
A New Indirect Spectrofluorimetric Method for Determination of Ascorbic Acid with 2,4,6-Tripyridyl-S-Triazine in Pharmaceutical Samples


Abstract
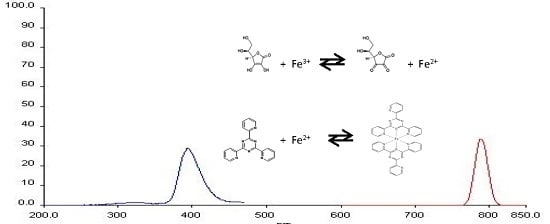
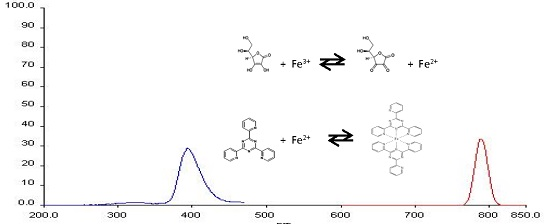
:
1. Introduction
2. Results and Discussion
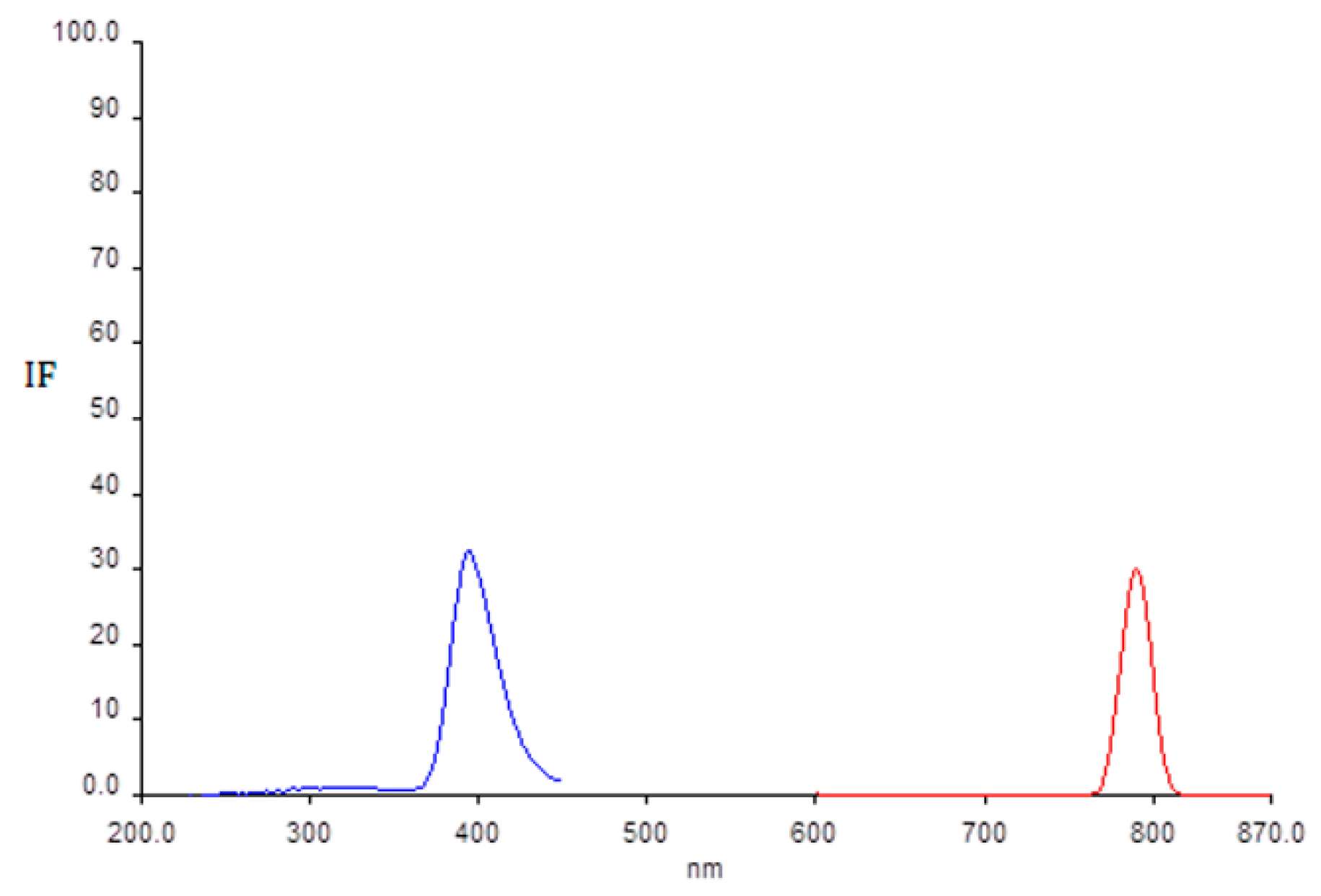
2.1. Determination of Wavelength for Maximum of Excitation and Emission

2.2. Optimization of Experimental Condition for the Proposed Method
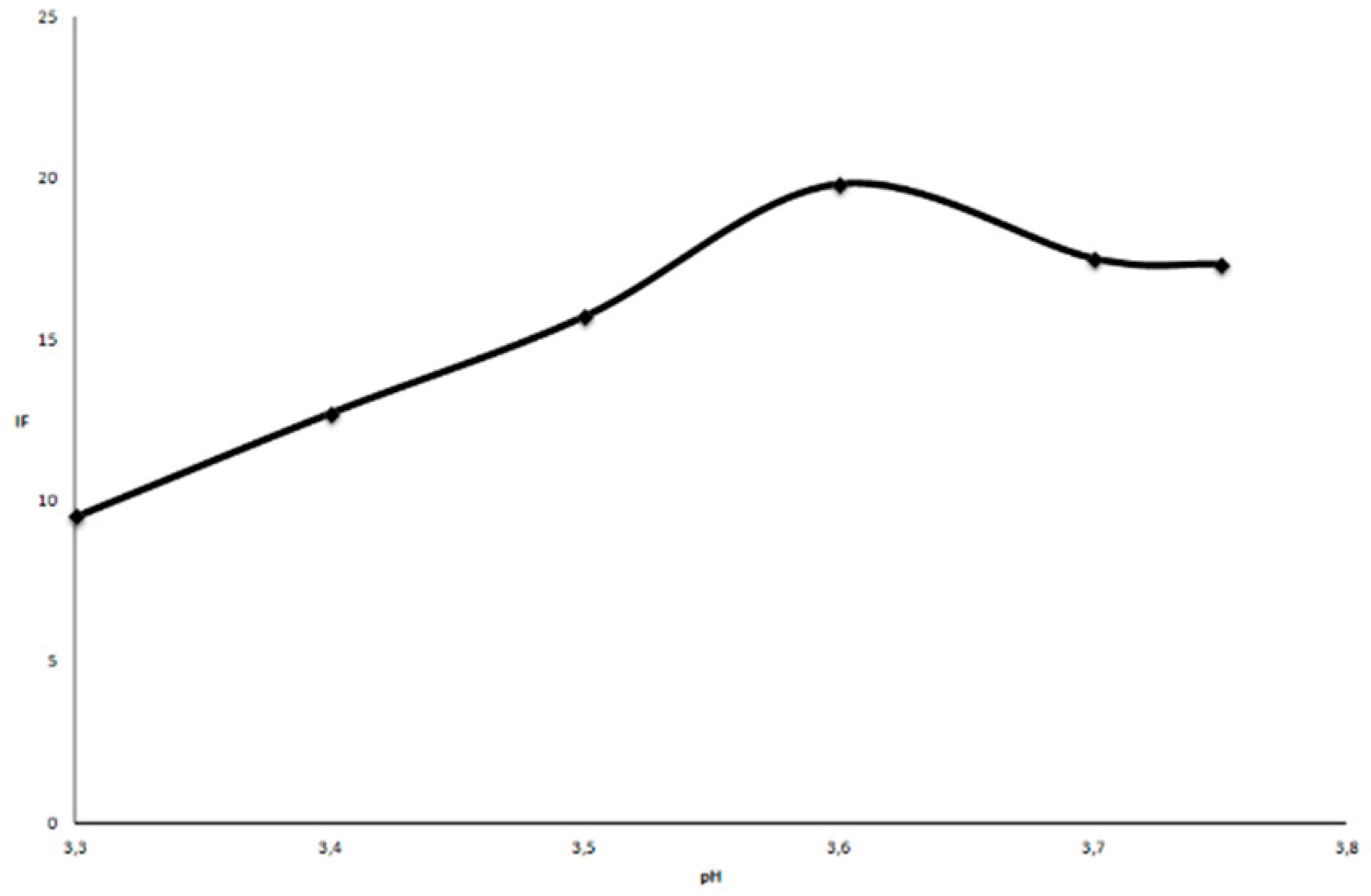
2.2.1. Influence of pH

2.2.2. Influence of the Concentrations of Iron(III) and TPTZ
2.2.3. Reaction Time
2.2.4. Calibration Curve, LOD and LOQ
2.2.5. Accuracy, Precision and Repeatability

{kind=link}
{kind=link}
{kind=link}
| A Standard Solution of AA mmol·L−1 | IF * ± SD | Calculated Concentration mmol·L−1 | %RSD | Recovery % |
|---|---|---|---|---|
| 2.99 | 32.414 ± 0.237 | 2.989 | 0.73 | 99.98 |
| 2.99 | 32.369 ± 0.175 | 2.984 | 0.54 | 99.80 |
| 2.99 | 32.695 ± 0.245 | 3.020 | 0.75 | 101.00 |
| 0.297 | 16.657 ± 0.050 | 0.293 | 0.66 | 98.32 |
| 0.297 | 16.200 ± 0.106 | 0.280 | 0.98 | 94.43 |
| 0.297 | 16.631 ± 0.165 | 0.292 | 0.67 | 98.17 |
| 0.0595 | 7.508 ± 0.096 | 0.057 | 1.28 | 95.41 |
| 0.0595 | 7.668 ± 0.086 | 0.061 | 1.12 | 102.34 |
| 0.0595 | 7.459 ± 0.084 | 0.056 | 1.13 | 94.85 |
| # | Concentration mmol·L−1 | IF * ± SD | %RSD | Avarage IF ± SD | Avarage %RSD |
|---|---|---|---|---|---|
| 1 | 0.119 | 10.667 ± 0.141 | 1.33 | 10.364 ± 0.119 | 1.13 |
| 2 | 0.119 | 10.282 ± 0.156 | 1.52 | ||
| 3 | 0.119 | 10.340 ± 0.057 | 0.55 | ||
| 4 | 0.119 | 10.350 ± 0.132 | 1.27 | ||
| 5 | 0.119 | 10.180 ± 0.102 | 1.00 |
| Concentration mmol·L−1 | IF * | Concentration mmol·L−1 Added | Total Concentration mmol·L−1 | IF * | Concentration Recovered mmol·L−1 | Recovery % |
|---|---|---|---|---|---|---|
| 0.3 | 21.217 | 0.075 | 0.375 | 24.151 | 0.341 | 91.06 |
| 0.3 | 21.178 | 0.075 | 0.375 | 23.827 | 0.338 | 90.00 |
| 0.3 | 21.025 | 0.075 | 0.375 | 23.628 | 0.337 | 89.90 |
| 0.15 | 9.256 | 0.075 | 0.225 | 13.846 | 0.220 | 97.77 |
