
Treatment with Akebia Saponin D Ameliorates Aβ1–42-Induced Memory Impairment and Neurotoxicity in Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
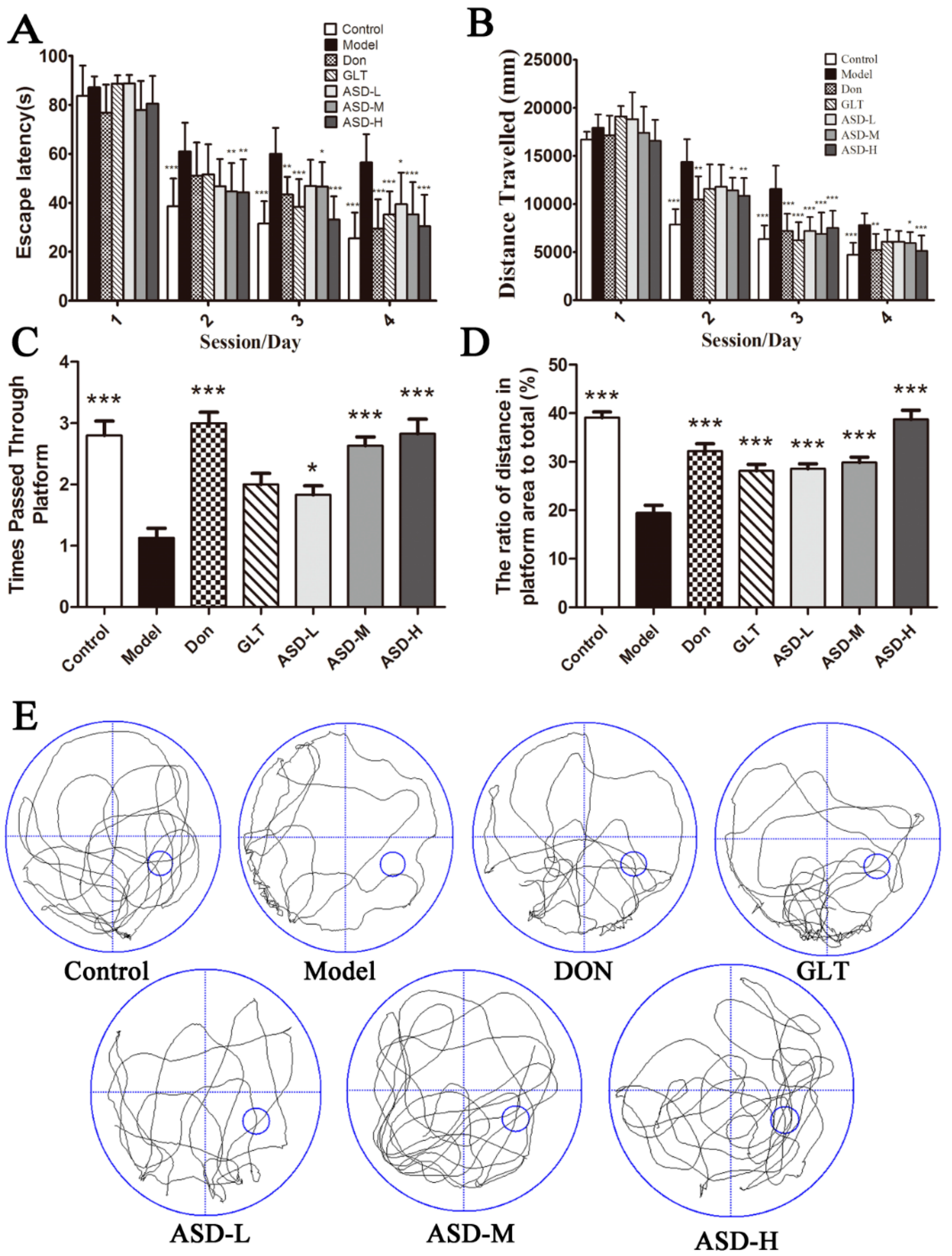
2.1. ASD Reversed Spatial Learning and Memory Impairment Induced by Aβ1–42
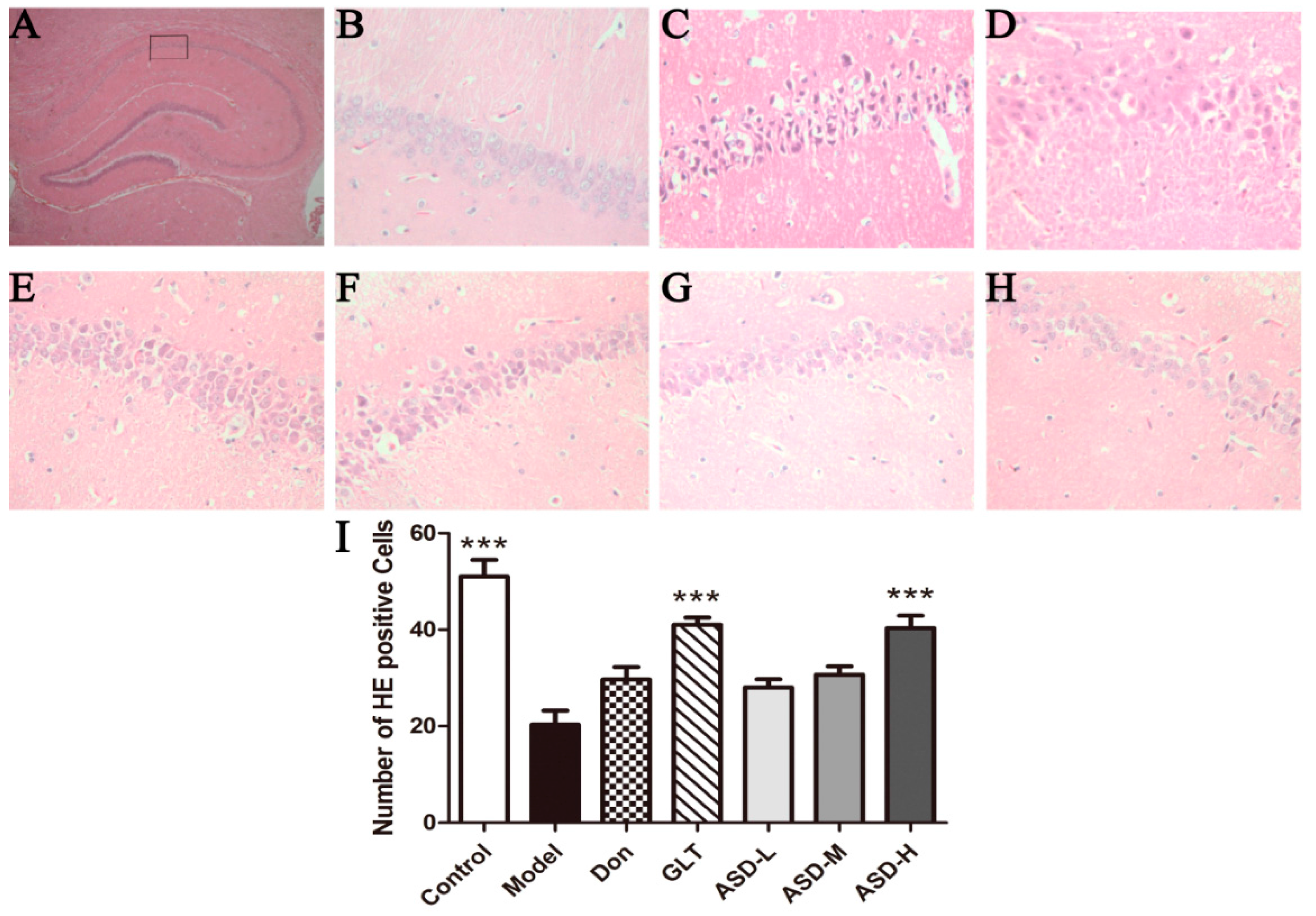
2.2. HE Staining
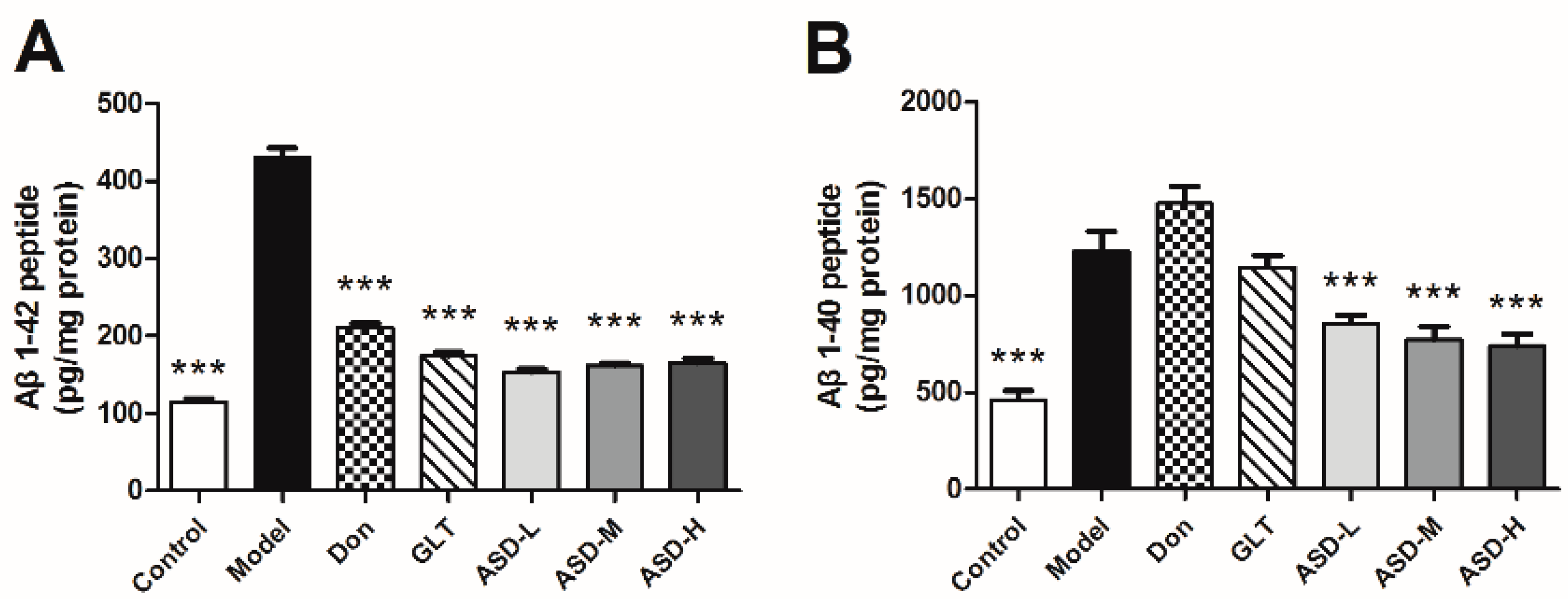
2.3. ASD Blocked Aβ1–42-Induced Production of Aβ
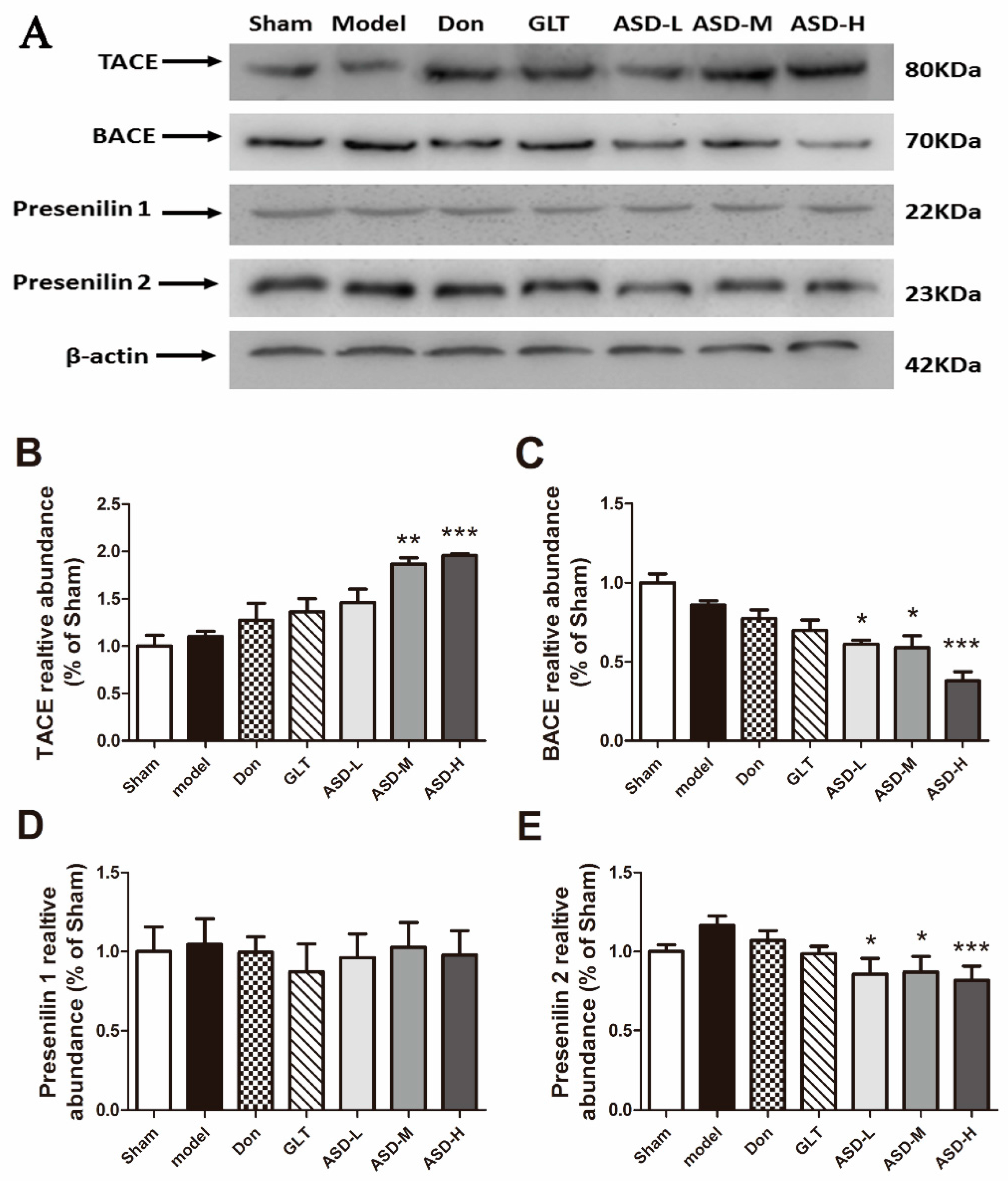
2.4. Effects of ASD on the Expression of Aβ Generation Proteins
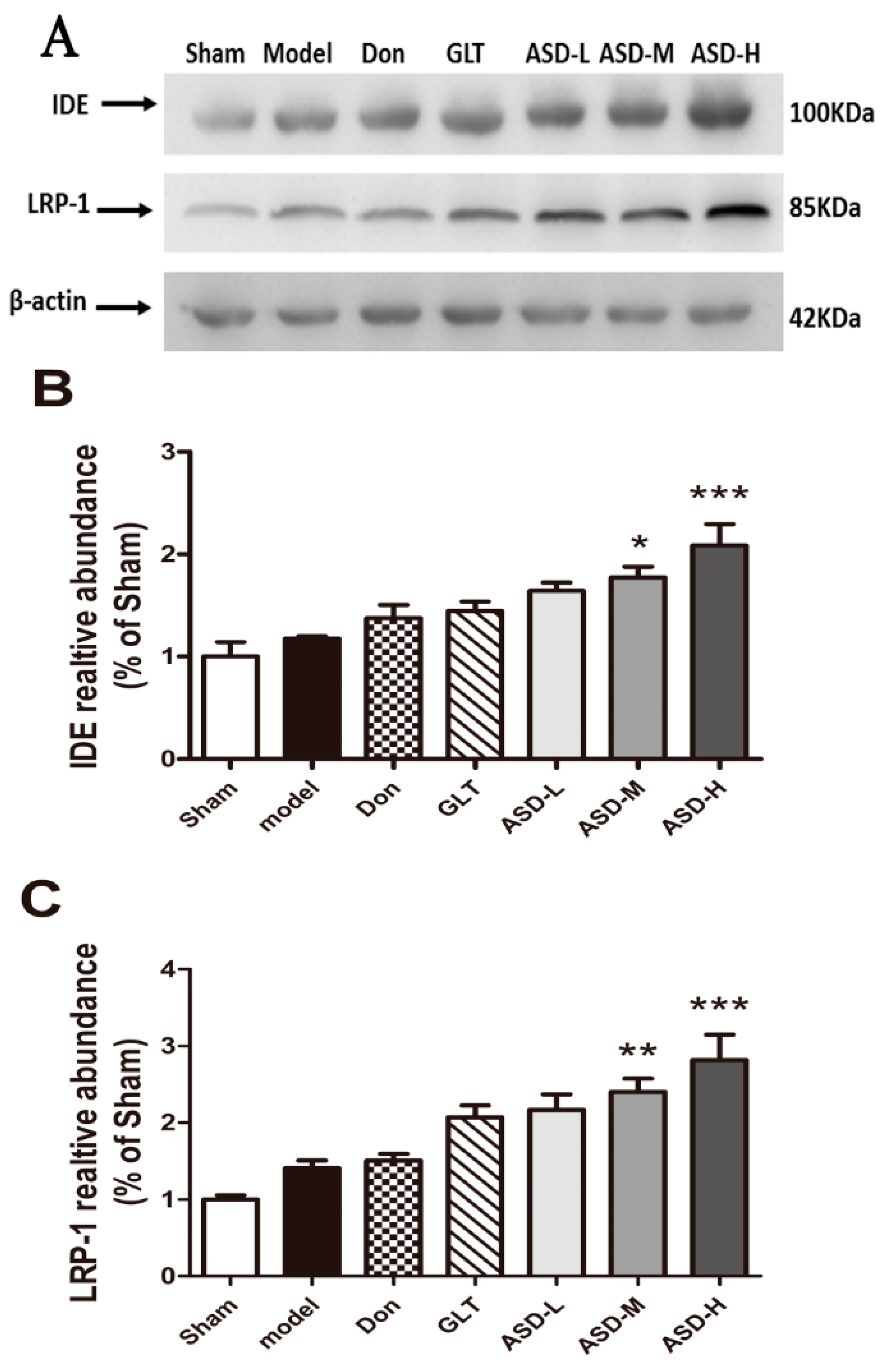
2.5. Effects of ASD on the Expression of Aβ Degradation-Related and Transshipment-Related Proteins
3. Materials and Methods
3.1. Animals and Housing
3.2. Drugs and Materials
3.3. Stereotaxic Intracerebroventricular (ICV) Aβ1–42 Injection and Drug Treatment
3.4. Morris Water Maze Task
3.5. Hematoxylin-Eosin Staining of Neurons in the Hippocampus of Rats
3.6. Western Blot Analysis
3.7. Measurement of Aβ1–40 and Aβ1–42
3.8. Statistical Analysis
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mueller, S.G.; Dickerson, B.C. Atrophy accelerates with conversion from mild cognitive impairment to Alzheimer disease. Neurology 2008, 70, 1728–1729. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Callahan, P.M.; Hall, B.; Webster, S.J. Alzheimer’s disease and age-related memory decline (preclinical). Pharmacol. Biochem. Behav. 2011, 99, 190–210. [Google Scholar] [CrossRef] [PubMed]
