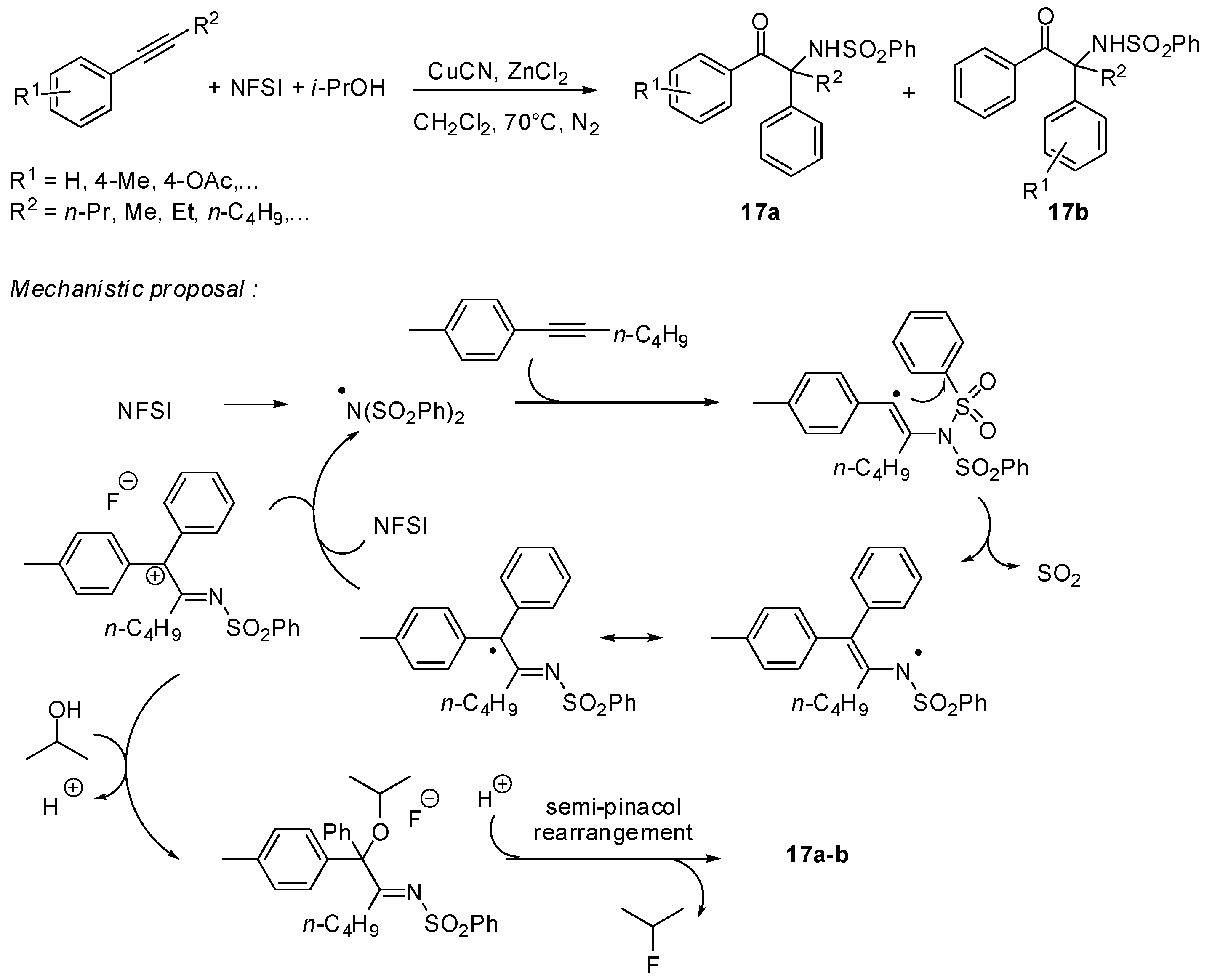
- Zheng, G.; Li, Y.; Han, J.; Xiong, T.; Zhang, Q. Radical cascade reaction of alkynes with N-fluoroarylsulfonimides and alcohols. Nat. Commun. 2015, 6, article 7011. [Google Scholar] [CrossRef] [PubMed]
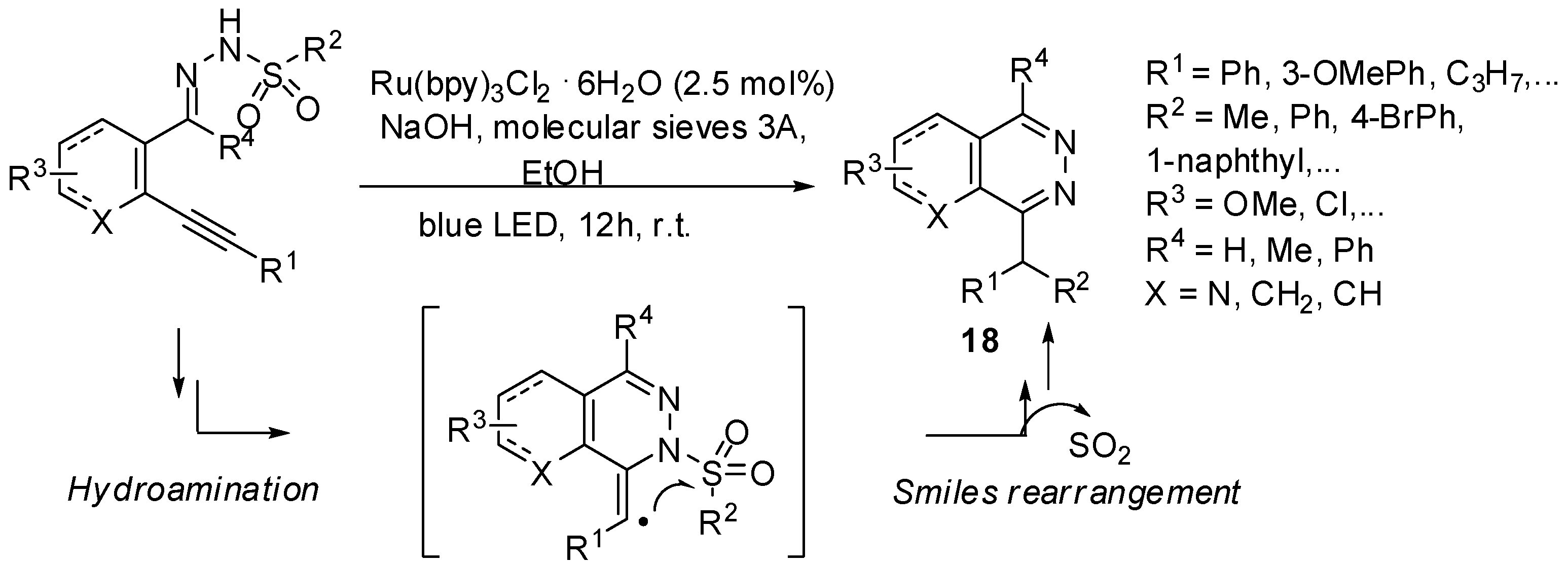
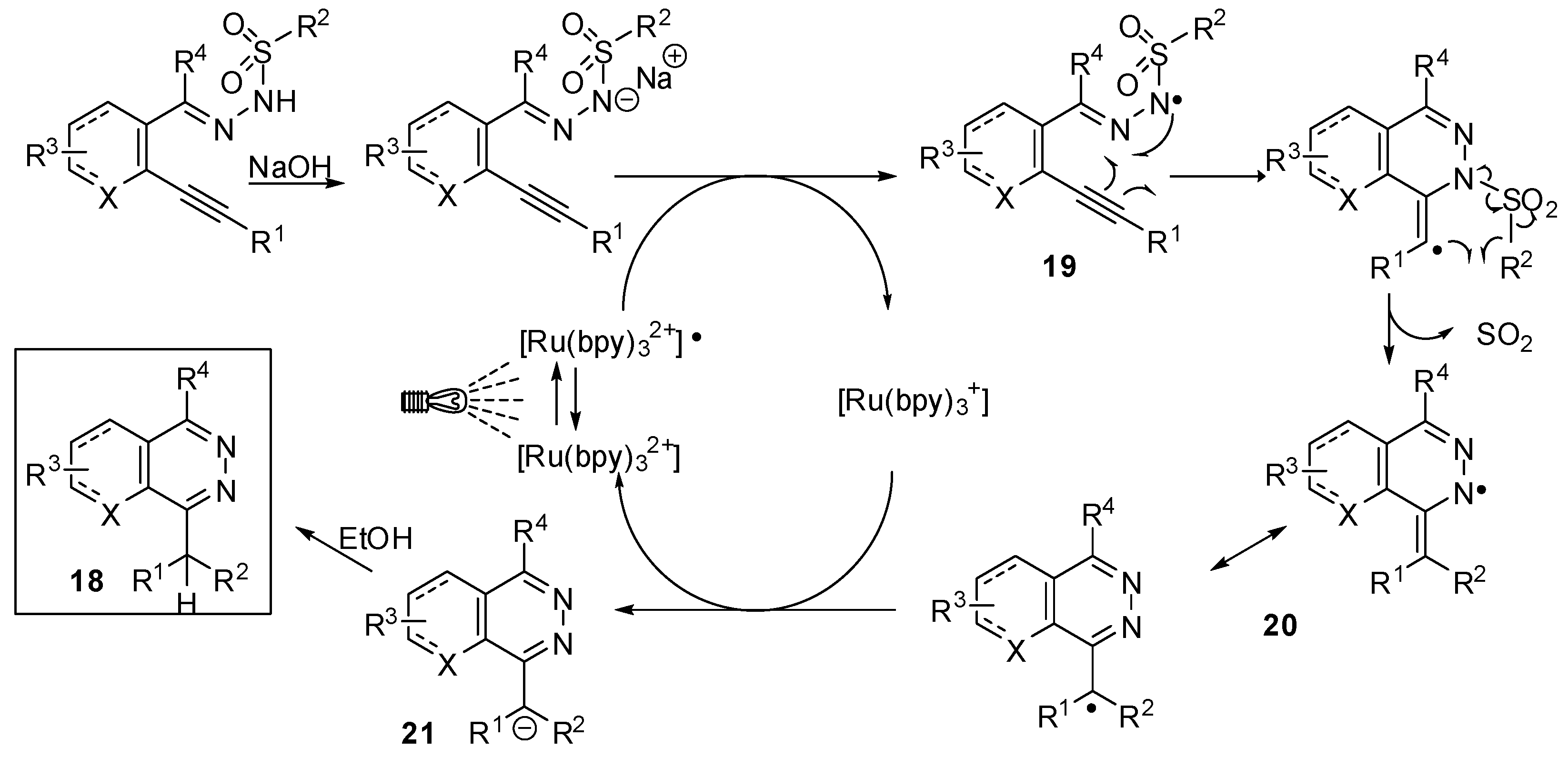
- Brachet, E.; Marzo, L.; Selkti, M.; König, B.; Belmont, P. Visible light amination/Smiles cascade: Access to phthalazine derivatives. Chem. Sci. 2016. [Google Scholar] [CrossRef]

















© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allart-Simon, I.; Gérard, S.; Sapi, J. Radical Smiles Rearrangement: An Update. Molecules 2016, 21, 878. https://doi.org/10.3390/molecules21070878
Allart-Simon I, Gérard S, Sapi J. Radical Smiles Rearrangement: An Update. Molecules. 2016; 21(7):878. https://doi.org/10.3390/molecules21070878
Chicago/Turabian StyleAllart-Simon, Ingrid, Stéphane Gérard, and Janos Sapi. 2016. "Radical Smiles Rearrangement: An Update" Molecules 21, no. 7: 878. https://doi.org/10.3390/molecules21070878