| 0.15 | 9.315 | 0.075 | 0.225 | 13.497 | 0.217 | 96.60 |
| 0.15 | 9.346 | 0.075 | 0.225 | 13.827 | 0.222 | 98.63 |
2.2.6. Sample Preparation
| Sample | Labeled Value (mg) | Proposed Method (mg) | Titration with Iodine (mg) |
|---|---|---|---|
| Vitamin C (Alkaloid) | 500 | 543 ± 0.12 | 513 |
| Plivit C (Pliva) | 500 | 449 ± 0.18 | 515 |
| Plivit C (Pliva) | 50 | 46 ± 0.16 | 49 |
| Day by day (Krüger) | 160 | 157 ± 0.25 | 124 |
| Vitamin C (Schneekoppe) | 180 | 178 ± 0.23 | 200 |
| Vitamin C (DM) | 240 | 261 ± 0.23 | 260 |
| Vitamin C K Plus (Krüger) | 180 | 189 ± 0.27 | 183 |
| Vitamin C (ZADA) | 500 | 460 ± 0.14 | 504 |
| Interferences | Concentration (mg·mL−1) | Change of Signal(%) a | Change of Signal (%) b |
|---|---|---|---|
| Rutin | 1.45 | −95.68 | −89.99 |
| 0.145 | −16.35 | −20.49 | |
| 0.0145 | +25.53 | +82.43 | |
| Hesperidin | 1.04 | +465.91 | +109.63 |
| 0.104 | +86.89 | −1.86 | |
| 0.0104 | +43.79 | +5.58 | |
| Quercetin | 0.144 | +56.09 | +1.37 |
| 0.0144 | +48.49 | −5.96 | |
| 0.0144 | +31.06 | −14.15 | |
| Paracetamol | 1.53 | +333.45 | +145.94 |
| 0.153 | +455.09 | +65.96 | |
| 0.0153 | +487.0 | +32.14 | |
| Sorbitol | 1.49 | −2.79 | +12.87 |
| 0.149 | −0.89 | +13.80 | |
| 0.0149 | −1.04 | +7.84 | |
| Zinc chloride | 2.70 | +49.63 | +135.71 |
| 0.27 | −9.09 | +108.93 | |
| 0.027 | −3.31 | +24.97 | |
| Magnesium carbonate | 1.21 | +4.56 | +20.87 |
| 0.121 | −12.71 | +1.48 | |
| 0.0121 | +0.63 | −0.43 | |
| Iron(II) sulfate | 2.86 | −35.6 | +111.38 |
| 0.286 | −18.18 | +56.87 | |
| 0.0286 | −4.74 | +14.72 | |
| Citric acid | 1.015 | −38.59 | +192.5 |
| 0.1015 | −16.37 | +41.99 | |
| 0.010 | −0.77 | +11.02 | |
| Calcium carbonate | 1.50 | +49.18 | +114.13 |
| 0.15 | −3.33 | −1.81 |
2.2.7. Interferences
| Sample | Labeled Value (mg) | Proposed Method (mg) | Titration with Iodine (mg) |
|---|---|---|---|
| Fabricet (Hemofarm) | 200 | 240 ± 0.30 | 210 |
| Gripostad (Stada) | 180 | 174 ± 0.38 | 157 |
| Eisen + vitamin C (Fit + vital) | 160 | 171 ± 0.21 | 164 |