- Bero, A.W.; Bauer, A.Q.; Stewart, F.R.; White, B.R.; Cirrito, J.R.; Raichle, M.E.; Culver, J.P.; Holtzman, D.M. Bidirectional relationship between functional connectivity and amyloid-beta deposition in mouse brain. J. Neurosci. 2012, 32, 4334–4340. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The role of amyloid precursor protein processing by bace1, the beta-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef] [PubMed]
- Vahidi, A.; Glenn, G.; van der Geer, P. Identification and mutagenesis of the tace and gamma-secretase cleavage sites in the colony-stimulating factor 1 receptor. Biochem. Biophys. Res. Commun. 2014, 450, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, H. Research on regulation function of gamma-secretase inhibitor dapt on the differentiation of neural precursor cell line. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2015, 32, 126–130. [Google Scholar] [PubMed]
- Del Turco, D.; Schlaudraff, J.; Bonin, M.; Deller, T. Upregulation of APP, ADAM10 and ADAM17 in the denervated mouse dentate gyrus. PLoS ONE 2014, 9, e84962. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Stricker, R.; Lendeckel, U.; Bertram, I.; Dobrowolny, H.; Steiner, J.; Bogerts, B.; Reiser, G. Reduced neuronal co-localisation of nardilysin and the putative α-secretases ADAM10 and ADAM17 in Alzheimer’s disease and down syndrome brains. AGE 2009, 31, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Klatt, S.; Roch, M.; Konthur, Z.; Klose, J.; Willnow, T.E.; Rohe, M. Soluble Alpha-APP (sAPPalpha) regulates CDK5 expression and activity in neurons. PLoS ONE 2013, 8, e65920. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Chow, K.M.; Shah, R.; Rhodes, C.J.; Hersh, L.B. Degradation of islet amyloid polypeptide by neprilysin. Diabetologia 2012, 55, 2989–2998. [Google Scholar] [CrossRef] [PubMed]
- Kakiya, N.; Saito, T.; Nilsson, P.; Matsuba, Y.; Tsubuki, S.; Takei, N.; Nawa, H.; Saido, T.C. Cell surface expression of the major amyloid-βpeptide (Aβ)-degrading enzyme, neprilysin, depends on phosphorylation by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (mek) and dephosphorylation by protein phosphatase 1a. J. Biol. Chem. 2012, 287, 29362–29372. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Belbin, O.; Zou, F.; Allen, M.; Ertekin-Taner, N.; Ansari, M.; Wilcox, S.L.; Kashino, M.R.; Ma, L.; Younkin, L.H.; et al. Concordant association of insulin degrading enzyme gene (IDE) variants with IDE mRNA, Aβ, and Alzheimer’s Disease. PLoS ONE 2010, 5, e8764. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Magnani, N.; Villordo, S.; Buslje, C.M.; Evelson, P.; Castano, E.M.; Morelli, L. Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid β (Aβ) peptide catabolism and functionality. J. Biol. Chem. 2013, 288, 12920–12931. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Quinto, J.; Herdt, A.; Eckman, C.B.; Eckman, E.A. Endothelin-converting enzymes and related metalloproteases in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S101–S110. [Google Scholar] [PubMed]
