
Pharmacokinetic and Metabolic Characteristics of Herb-Derived Khellactone Derivatives, A Class of Anti-HIV and Anti-Hypertensive: A Review


Abstract
:1. Introduction
2. Pharmacokinetic Properties of Khellactone Derivatives
2.1. Absorption
2.2. Distribution
2.3. Excretion
3. Metabolism of Khellactone Derivatives
3.1. Metabolic Pathways of Khellactone Derivatives
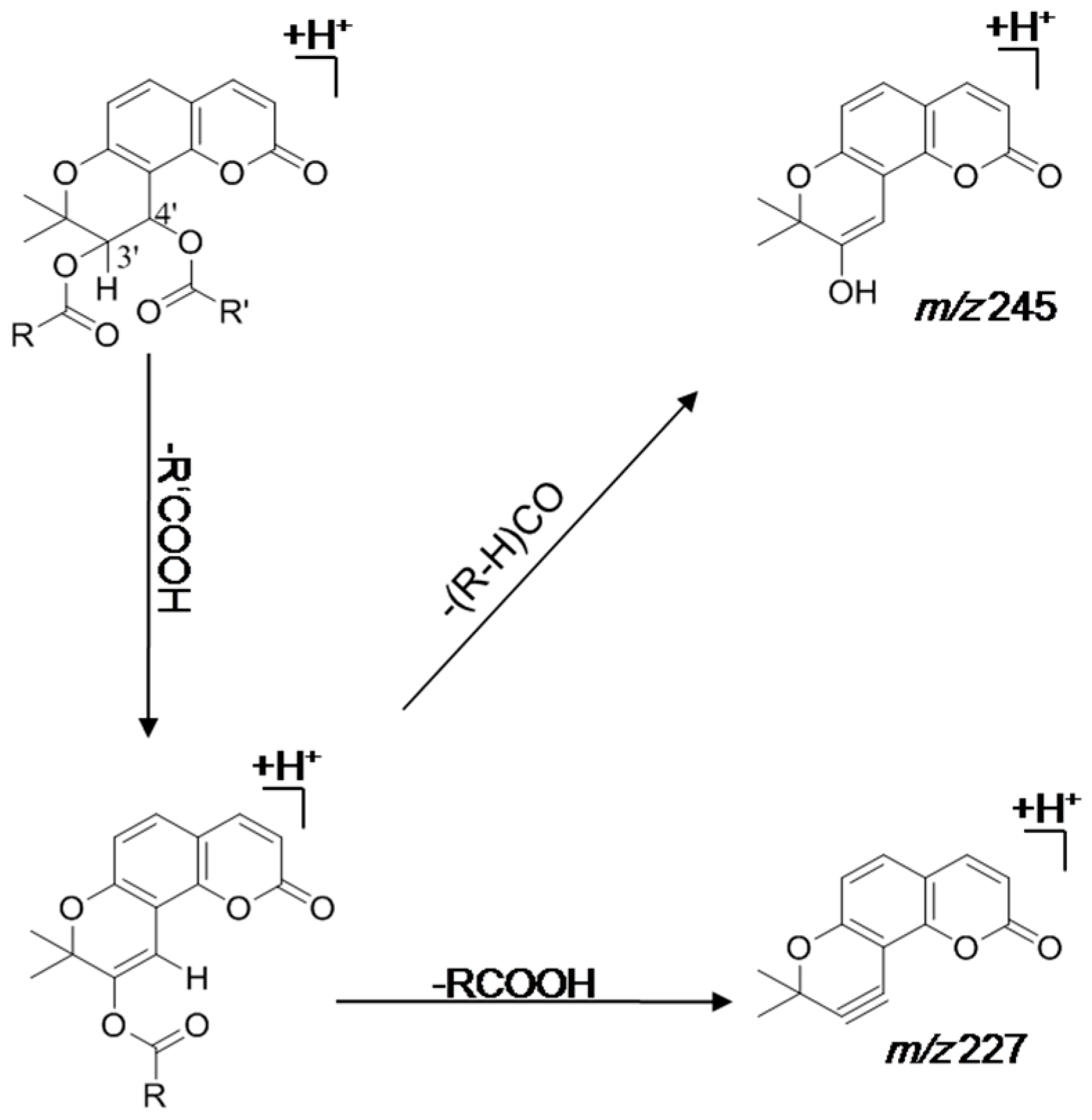
3.1.1. Hydrolysis of Khellactone Derivatives
3.1.2. Oxidation of Khellactone Derivatives
3.2. Enzyme Involved in the Metabolism of KDs
3.3. Structure-Metabolism Relationship
4. Potential DDIs
4.1. CYP450s and UGTs Mediated DDIs
4.2. Transporter-Mediated DDIs
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lin, J.H.; Lu, A.Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar] [PubMed]
- Song, Y.L.; Jing, W.H.; Yan, R.; Wang, Y.T. Research progress of the studies on the roots of Peucedanum praeruptorum Dunn (Peucedani Radix). Pak. J. Pharm. Sci. 2014, 28, 71–81. [Google Scholar]
- Xie, L.; Zhao, C.H.; Zhou, T.; Chen, H.F.; Fan, B.T.; Chen, X.H.; Ma, J.Z.; Li, J.Y.; Bao, Z.Y.; Lo, Z.; et al. Molecular modeling, design, synthesis, and biological evaluation of novel 3′,4′-dicamphanoyl-(+)-cis-khellactone (DCK) analogs as potent anti-HIV agents. Bioorg. Med. Chem. 2005, 13, 6435–6449. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Huang, W.L.; Peng, S.X.; Hua, W.Y. Synthesis and bioactivity of some 3,4-diacyloxybenzopyran. Yao Xue Xue Bao 1997, 32, 97–105. [Google Scholar] [PubMed]
- Fong, W.F.; Shen, X.L.; Globisch, C.; Wiese, M.; Chen, G.Y.; Zhu, G.Y.; Yu, Z.L.; Tse, A.K.; Hu, Y.J. Methoxylation of 3′,4′-aromatic side chains improves P-glycoprotein inhibitory and multidrug resistance reversal activities of 7,8-pyranocoumarin against cancer cells. Bioorg. Med. Chem. 2008, 16, 3694–3703. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Du, G.; Yang, F.Q.; Yan, R.; Wang, Y.T. Qualitative analysis and enantiospecific determination of angular-type pyranocoumarins in Peucedani Radix using achiral and chiral liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. A 2014, 1338, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Tu, P.F.; Wang, Y.T. Enantiomeric separation of angular-type pyranocoumarins from Peucedani Radix using AD-RH chiral column. Nat. Prod. Res. 2014, 28, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Jing, W.; Yang, F.; Shi, Z.; Yao, M.; Yan, R.; Wang, Y. Simultaneously enantiospecific determination of (+)-trans-khellactone, (+/−)-praeruptorin A, (+/−)-praeruptorin B, (+)-praeruptorin E, and their metabolites, (+/−)-cis-khellactone, in rat plasma using online solid phase extraction-chiral LC-MS/MS. J. Pharm. Biomed. Anal. 2014, 88, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Zhao, H.Y.; Yan, R.; Li, P.T.; Wang, Y.T. Stereoselective metabolism of (+/−)-praeruptorin A, a calcium channel blocker from Peucedani Radix, in pooled liver microsomes of rats and humans. Xenobiotica 2012, 42, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Zhang, Q.W.; Li, Y.P.; Yan, R.; Wang, Y.T. Enantioseparation and absolute configuration determination of angular-type pyranocoumarins from Peucedani Radix using enzymatic hydrolysis and chiral HPLC-MS/MS analysis. Molecules 2012, 17, 4236–4251. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.; Kashiwada, Y.; Huang, L.; Snider, J.; Cosentino, M.; Lee, K.H. Suksdorfin: An anti-HIV principle from Lomatium suksdorfii, its structure-activity correlation with related coumarins, and synergistic effects with anti-AIDS nucleosides. Bioorg. Med. Chem. 1994, 2, 1051–1056. [Google Scholar] [CrossRef]