| Vitamin A + C + E + Se (Krüger) | 100 | 101 ± 0.24 | 102 |
| Vitamin C + Zn (DM) | 400 | 421 ± 0.09 | 405 |
| Bio-C (Dietpharm) | 500 | 625 ± 0.20 | 510 |
2.2.8. Applications
| Reagent(s) Used | λex and λem (nm) | Linearity Range (µmol·L−1) | LOD (µmol·L−1) | Sample | References |
|---|---|---|---|---|---|
| 2,3-Diaminonaphthalene | 400, 520 | 11–1700 | 2.27 | Pharmaceutical samples | [22] |
| 1,2-Diamino-4,5-dimethoxybenzene | 370, 458 | 7.3–1130 | 0.0051 | Blood serum | [23] |
| Cerium(IV) ion | 303, 340 | 0.1–8.0 | 0.016 | Pharmaceutical samples | [24] |
| Mono-3-[6-N(4-carboxyphenyl)]-β-cyclodextrin | 280, 352 | 0.28–450 | 0.068 | Real samples | [12] |
| o-Phenylenediamine | 360, 430 | 1.0–5.6 | No data | Beer, wine, urine and pharmaceutical samples | [25] |
| o-Phenylenediamine | 356, 440 | 11–567 | 7.38 | Pharmaceutical samples | [26] |
| o-Phenylenediamine | 360, 430 | 0.5–170 0.3–230 | 0.113 0.034 | Pharmaceutical samples, fruit juice, non-alcoholic beverages and blood serum | [27,28,29] |
| Ti(III) | 227, 419 | 1–10 | 0.8 | Real samples | [30] |
| Methylene blue | 664, 682 | 0.3–6 | 0.25 | Pharmaceutical samples | [11,31] |
| Methylene blue | 660 ,694 | 0.6–230 | 1.3 | Non-alcoholic beverages | [32] |
| Iron(III) and TPTZ | 393,790 | 5.4–540 | 0.77 | Pharmaceutical samples | Present work |
3. Experimental Section
3.1. Reagents
3.2. Apparatus
3.3. Procedure
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salkić, M.; Selimović, A. Spectrophotometric Determination of l-Ascorbic Acid in Pharmaceuticals Based on Its Oxidation by Potassium Peroxymonosulfate and Hydrogen Peroxide. Croat. Chem. Acta 2015, 88, 73–79. [Google Scholar] [CrossRef]
- Fujita, Y.; Mori, I.; Yamaguchi, T.; Hoshino, M.; Shigemura, Y.; Shimano, M. Spectrophotometric determination of ascorbic acid with iron(III) and p-carboxyphenylfluorone in cationic surfactant micellar medium. Anal. Sci. 2001, 17, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Njoku, P.C.; Ayuk, A.A.; Okoye, C.V. Temperature effects on vitamin C content in citrus fruits. Pak. J. Nutr. 2011, 10, 1169–2011. [Google Scholar] [CrossRef]
