
Ultrasoft Classical Systems at Zero Temperature


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Analytical Description
2.1. Triangular Cluster-Crystal in Two Dimensions
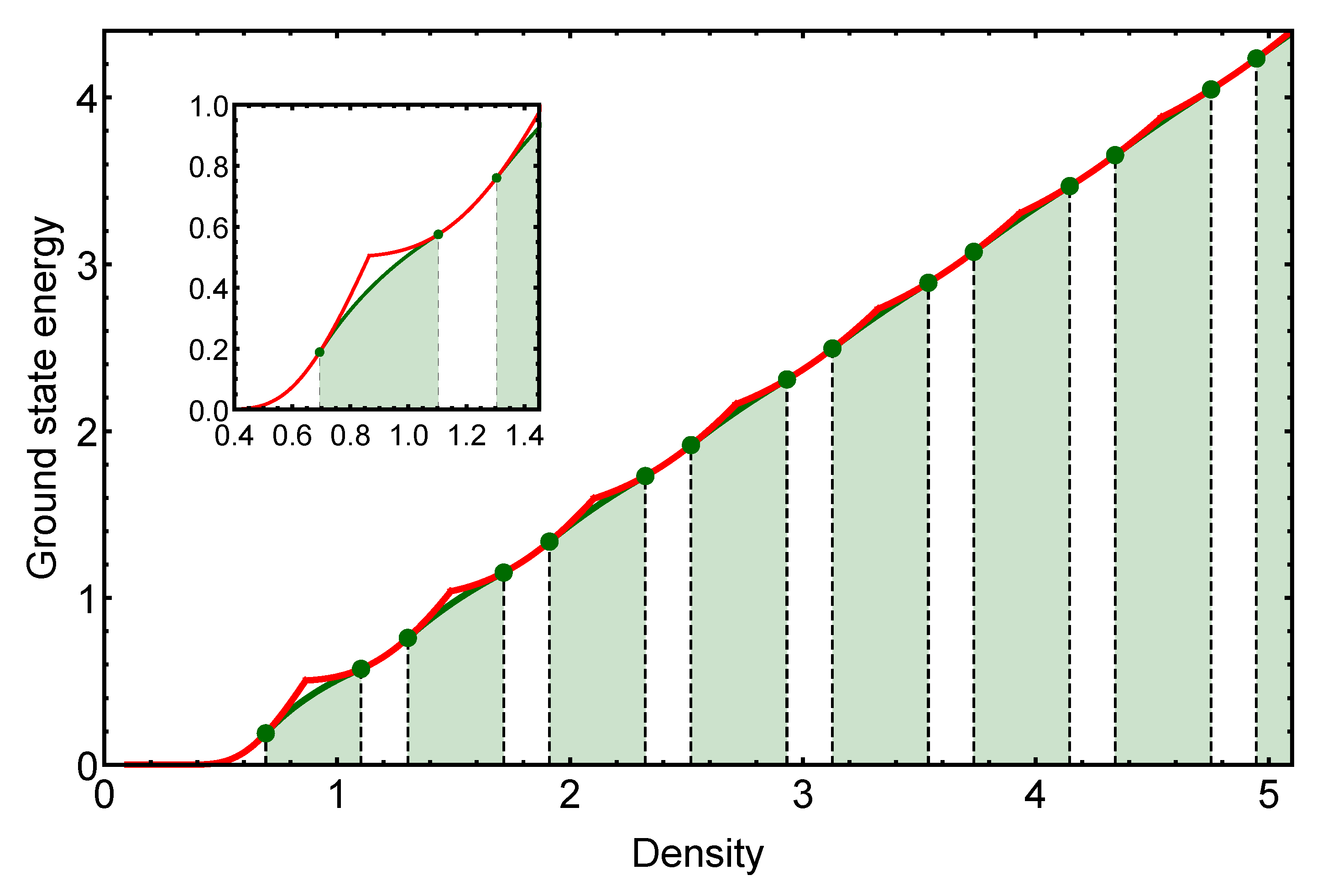
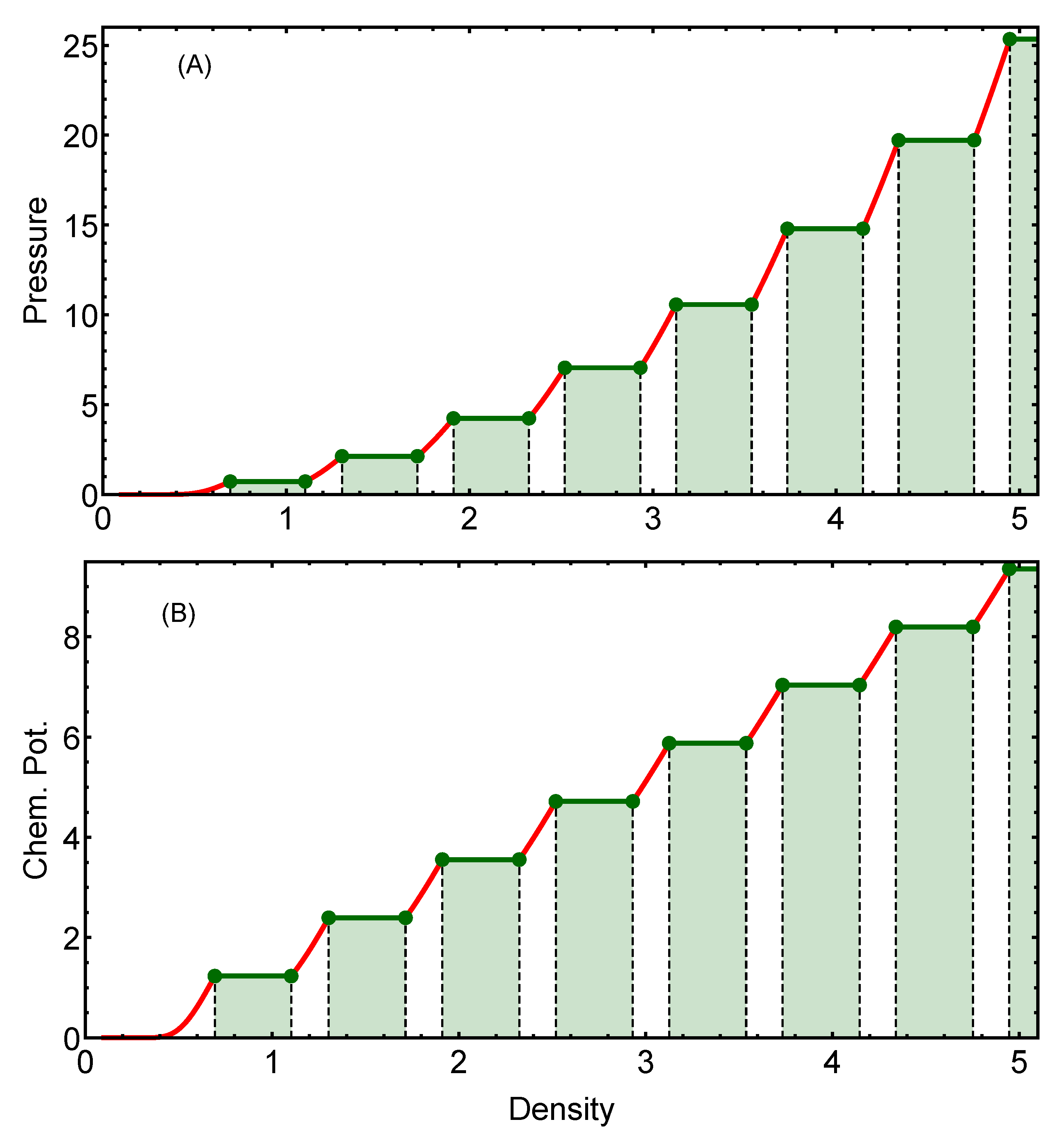
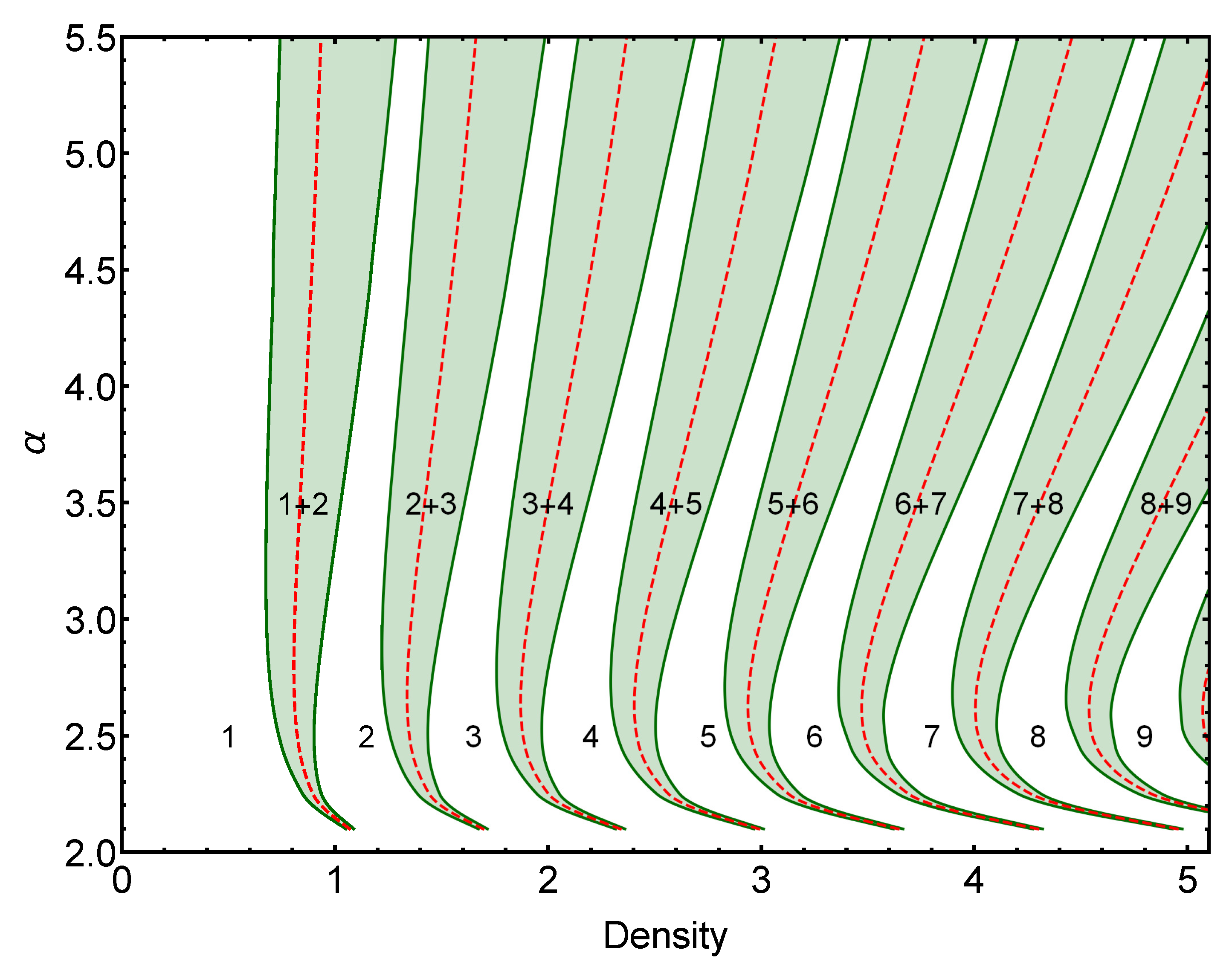
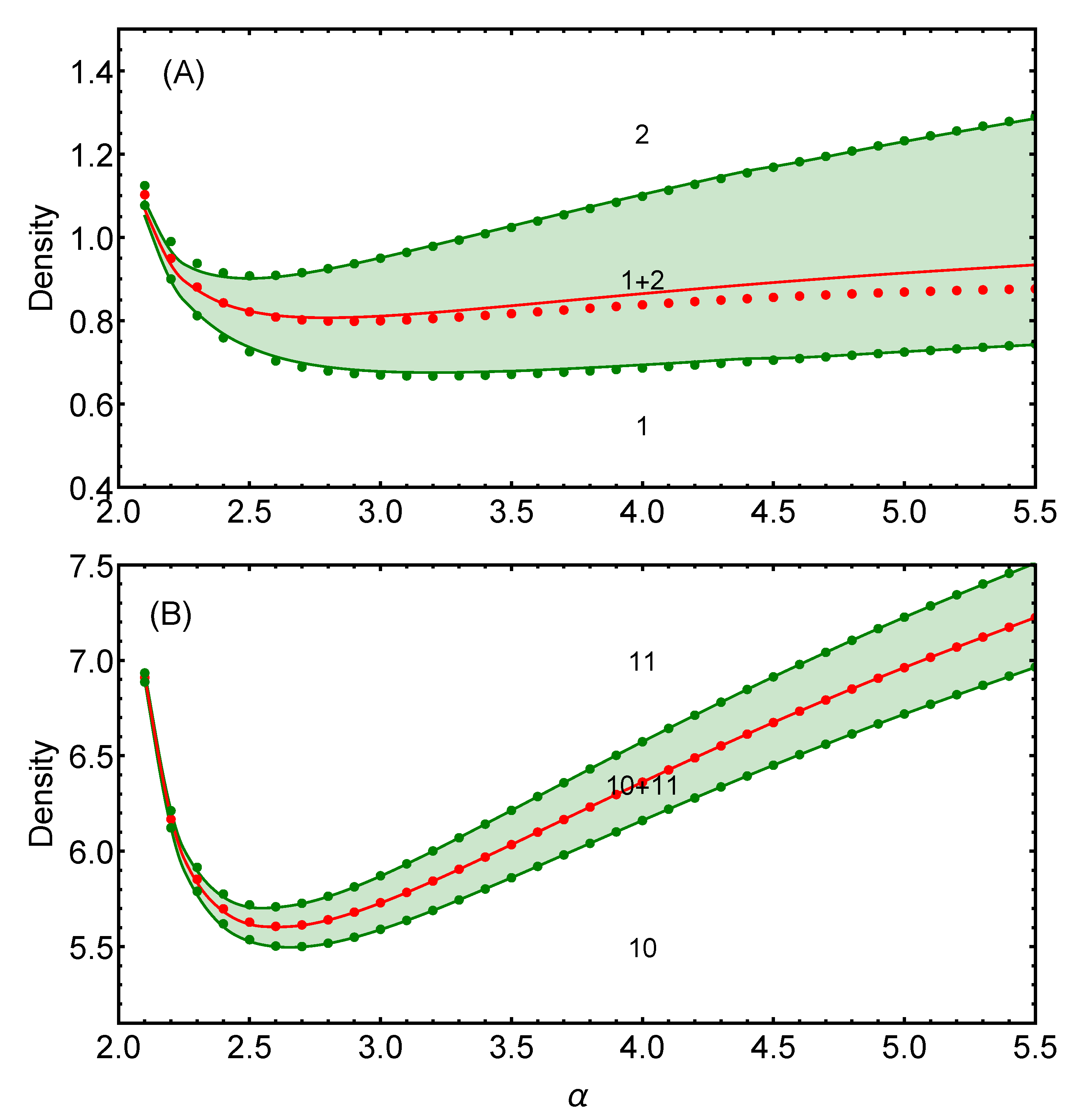
2.1.1. Energy Crossing Densities
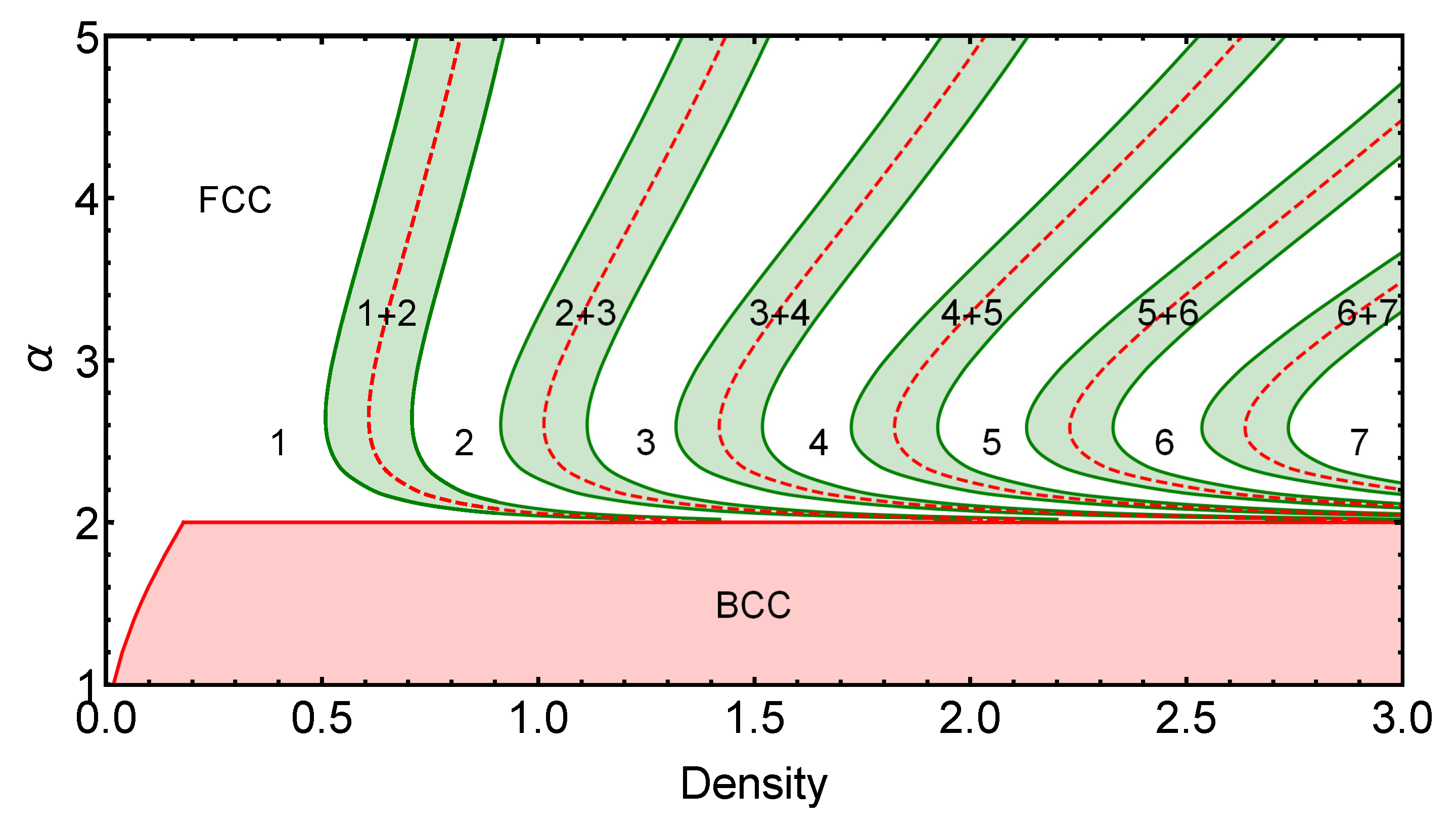
2.1.2. Coexistence Regions
2.2. FCC Cluster-Crystal in Three Dimensions
3. Numerical Results with GEM-
3.1. Two-Dimensional Case
3.1.1. Comparison between Analytical and Numerical Results
3.2. Three-Dimensional Case