- Huang, L.; Kashiwada, Y.; Cosentino, L.M.; Fan, S.; Chen, C.H.; McPhail, A.T.; Fujioka, T.; Mihashi, K.; Lee, K.H. Anti-AIDS agents. 15. Synthesis and anti-HIV activity of dihydroseselins and related analogs. J. Med. Chem. 1994, 37, 3947–3955. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yuan, X.; Yu, D.; Lee, K.H.; Chen, C.H. Mechanism of action and resistant profile of anti-HIV-1 coumarin derivatives. Virology 2005, 332, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Morris-Natschke, S.L.; Lee, K.H. New developments in natural products-based anti-AIDS research. Med. Res. Rev. 2007, 27, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S.L.; Lee, K.H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003, 23, 322–345. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Takeuchi, Y.; Cosentino, L.M.; Lee, K.H. Anti-AIDS agents. 37. Synthesis and structure-activity relationships of (3′R,4′R)-(+)-cis-khellactone derivatives as novel potent anti-HIV agents. J. Med. Chem. 1999, 42, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Guo, H.F.; Lu, H.; Zhuang, X.M.; Zhang, A.M.; Wu, G.; Ruan, J.X.; Zhou, T.; Yu, D.; Qian, K.; et al. Development and preclinical studies of broad-spectrum anti-HIV agent (3′R,4′R)-3-cyanomethyl-4-methyl-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone (3-cyanomethyl-4-methyl-DCK). J. Med. Chem. 2008, 51, 7689–7696. [Google Scholar] [CrossRef] [PubMed]
- Usach, I.; Melis, V.; Peris, J.E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sarkhail, P.; Shafiee, A.; Sarkheil, P. Biological activities and pharmacokinetics of praeruptorins from Peucedanum species: A systematic review. BioMed Res. Int. 2013, 2013, 343808. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Chen, Y.G.; Yuan, Y.F.; Yan, R.; Wang, Y.T. 1H nuclear magnetic resonance based-metabolomic characterization of Peucedani Radix and simultaneous determination of praeruptorin A and praeruptorin B. J. Pharm. Biomed. Anal. 2014, 93, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, X.; Dai, Y.; Kong, L.; Wang, F.; Xu, H.; Lu, D.; Song, J.; Hou, Z. (+/−)-Praeruptorin A enantiomers exert distinct relaxant effects on isolated rat aorta rings dependent on endothelium and nitric oxide synthesis. Chem.-Bio. Int. 2010, 186, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.Y.; Wu, X.L.; Min, Z.D. Semi-synthesis of derivatives with C-3′ and C-4′ trans-configuration from (+)-praeruptorin A. Yao Xue Xue Bao 2003, 38, 358–363. [Google Scholar] [PubMed]
- Wu, X.L.; Kong, L.Y.; Min, Z.D. Studies on structure modification of (+)-praeruptorin A. Yao Xue Xue Bao 2002, 37, 527–534. [Google Scholar] [PubMed]