- Hormozi Nezhad, M.R.; Karimi, M.A.; Shahheydari, F. A sensitive colorimetric detection of ascorbic acid in pharmaceutical products based on formation of anisotropic Silver nanoparticles. Sci. Iran. Trans. F Nano 2010, 17, 148–153. [Google Scholar]
- Skrovankova, S.; Mlcek, J.; Sochor, J.; Baron, M.; Kynicky, J.; Jurikova, T. Determination of Ascorbic Acid by Electrochemical Techniques and other Methods. Int. J. Electrochem. Sci. 2015, 10, 2421–2431. [Google Scholar]
- Arya, S.P.; Mahajan, M.; Jain, P. Non-spectrophotometric methods for the determination of Vitamin C. Anal. Chim. Acta 2000, 417. [Google Scholar] [CrossRef]
- Shintani, H. HPLC Analysis of Ascorbic Acid (Vitamin C). Pharm. Anal. Acta 2013, 4. [Google Scholar] [CrossRef]
- Abbaspour, A.; Khajehzadeh, A.; Noori, A. A simple and selective sensor for the determination of ascorbic acid in vitamin C tablets based on paptode. Anal. Sci. 2008, 24, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Revanasiddappa, H.D.; Veena, M.A. Sensitive spectrophotometric methods for the determination of ascorbic acid. Eur. J. Chem. 2008, 5, 10–15. [Google Scholar] [CrossRef]
- Kukoc-Modun, L.; Biocic, M.; Radic, N.J. Indirect method for spectrophotometric determination of ascorbic acid in pharmaceutical preparations with 2,4,6-tripyridyl-S-triazine by flow-injection analysis. Talanta 2012, 96, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Dilgin, Y.; Nisli, G. Fluorimetric Determination of Ascorbic Acid in Vitamin C Tablets Using Methylene Blue. Chem. Pharm. Bull. 2005, 53, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L.; She, S.; Gao, F. Direct fluorimetric determination of ascorbic acid by the supramolecular system of AA with beta-cyclodextrin derivative. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 2737–2740. [Google Scholar] [CrossRef] [PubMed]
- Holm, D.J.; Hazel, P. Analytical Biochemistry, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1998; p. 66. [Google Scholar]