- Wang, R.; Wang, S.; Malter, J.S.; Wang, D.S. Effects of 4-hydroxy-nonenal and amyloid-β on expression and activity of endothelin converting enzyme and insulin degrading enzyme in SH-SY5Y cells. J. Alzheimers Dis. 2009, 17, 489–501. [Google Scholar] [PubMed]
- Vepsalainen, S.; Hiltunen, M.; Helisalmi, S.; Wang, J.; van Groen, T.; Tanila, H.; Soininen, H. Increased expression of Aβ degrading enzyme IDE in the cortex of transgenic mice with Alzheimer’s disease-like neuropathology. Neurosci. Lett. 2008, 438, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; Teplow, D.B.; Linse, S. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid β-peptide. FEBS J. 2009, 276, 1266–1281. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedros, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurologia 2015. [Google Scholar] [CrossRef]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A. What’s next for alzheimer treatment?: While Aβ isn’t out of the picture yet, several other therapeutic routes are being explored. Ann. Neurol. 2013, 73, A7–A9. [Google Scholar] [CrossRef] [PubMed]
- Ginty, D.D.; Bonni, A.; Greenberg, M.E. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 1994, 77, 713–725. [Google Scholar] [CrossRef]
- Hung, T.M.; Na, M.; Thuong, P.T.; Su, N.D.; Sok, D.; Song, K.S.; Seong, Y.H.; Bae, K. Antioxidant activity of caffeoyl quinic acid derivatives from the roots of dipsacus asper wall. J. Ethnopharmacol. 2006, 108, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.Y.; Do, J.C.; Son, K.H. Triterpene glycosides from the roots of dipsacus asper. J. Nat. Prod. 1993, 56, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Kim, Y.C.; Zadeh, H.; Park, Y.J.; Pi, S.H.; Shin, H.S.; You, H.K. Effects of the dichloromethane fraction of Dipsaci Radix on the osteoblastic differentiation of human alveolar bone marrow-derived mesenchymal stem cells. Biosci. Biotechnol. Biochem. 2011, 75, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Li, Y.; Huang, H.; Kong, X.; Zhang, R.; Liu, L.; Sun, Y.; Wang, T.; Mei, Q. Asperosaponin VI, a saponin component from Dipsacus asper wall, induces osteoblast differentiation through bone morphogenetic protein-2/p38 and extracellular signal-regulated kinase 1/2 pathway. Phytother. Res. 2011, 25, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.X.; Zhang, L.N.; Jin, S.M.; Zhang, J. Effects of traditional chinese medicine on bone remodeling during orthodontic tooth movement. J. Ethnopharmacol. 2012, 141, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Qian, Y.H.; Hu, H.T.; Yang, J.; Yang, G.D. The herbal medicine dipsacus asper wall extract reduces the cognitive deficits and overexpression of β-amyloid protein induced by aluminum exposure. Life Sci. 2003, 73, 2443–2454. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Yang, Z.L.; Xu, L.; Li, P.; Hu, Y.Z. Akebia saponin D, a saponin component from Dipsacus asper wall, protects PC 12 cells against amyloid-beta induced cytotoxicity. Cell Biol. Int. 2009, 33, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Caracciolo, L.; Tweedie, D.; Choi, S.H.; Greig, N.H.; Barlati, S.; Bosetti, F. 3,6’-dithiothalidomide, a new TNF-α synthesis inhibitor, attenuates the effect of Aβ–42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J. Neurochem. 2012, 122, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, L.N.; Du, Q.M.; Ma, L.; Chen, L.; You, R.; Liu, L.; Ling, J.J.; Yang, Z.L.; Ji, H. Akebia saponin d attenuates amyloid β-induced cognitive deficits and inflammatory response in rats: Involvement of akt/nf-kappab pathway. Benav. Brain Res. 2012, 235, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Geldmacher, D.; Farlow, M.; Sabbagh, M.; Christensen, D.; Betz, P.; Grp, D.M.E.W. High-dose donepezil (23 mg/day) for the treatment of moderate and severe Alzheimer’s disease: Drug profile and clinical guidelines. CNS Neurosci. Ther. 2013, 19, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Lei, Y.; Tang, X.C.; Zhang, H.Y. Donepezil attenuates abeta-associated mitochondrial dysfunction and reduces mitochondrial abeta accumulation in vivo and in vitro. Neuropharmacology 2015, 95, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liu, Q.; Wang, Y.; Luo, G. Coadministration of huperzine a and ligustrazine phosphate effectively reverses scopolamine-induced amnesia in rats. Pharmacol. Biochem. Behav. 