- Lu, H. Stereoselectivity in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2007, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Chen, G.; Zhu, G.; Cai, J.; Wang, L.; Hu, Y.; Fong, W.F. 3′-O, 4′-O-aromatic acyl substituted 7,8-pyranocoumarins: A new class of P-glycoprotein modulators. J. Pharm. Pharm. 2012, 64, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Chen, G.; Zhu, G.; Fong, W.F. (+/−)-3′-O, 4′-O-dicynnamoyl-cis-khellactone, a derivative of (+/−)-praeruptorin A, reverses P-glycoprotein mediated multidrug resistance in cancer cells. Bioorg. Med. Chem. 2006, 14, 7138–7145. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Fong, W.F.; Zhang, J.X.; Leung, C.H.; Kwong, H.L.; Yang, M.S.; Li, D.; Cheung, H.Y. Reversal of multidrug resistance in cancer cells by pyranocoumarins isolated from Radix Peucedani. Eur. J. Pharm. 2003, 473, 9–17. [Google Scholar] [CrossRef]
- Hsiao, G.; Ko, F.N.; Jong, T.T.; Teng, C.M. Antiplatelet action of 3′,4′-diisovalerylkhellactone diester purified from Peucedanum japonicum Thunb. Biol. Pharm. Bull. 1998, 21, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Aida, Y.; Kasama, T.; Takeuchi, N.; Tobinaga, S. The antagonistic effects of khellactones on platelet-activating factor, histamine, and leukotriene D4. Chem. Pharm. Bull. 1995, 43, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.J.; Li, J.R.; Zhu, Z.G.; Kong, H.Y.; Jin, H.; Zhang, J.Y.; Tian, Y.X.; Li, Z.H.; Wu, X.Y.; Zhang, J.J.; et al. Praeruptorin D and E attenuate lipopolysaccharide/hydrochloric acid induced acute lung injury in mice. Eur. J. Pharm. 2013, 710, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Y.; Wang, J.S.; Wu, F.H.; Li, J.; Kong, L.Y. The effects of (+/−)-Praeruptorin A on airway inflammation, remodeling and transforming growth factor-beta1/Smad signaling pathway in a murine model of allergic asthma. Int. Immun. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, J.; Wu, F.; Li, J.; Zhou, L.; Kong, L. Effects of (+/−)-praeruptorin A on airway inflammation, airway hyperresponsiveness and NF-κB signaling pathway in a mouse model of allergic airway disease. Eur. J. Pharm. 2012, 683, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Fong, W.F.; Wu, J.Y.; Yang, M.; Cheung, H.Y. Pyranocoumarins isolated from Peucedanum praeruptorum as differentiation inducers in human leukemic HL-60 cells. Planta Med. 2003, 69, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.F.; Zhang, J.X.; Wu, J.Y.; Tse, K.W.; Wang, C.; Cheung, H.Y.; Yang, M.S. Pyranocoumarin (+/−)-4′-O-acetyl-3′-O-angeloyl-cis-khellactone induces mitochondrial-dependent apoptosis in HL-60 cells. Planta Med. 2004, 70, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Yue, W.; Li, Q. Chemopreventive effects of Peucedanum praeruptorum Dunn and its major constituents on SGC7901 gastric cancer cells. Molecules 2010, 15, 8060–8071. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Yue, W.; Du, X.; Ren, L.; Li, Q. Pharmacokinetics and tissue distribution study of praeruptorin d from radix peucedani in rats by high-performance liquid chromatography (HPLC). Int. J. Mol. Sci. 2012, 13, 9129–9141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liang, X.; Su, M.; Liang, Q.; Li, L.; Zhang, X.; Wang, X.; Zhu, X. Pharmacokinetics of dl-praeruptorin A after single-dose intravenous administration to rats with liver cirrhosis. Daru 2011, 19, 210–215. [Google Scholar] [PubMed]
- Ma, Y.F.; Hu, F.D.; Li, W.; Feng, S.L.; Sun, Q.; Shi, Y.K.; Bi, Y.Y.; Hu, F.; Wang, Z. Pharmacokinetics study and its main metabolite of pteryxin in rats by LC-MS/MS. Zhongguo Shiyan Fangjixue Zazhi 2011, 17, 112–115. [Google Scholar]