- Liu, T.Z.; Chin, N.; Kiser, M.D.; Bigler, W.N. Specific spectrophotometry of ascorbic acid in serum or plasma by use of ascorbate oxidase. Clin. Chem. 1982, 28, 2225–2228. [Google Scholar] [PubMed]
- Annegowda, H.V.; Ween Nee, C.; Mord, M.N.; Ramanathan, S.; Mansor, S.M. Evaluation of phenolic content and antioxidant property of hydrolysed extracts of Terminalia catappa L. Leaf. Asian. J. Plant. Sci. 2010, 9, 479–485. [Google Scholar] [CrossRef]
- Collins, P.H.; Diehl, H.; Smith, G.F. 2,4,6-Tripyridyl-S-triazine as reagent for iron. Determination of iron in limestone, silicates, and refractories. Anal. Chem. 1959, 31, 1862–1867. [Google Scholar] [CrossRef]
- Kukoc-Modun, L.; Radic, N. Novel kinetic spectrophotometric method for determination of tiopronin N-(2-Mercaptopropionyl)-Glycine. Croat. Chem. Acta 2010, 83, 189–195. [Google Scholar]
- Szôllôsi, R.; Varga, I.S. Total antioxidant power in some species of Labiatae (Adaptation of FRAP method). Acta Biol. Szeged. 2002, 46, 125–127. [Google Scholar]
- Pagenkopf, G.K.; Margerum, D.W. Formation and dissociation kinetics of bis(2,4,6-tripyridyl-S-triazine)iron(II). Inorg. Chem. 1968, 7, 2514–2519. [Google Scholar] [CrossRef]
- Ozyurek, M.; Guclu, K.; Bektasoglu, B.; Apak, R. Spectrophotometric determination of ascorbic acid by the modified CUPRAC method with extractive separation of flavonoids-La(III) complexes. Anal. Chim. Acta 2007, 588, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Olgun, F.A.O.; Ozyurt, D.; Berker, K.I.; Demirata, B.; Apak, R. Folin-Ciocalteu spectrophotometric assay of ascorbic acid in pharmaceutical tablets and orange juice with pH adjustment and pre-extraction of lanthanum(III)-flavonoid complexes. J. Sci. Food Agric. 2014, 94, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tong, C.; Jie, N.; Zhang, G.; Ren, X.; Hu, J. Fluorescent reaction between ascorbic acid and DAN and its analytical application. Talanta 1997, 44, 855–858. [Google Scholar] [CrossRef]