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Appendix A. Lattice Sum Identity
References
- Likos, C.N. Effective interactions in soft condensed matter physics. Phys. Rep. 2001, 348, 267–439. [Google Scholar] [CrossRef]
- Likos, C.N.; Mladek, B.M.; Gottwald, D.; Kahl, G. Why do ultrasoft repulsive particles cluster and crystallize? Analytical results from density-functional theory. J. Chem. Phys. 2007, 126, 224502. [Google Scholar] [CrossRef] [Green Version]
- Coslovich, D.; Bernabei, M.; Moreno, A.J. Cluster glasses of ultrasoft particles. J. Chem. Phys. 2012, 137, 184904. [Google Scholar] [CrossRef] [Green Version]
- Prestipino, S.; Saija, F. Hexatic phase and cluster crystals of two-dimensional GEM4 spheres. J. Chem. Phys. 2014, 141, 184502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, F.; Macrì, T.; Lechner, W.; Pupillo, G.; Pohl, T. Defect-induced supersolidity with soft-core bosons. Nat. Commun. 2014, 5, 3235. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Méndez, R.; Mezzacapo, F.; Cinti, F.; Lechner, W.; Pupillo, G. Monodisperse glassy crystals: Classical and quantum dynamics. Phys. Rev. E 2015, 92, 052307. [Google Scholar] [CrossRef] [Green Version]
- Pupillo, G.; Ziherl, P.; Cinti, F. Quantum cluster quasicrystals. Phys. Rev. B 2020, 101, 134522. [Google Scholar] [CrossRef]