- Zhang, Z.; Liu, Y.Y.; Su, M.Q.; Liang, X.F.; Wang, W.F.; Zhu, X. Pharmacokinetics, tissue distribution and excretion study of dl-praeruptorin A of Peucedanum praeruptorum in rats by liquid chromatography tandem mass spectrometry. Phytomedicine 2011, 18, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Zhou, R.N.; Yan, R.; Wang, Y.T. Stereoselective pharmacokientics of (+/−)-praeruptorin A in rats. Drug Metab. Rev. 2011, 43, 281–282. [Google Scholar]
- Wang, J.; Ma, Y.; Li, W.; Hu, F.; Chen, T.; Shen, X.; Feng, S. Study on pharmacokinetics and tissue distribution of pteryxin in mice by ultra-pressure liquid chromatography with tandem mass spectrometry. Biomed. Chromatogr. 2012, 26, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Tu, P.F.; Wang, Y.T. Characterization of Peucedani Radix extract-derived angular-type pyranocoumarins in rats using ultra high performance liquid chromatography coupled with hybrid triple quadrupole-linear ion trap mass spectrometry. Anal. Methods 2014, 6, 5198–5206. [Google Scholar] [CrossRef]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man-fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yang, X.B.; Yang, X.W.; Wu, Q.; Wang, Y.; Zhang, L.X.; Xu, W. Intestinal permeability of the constituents from the roots of Saposhnikovia divaricata in the human Caco-2 cell monolayer model. Planta Med. 2011, 77, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Chuan, X.X.; Hong, X.X.; Jin, M.H.; Zhu, X.; Hong, Y.; Meng, F.H. Influence of penetration enhancers on in vitro transdermal permeation of dl-praeruptorin A. Chin. J. New Drugs 2011, 21, 926–930. [Google Scholar]
- Jing, W.H.; Song, Y.L.; Yan, R.; Bi, H.C.; Li, P.T.; Wang, Y.T. Transport and metabolism of (+/−)-praeruptorin A in Caco-2 cell monolayers. Xenobiotica 2011, 41, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.M.; Shen, G.L.; Yuan, M.; Li, H. Investigation of the pharmacokinetic interaction between ritonavir and CMDCK, a new non-nucleoside reverse transcriptase inhibitor. Drug Res. 2013, 63, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhuang, X.; Qian, K.; Sun, L.; Wang, X.; Li, H.; Lee, K.; Xie, L. Prodrug design, synthesis and pharmacokinetic evaluation of (3′,4′)-3-hydroxymethyl-4-methyl-3′,4′-di-(S)-camphanoyl-(+)-cis-khellactone. Acta Pharm. Sin. B 2012, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Zhang, Z.; Zhu, X. Liquid chromatography tandem mass spectrometry pharmacokinetic study of DL-praeruptorin A in rat plasma. Biomed. Chromatogr. 2010, 24, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.Y.; Liang, T.G.; Du, X.; Li, Q.S. The absorption and transport characteristics of praeruptorin in Caco-2 cell model. Zhongguo Yaowu Yu Linchuang 2012, 12, 13–15. [Google Scholar]
- Zhuo, G.; Chen, G.; Liu, H. Simultaneous quantification of three pyranocoumarins of Peucedanum praeruptorum in rat plasma by liquid chromatography-tandem mass spectrometry: Application to pharmacokinetic study. J. Chromatogr. Sci. 2015, 53, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Jing, W.H.; Yan, R.; Wang, Y.T. A pretreatment free method for the determination of seven natural products in a high-salt matrix by online guard column extraction coupled with tandem mass spectrometry. Anal. Method. 2014, 6, 623–628. [Google Scholar] [CrossRef]