- Iwata, T.; Hara, S.; Yamaguchi, M.; Nakamura, M.; Ohkura, Y. An Ultramicro Fluorimetric Determination of Total Ascorbic Acid in Human Serum using 1,2-diamino-4,5-dimetoxybenzene. Chem. Pharm. Bull. 1985, 33, 3499–3502. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.L.; Xiang, G.H.; Liu, W.P. Determination of ascorbic acid by indirect fluorimetry. Guang Pu Xue Yu Guang Pu Fen Xi 2005, 25, 598–600. [Google Scholar] [PubMed]
- Huang, H.; Cai, R.; Du, Y.; Zeng, Y. Flow-injection stopped-flow spectrofluorimetric kinetic determination of total ascorbic acid based on an enzyme-linked coupled reaction. Anal. Chim. Acta 1995, 309, 271–275. [Google Scholar] [CrossRef]
- Pérez-Ruiz, T.; Martınez-Lozano, C.; Sanz, A.; Guillén, A. Successive determination of thiamine and ascorbic acid inpharmaceuticals by flow injection analysis. J. Pharm. Biomed. Anal. 2004, 34, 551–557. [Google Scholar] [CrossRef]
- Chung, H.K.; Ingle, J.D. Fluorimetric kinetic method for the determination of total ascorbic acid with O-phenylenediamine. Anal. Chim. Acta 1991, 243, 89–95. [Google Scholar] [CrossRef]
- Pérez-Ruiz, T.; Martínez-Lozano, C.; Tomás, V.; Fenol, J. Fluorimetric determination of total ascorbic acid by a stopped-flow mixing technique. Analyst 2001, 126, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Diao, Y.; Sun, C.; Yang, J.; Wang, Y.; Sun, S. Fluorimetric determination of ascorbic acid with O-phenylenediamine. Talanta 2003, 59, 95–99. [Google Scholar] [CrossRef]
- Rezaei, B.; Ensafi, A.; Nouroozi, S. Flow-injection determination of ascorbic acid and cysteine simultaneously with spectrofluorometric detection. Anal. Sci. 2005, 21, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Briga, M.; Delic, D.; Copra-Janicijevic, A.; Klepo, L.; Sofic, E.; Topcagic, A.; Tahirovic, I. Fluorimetric determination of ascorbic acid using methylene blue. In Proceedings of the 7th CMAPSEEC, Conference on Medicinal and Aromatic Plants of Southeast European Countries, Subotica, Serbia, 27–30 May 2012; pp. 104–109.