2010, 96, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease and down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Olkhov, V.K.; Rayburn, S.N. Leading-Edge Research in Alzheimer’s Disease; Nova Science Publishers: Hauppauge, NY, USA, 2008; pp. 125–131. [Google Scholar]
- Candore, G.; Bulati, M.; Caruso, C.; Castiglia, L.; Colonna-Romano, G.; di Bona, D.; Duro, G.; Lio, D.; Matranga, D.; Pellicano, M.; et al. Inflammation, cytokines, immune response, apolipoprotein E, cholesterol, and oxidative stress in alzheimer disease: Therapeutic implications. Rejuv. Res. 2010, 13, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, A.; Sadraie, H.; Dargahi, L.; Khalaj, L.; Ahmadiani, A. Apoptosis inhibition can be threatening in Aβ-induced neuroinflammation, through promoting cell proliferation. Neurochem. Res. 2011, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. C-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death. Dis. 2014, 5, e1019. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhao, D.; Xie, C.W. Neurotrophins enhance camkii activity and rescue amyloid-beta-induced deficits in hippocampal synaptic plasticity. J. Alzheimers Dis. 2010, 21, 823–831. [Google Scholar] [PubMed]
- Gao, C.; Liu, Y.; Jiang, Y.; Ding, J.; Li, L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3β pathway in streptozotocin-induced alzheimer rat model. Brain Pathol. 2014, 24, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, M.; Delobel, P.; Sambo, A.V.; Smet, C.; Begard, S.; Violleau, A.; Landrieu, I.; Delacourte, A.; Lippens, G.; Flament, S.; et al. Neurofibrillary degeneration of the Alzheimer-type: an alternate pathway to neuronal apoptosis? Biochem. Pharmacol. 2003, 66, 1619–1625. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef] [PubMed]
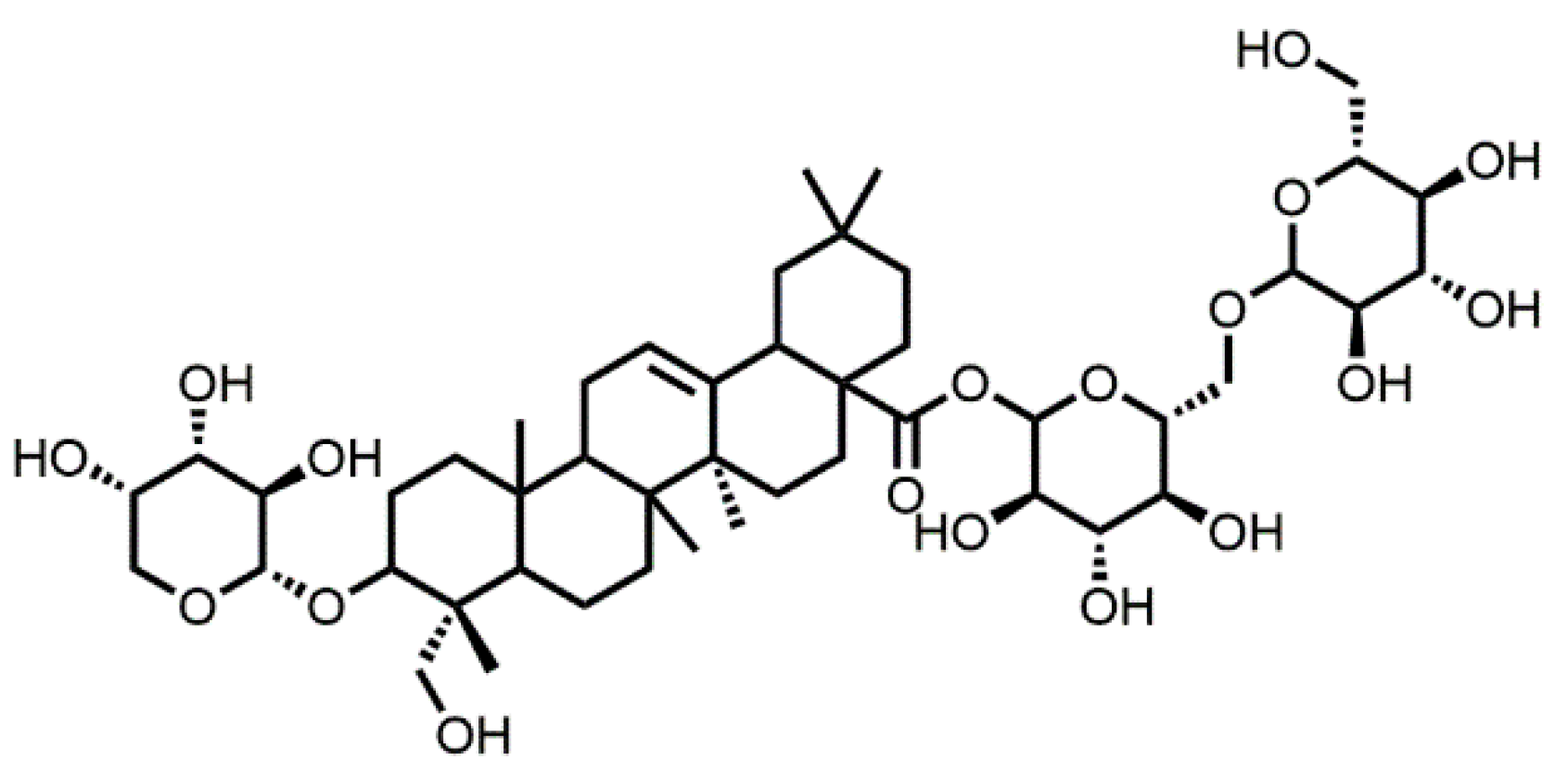
- Sample Availability: Samples of the compound Akebia saponin D is available from Zhong-Lin Yang.






© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Yang, X.; Chen, T.; Ji, J.; Lan, L.; Hu, R.; Ji, H. Treatment with Akebia Saponin D Ameliorates Aβ1–42-Induced Memory Impairment and Neurotoxicity in Rats. Molecules 2016, 21, 323. https://doi.org/10.3390/molecules21030323
Chen Y, Yang X, Chen T, Ji J, Lan L, Hu R, Ji H. Treatment with Akebia Saponin D Ameliorates Aβ1–42-Induced Memory Impairment and Neurotoxicity in Rats. Molecules. 2016; 21(3):323. https://doi.org/10.3390/molecules21030323
Chicago/Turabian StyleChen, Yongde, Xiaolin Yang, Tong Chen, Jing Ji, Li Lan, Rong Hu, and Hui Ji. 2016. "Treatment with Akebia Saponin D Ameliorates Aβ1–42-Induced Memory Impairment and Neurotoxicity in Rats" Molecules 21, no. 3: 323. https://doi.org/10.3390/molecules21030323