- Lin, J.H. Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv. Drug Deliv. Rev. 2003, 55, 53–81. [Google Scholar] [CrossRef]
- Song, Y.L.; Jing, W.H.; Yan, R.; Wang, Y.T. Metabolic characterization of (+/−)-praeruptorin A in vitro and in vivo by high performance liquid chromatography coupled with hybrid triple quadrupole-linear ion trap mass spectrometry and time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2014, 90, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Toon, S.; Rowland, M. Structure-pharmacokinetic relationships among the barbiturates in the rat. J. Pharm. Exp. Ther. 1983, 225, 752–763. [Google Scholar]
- Zhuang, X.M.; Wen, Y.Y.; Li, H.; Deng, J.T.; Kong, W.L.; Tian, X.T.; Cui, S.L.; Xie, L. Metabolism of 3-cyanomethyl-4-methyl-DCK, a new anti-HIV candidate, in human intestinal microsomes. Yao Xue Xue Bao 2010, 45, 1116–1122. [Google Scholar] [PubMed]
- Ruan, H.; Zhang, Z.; Liang, X.F.; Fu, Y.; Su, M.Q.; Liu, Q.L.; Wang, X.M.; Zhu, X. Metabolism of dl-praeruptorin A in rat liver microsomes using HPLC-electrospray ionization tandem mass spectrometry. Arch. Pharm. Res. 2011, 34, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.M.; Deng, J.T.; Li, H.; Kong, W.L.; Ruan, J.X.; Xie, L. Metabolism of novel anti-HIV agent 3-cyanomethyl-4-methyl-DCK by human liver microsomes and recombinant CYP enzymes. Acta Pharm. Sin. 2011, 32, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Li, Y.; Smith, P.C.; Swenberg, J.A.; Martin, D.E.; Morris-Natschke, S.L.; Lee, K.H. Anti-AIDS agents 65: Investigation of the in vitro oxidative metabolism of 3′,4′-Di-O-(−)-camphanoyl-(+)-cis-khellactone derivatives as potent anti-hiv agents. Drug Metab. Dispos. 2005, 33, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.H.; Song, Y.L.; Yan, R.; Wang, Y.T. Identification of cytochrome P450 isoenzymes involved in metabolism of (+)-praeruptorin A, a calcium channel blocker, by human liver microsomes using ultra high-performance liquid chromatography coupled with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2013, 77, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Yan, R.; Jing, W.H.; Zhao, H.Y.; Wang, Y.T. Characterization of metabolism of (+)-praeruptorin B and (+)-praeruptorin E in human and rat liver microsomes by liquid chromatography coupled with ion trap mass spectrometry and time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, G.; Zuo, Z. Position preference on glucuronidation of mono-hydroxylflavones in human intestine. Life Sci. 2006, 78, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.G. Coumarin metabolism, toxicity and carcinogenicity: Relevance for human risk assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]
- Lewis, D.F.; Ito, Y.; Lake, B.G. Metabolism of coumarin by human P450s: A molecular modelling study. Toxicol. in Vitro 2006, 20, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Born, S.L.; Caudill, D.; Fliter, K.L.; Purdon, M.P. Identification of the cytochromes P450 that catalyze coumarin 3,4-epoxidation and 3-hydroxylation. Drug Metab. Dispos. 2002, 30, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Gu, J.; Zhang, Q.Y.; Spink, D.C.; Kaminsky, L.S.; Ding, X. Biotransformation of coumarin by rodent and human cytochromes P-450: Metabolic basis of tissue-selective toxicity in olfactory mucosa of rats and mice. J. Pharm. Exp. Ther. 1999, 288, 463–471. [Google Scholar]