- Sheng, L.; Wang, H.; Han, X. Determination of ascorbic acid by fluorescence quenching method with methylene blue. Chin. J. Anal. Lab. 2008, 27, 26–29. [Google Scholar]
- Shrivas, K; Agrawala, K.; Kumar Patel, D. A Spectrophotometric Determination of Ascorbic Acid. J. Chin. Chem. Soc. 2005, 52, 503–506. [Google Scholar]
- Aydogmus, Z.; Cetin, S.M. Determination of Ascorbic Acid in Vegetables by Derivative Spectrophotometry. Turk. J. Chem. 2002, 26, 697–704. [Google Scholar]
- Tiwari, K.K. A New Spectrophotometric Method for the Determination of Ascorbic Acid Using Leuco Malachite Green. J. Chin. Chem. Soc. 2010, 57, 105–110. [Google Scholar]
- Farajzadeh, M.A.; Nagizadeh, S. A Simple and Reliable Spectrophotometric Method for the Determination of Ascorbic Acid in Pharmaceutical Preparations. J. Anal. Chem. 2003, 58, 927–932. [Google Scholar] [CrossRef]
- Bulatova, A.V.; Strashnova, U.M.; Vishnikin, A.B.; Alekseeva, G.M.; Sineva, T.D.; Moskvin, A.L.; Moskvin, L.N. Stepwise Injection Photometric Determination of scorbic Acid in Drugs. J. Anal. Chem. 2011, 66, 275–279. [Google Scholar] [CrossRef]
- Noroozifar, M.; Khorasani-Motlagh, M.; Farahmand, A.R. Automatic spectrophotometric procedure for determination of l-ascorbic acid based on reduction of iron(III)-thiocyanate complex. Acta Chim. Slov. 2004, 51, 717–727. [Google Scholar]
- Zenki, M.; Tanishita, A.; Yokoyama, T. Repetitive determination of ascorbic acid using iron(III)-1,10-phenanthroline-peroxodisulfate system in a circulatory flow injection method. Talanta 2004, 64, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Council of Europe. European Directorate for the Quality of Medicines; European Pharmacopoeia: Nordlingen, Germany, 2008. [Google Scholar]
- Thaipong, K.; Boonprakob, U.; Crosby, K.; Cisneros-Zevallos, L.; Byrne, D.H. Comparison of ABTS, DPPH, FRAP, and ORAC assays for estimating antioxidant activity from guava fruit extracts. J. Food Comp. Anal. 2006, 19, 669–675. [Google Scholar] [CrossRef]
- Lim, S.H.; Lim, S.L. Ferric reducing capacity vs. ferric reducing antioxidant power for measuring total antioxidant capacity. Lab. Med. 2013, 44, 51–55. [Google Scholar] [CrossRef]
- Sample Availability: All chemicals and pharmaceutical samples are available, but they have limited expiration date (specially pharmaceutical samples).
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klepo, L.; Copra-Janicijevic, A.; Kukoc-Modun, L. A New Indirect Spectrofluorimetric Method for Determination of Ascorbic Acid with 2,4,6-Tripyridyl-S-Triazine in Pharmaceutical Samples. Molecules 2016, 21, 101. https://doi.org/10.3390/molecules21010101
Klepo L, Copra-Janicijevic A, Kukoc-Modun L. A New Indirect Spectrofluorimetric Method for Determination of Ascorbic Acid with 2,4,6-Tripyridyl-S-Triazine in Pharmaceutical Samples. Molecules. 2016; 21(1):101. https://doi.org/10.3390/molecules21010101
Chicago/Turabian StyleKlepo, Lejla, Amira Copra-Janicijevic, and Lea Kukoc-Modun. 2016. "A New Indirect Spectrofluorimetric Method for Determination of Ascorbic Acid with 2,4,6-Tripyridyl-S-Triazine in Pharmaceutical Samples" Molecules 21, no. 1: 101. https://doi.org/10.3390/molecules21010101