- Wang, D.C.; Zhao, X.M.; Li, T.D.; Ma, J.; Kong, Z.F.; Li, K.; Gao, Y.S. Effects of total coumarins from Peucedanum praeruptorum Dunn on the activity of hepatic drug-metablizing enzymes in mice. Yi Yao Dao Bao 2004, 23, 34–36. [Google Scholar]
- Iwata, H.; Tezuka, Y.; Usia, T.; Kadota, S.; Hiratsuka, A.; Watabe, T. Inhibition of human liver microsomal CYP3A4 and CYP2D6 by extracts from 78 herbal medicines. J. Tradit. Med. 2004, 21, 42–50. [Google Scholar]
- Iwata, H.; Tezuka, Y.; Kadota, S.; Hiratsuka, A.; Watabe, T. Metabolism-dependent inhibition of CYP3A4 and CYP2D6 by extracts from 26 herbal medicines. J. Tradit. Med. 2004, 21, 281–286. [Google Scholar]
- Huang, L.; Bi, H.C.; Li, Y.H.; Zhang, J.Q.; Kuang, S.Y.; Zhang, L.; Wang, Y.T.; Huang, M. Regulation of human pregnane X receptor and its target gene cytochrome P450 3A by praeruptorin A isolated from the herbal medicine Peucedanum praeruptorum. Planta Med. 2013, 79, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bi, H.C.; Liu, Y.H.; Wang, Y.T.; Xue, X.P.; Huang, M. CAR-mediated up-regulation of CYP3A4 expression in LS174T cells by Chinese herbal compounds. Drug Metab. Pharm. 2011, 26, 331–340. [Google Scholar] [CrossRef]
- Huang, L.; Huang, M.; Li, Y.H.; Li, R.M.; Zeng, Y.; Kuang, S.Y.; Zhang, L.; Wang, Y.T.; Bi, H.C. Up-regulatation of CYP3A expression through pregnent X receptor by praeruptorin D isolated from Peucedanum praeruptorum Dunn. J. Ethnopharmacol. 2013, 148, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, Q.; Li, Y.H.; Wang, Y.T.; Bi, H.C. PXR-Mediated Upregulation of CYP3A Expression by Herb Compound Praeruptorin C from Peucedanum praeruptorum Dunn. Evid. Based Complement. Altern. Med. 2013, 2013, 156574. [Google Scholar] [CrossRef]
- Liu, Y.H.; Mo, S.L.; Bi, H.C.; Hu, B.F.; Li, C.G.; Wang, Y.T.; Huang, L.; Huang, M.; Duan, W.; Liu, J.P.; et al. Regulation of human pregnane X receptor and its target gene cytochrome P450 3A4 by Chinese herbal compounds and a molecular docking study. Xenobiotica 2011, 41, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Zeng, S.; Xie, W. Nuclear receptors PXR and CAR: Implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin. Drug Metab. Toxicol. 2013, 9, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.X.; Lambert, M.H.; Wisely, B.B.; Warren, E.N.; Weinert, E.E.; Waitt, G.M.; Williams, J.D.; Collins, J.L.; Moore, L.B.; Willson, T.M.; et al. A structural basis for constitutive activity in the human CAR/RXRα heterodimer. Mol. Cell 2004, 16, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Vincent, J.; Brunzelle, J.S.; Dussault, I.; Lin, M.; Ianculescu, I.; Sherman, M.A.; Forman, B.M.; Fernandez, E.J. Structure of the murine constitutive androstane receptor complexed to androstenol: A molecular basis for inverse agonism. Mol. Cell 2004, 16, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xie, W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab. Dispos. 2010, 38, 2091–2095. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Route | CL (mL/min/kg) | Vd (L/kg) | AUC (μg·min/L) | t1/2 (min) | Cmax (ng/mL) | tmax (h) | MRT0–t (h) | F (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Ref. 13-4 a | i.v. | 0.01 ± 0.003 | 2.34 ± 0.31 | 195,270.08 | 97 ± 11 | N.A. | N.A. | N.A. | N.A. | [17] |
| Ref. 13-4 | i.g. | 0.058 ± 0.01 | 8.19 ± 2.60 | 348,067.29 | 167 ± 30 | N.A. | N.A. | N.A. | 17.8 | [17] |
| CMDCK | i.v. | 0.044 ± 0.010 | 8.14 ± 5.88 | 47,082 ± 10,464 | 120 ± 60 | N.A. | N.A. | N.A. | N.A. | [47] |
| CMDCK | i.g. | N.A. | N.A. | 29,874 ± 4524 | 200.4 ± 31.8 | 105.7 ± 73.3 | 0.4 ± 0.1 | N.A. | 15.8 ± 2.1 | [47] |
| HMDCK b | i.v. | 0.050 ± 0.0017 | 0.008 ± 0.002 | 558.4 ± 8.71 | N.A. | N.A. | N.A. | 1.235 ± 0.618 | N.A. | [48] |
| HMDCK b | i.g. | 0.33 ± 0.083 | 0.079 ± 0.039 | 57,978 ± 13,182 | N.A. | 530.7 ± 120.8 | 0.58 | 1.983 ± 0.478 | 17.3 | [48] |
| Ref. 48-5 | i.g. | 0.55 ± 0.10 | 0.252 ± 0.130 | 31,302 ± 8016 | N.A. | 284.1 ± 74.73 | 0.50 | 3.092 ± 0.454 | 10.3 | [48] |
| Ref. 48-6 | i.g. | 1.7 ± 0.33 | 0.669 ± 0.422 | 9474 ± 2576.4 | N.A. | 80.9 ± 18.80 | 0.38 | 2.658 ± 0.681 | 3.3 | [48] |
| Ref. 48-7 | i.g. | 0.77 ± 0.22 | 0.373 ± 0.193 | 21,252 ± 6336 | N.A. | 254.6 ± 32.45 | 0.25 | 3.769 ± 0.063 | 7.2 | [48] |
| Ref. 48-8 | i.g. | 0.57 ± 0.067 | 0.102 ± 0.011 | 35,334 ± 4323.6 | N.A. | 398.5 ± 29.00 | 0.50 | 1.517 ± 0.207 | 12.4 | [48] |
| Ref. 48-9 | i.g. | 0.62 ± 0.017 | 0.467 ± 0.063 | 23,046 ± 372.6 | N.A. | 163.6 ± 13.56 | 0.25 | 3.581 ± 0.307 | 7.5 | [48] |
| Ref. 48-10 | i.g. | 0.27 ± 0.12 | 0.045 ± 0.017 | 77,790 ± 26,256 | N.A. | 621.5 ± 79.67 | 0.75 | 1.793 ± 0.181 | 25.7 | [48] |
| Ref. 48-12 | i.g. | 0.43 ± 0.083 | 0.049 ± 0.020 | 46,536 ± 9774 | N.A. | 529.0 ± 192.9 | 0.50 | 1.286 ± 0.085 | 15.8 | [48] |
| Ref. 48-13 | i.g. | 0.67 ± 0.17 | 0.123 ± 0.058 | 28,668 ± 7722 | N.A. | 191.1 ± 2.48 | 0.75 | 2.136 ± 0.123 | 9.8 | [48] |
| PA | i.v. | N.A. | N.A. | 37,835.6 ± 5871.6 | 51.18 ± 9.02 | N.A. | N.A. | N.A. | N.A. | [37,39,49] |
| Pteryxin | i.g. | N.A. | N.A. | 4128.08 | 87.78 | 976.04 | 2.00 | 6.732 | N.A. | [38,41] |
| dPB | i.v. | 9.6 ± 3.2 | N.A. | 1,088,700 ± 375,900 | 7.14 ± 2.14 | N.A. | N.A. | N.A. | N.A. | [36] |
| dPA | i.v. | N.A. | N.A. | 80,346 ± 8724 | 109.2 ± 51.6 | N.A. | N.A. | 1.61 ± 0.58 | N.A. | [40] |
| lPA c | i.v. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | [40] |
| lCK d | i.g. | N.A. | N.A. | 191,172 ± 89,460 | 352.2 ± 169.8 | 345.6 ± 204.0 | 0.71 ± 0.19 | 10.0 ± 3.35 | N.A. | [40] |
| lCK e | i.g. | N.A. | N.A. | 512,820 ± 235,470 | 353.4 ± 157.2 | 1156.3 ± 637.6 | 0.72 ± 0.53 | 10.3 ± 3.95 | N.A. | [40] |
| dCK f | i.g. | N.A. | N.A. | 46,536 ± 22,890 | 409.2 ± 180.6 | 108.5 ± 38.2 | 0.43 ± 0.35 | 11.0 ± 4.46 | N.A. | [40] |
| lCK f | i.g. | N.A. | N.A. | 177,612 ± 98,910 | 427.8 ± 266.4 | 685.1 ± 254.3 | 0.36 ± 0.20 | 13.7 ± 5.84 | N.A. | [40] |
| dTK g | i.g. | N.A. | N.A. | 29,280 ± 6462 | 526.2 ± 69 | 55.5 ± 31.3 | 4.83 ± 1.83 | N.A. | N.A. | [8] |
| lCK g | i.g. | N.A. | N.A. | 213,414 ± 54,096 | 420 ± 85.2 | 468 ± 233 | 1.08 ± 1.14 | N.A. | N.A. | [8] |
| dCK g | i.g. | N.A. | N.A. | 50,982 ± 10,110 | 420.6 ± 84 | 362 ± 224 | 1.08 ± 1.14 | N.A. | N.A. | [8] |
| dPA g | i.g. | N.A. | N.A. | 3468 ± 1620 | 1666.2 ± 2049 | 19.8 ± 11.3 | 0.38 ± 0.56 | N.A. | N.A. | [8] |
| dPB g | i.g. | N.A. | N.A. | 7932 ± 2088 | 526.2 ± 69 | 10.3 ± 5.42 | 4.83 ± 1.83 | N.A. | N.A. | [8] |
| dPE g | i.g. | N.A. | N.A. | 7734 ± 954 | 526.2 ± 69 | 5.35 ± 0.41 | 4.83 ± 1.83 | N.A. | N.A. | [8] |
| Compound | Papp AP→BL (×10−6, cm/s) | Papp BL→AP (×10−6, cm/s) | Papp BL→AP/Papp AP→BL | Ref. |
|---|---|---|---|---|
| dPB | 1.25 ± 0.05 | 1.33 ± 0.13 | 1.06 | [50] |
| lPB | 15.27 ± 0.45 | 13.82 ± 1.37 | 0.90 | [51] |
| dPA | 22.5–30.3 | 16.5–19.7 | 0.6–0.8 | [46] |
| lPA | 20.1–28.2 | 15.8–18.8 | 0.6–0.8 | [46] |
| CMDCK | 23.2 ± 1.76 | 12.1 ± 1.37 | 0.52 | [17,47] |
| Comp. | t1/2 (min) | CLint (mL/min/mg) | CLh (mL/min/kg) | Qh (mL/min/kg) | Km (μmol/L) | Vmax (pmol/min/mg) | Ref. |
|---|---|---|---|---|---|---|---|
| Ref. 38-1 a | 2.4 | 2.9 | - | - | - | - | [59] |
| Ref. 38-2 | 5.1 | 1.4 | - | - | - | - | [59] |
| Ref. 38-3 | 1.5 | 4.6 | - | - | - | - | [59] |
| Ref. 38-4 | 2.1 | 3.3 | - | - | - | - | [59] |
| Ref. 38-5 | 3.7 | 1.9 | - | - | - | - | [59] |
| Ref. 38-6 | 2.0 | 3.5 | - | - | - | - | [59] |
| Ref. 38-7 | 3.4 | 2.0 | - | - | - | - | [59] |
| Ref. 38-8 | 5.2 | 1.3 | - | - | - | - | [59] |
| Ref. 38-9 | 2.2 | 3.2 | - | - | - | - | [59] |
| Ref. 38-10 | 2.0 | 3.5 | - | - | - | - | [59] |
| Ref. 38-11 | 2.1 | 3.3 | - | - | - | - | [59] |
| Ref. 38-12 | b | b | - | - | - | - | [59] |
| Ref. 38-13 | 5.7 | 1.2 | - | - | - | - | [59] |
| Ref. 38-14 | 4.3 | 1.6 | - | - | - | - | [59] |
| Ref. 48-3 | 17.92 | 0.39 | - | - | - | - | [48] |
| Ref. 48-5 | 34.61 | 0.20 | - | - | - | - | [48] |
| Ref. 48-6 | 35.27 | 0.20 | - | - | - | - | [48] |
| Ref. 48-7 | 29.07 | 0.24 | - | - | - | - | [48] |
| Ref. 48-8 | 25.93 | 0.27 | - | - | - | - | [48] |
| Ref. 48-9 | 3.90 | 1.78 | - | - | - | - | [48] |
| Ref. 48-10 | 49.26 | 0.14 | - | - | - | - | [48] |
| Ref.48-11 | 30.09 | 0.23 | - | - | - | - | [48] |
| Ref. 48-12 | 30.77 | 0.23 | - | - | - | - | [48] |
| Ref. 48-13 | 26.54 | 0.26 | - | - | - | - | [48] |
| CMDCK (HIM) | 25.7 | 0.012 | 3.3 | - | 45.6 | 0.33 | [56] |
| CMDCK (HLM) | 5.62 ± 0.57 | 0.31 ± 0.031 | 19.4 ± 0.12 | 20.7 | 14.3 | 1.78 | [58] |
| CMDCK (CYP3A4) | 6.84 ± 1.55 | - | - | - | 12.1 | 1.58 | [58] |
| PA (HLM) | 30.13 c | 0.27 | 0.12 | - | - | - | [54] |
| lCK | 1.29 | 0.02 ± 0.004 | 25.8 ± 2.70 | [60] | |||
| CAK-4 | 4.33 ± 1.40 | 0.402 ± 0.0715 | [40] | ||||
| CAK-3 | 9.97 ± 3.55 | 0.663 ± 0.165 | [40] | ||||
| PA(RLM) | 8.19 c | 0.24 ± 0.02 | - | - | 64.1 ± 4.22 | 0.26 ± 0.036 | [37,54,57] |
| dPA(HLM) | 22.65 c | 0.20 | - | - | 17.83 ± 15.02 | - | [9,60] |
| dPA(RLM) | 10.24 c | - | - | - | - | - | [9] |
| lPA(HLM) | 31.09 c | 0.28 | - | - | - | - | [9] |
| lPA(RLM) | 3.01 c | - | - | - | - | - | [9] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, W.; Liu, R.; Du, W.; Luo, Z.; Guo, P.; Zhang, T.; Zeng, A.; Chang, C.; Fu, Q. Pharmacokinetic and Metabolic Characteristics of Herb-Derived Khellactone Derivatives, A Class of Anti-HIV and Anti-Hypertensive: A Review. Molecules 2016, 21, 314. https://doi.org/10.3390/molecules21030314
Jing W, Liu R, Du W, Luo Z, Guo P, Zhang T, Zeng A, Chang C, Fu Q. Pharmacokinetic and Metabolic Characteristics of Herb-Derived Khellactone Derivatives, A Class of Anti-HIV and Anti-Hypertensive: A Review. Molecules. 2016; 21(3):314. https://doi.org/10.3390/molecules21030314
Chicago/Turabian StyleJing, Wanghui, Ruilin Liu, Wei Du, Zhimin Luo, Pengqi Guo, Ting Zhang, Aiguo Zeng, Chun Chang, and Qiang Fu. 2016. "Pharmacokinetic and Metabolic Characteristics of Herb-Derived Khellactone Derivatives, A Class of Anti-HIV and Anti-Hypertensive: A Review" Molecules 21, no. 3: 314. https://doi.org/10.3390/molecules21030314