- Mendoza-Coto, A.; Caetano, D.d.S.; Díaz-Méndez, R. Ground-state phase diagram of ultrasoft bosons. Phys. Rev. A 2021, 104, 013301. [Google Scholar] [CrossRef]
- Mendoza-Coto, A.; Turcati, R.; Zampronio, V.; Díaz-Méndez, R.; Macrì, T.; Cinti, F. Exploring quantum quasicrystal patterns: A variational study. Phys. Rev. B 2022, 105, 134521. [Google Scholar] [CrossRef]
- Varney, C.N.; Sellin, K.A.; Wang, Q.Z.; Fangohr, H.; Babaev, E. Hierarchical structure formation in layered superconducting systems with multi-scale inter-vortex interactions. J. Phys. Condens. Matter 2013, 25, 415702. [Google Scholar] [CrossRef]
- Díaz-Méndez, R.; Mezzacapo, F.; Lechner, W.; Cinti, F.; Babaev, E.; Pupillo, G. Glass transitions in monodisperse cluster-forming ensembles: Vortex matter in type-1.5 superconductors. Phys. Rev. Lett. 2017, 118, 067001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Díaz-Méndez, R.; Wallin, M.; Lidmar, J.; Babaev, E. Melting of a two-dimensional monodisperse cluster crystal to a cluster liquid. Phys. Rev. E 2019, 99, 042140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, C.J.; Pérez-García, M.A. Neutrino-pasta scattering: The opacity of nonuniform neutron-rich matter. Phys. Rev. C 2004, 69, 045804. [Google Scholar] [CrossRef] [Green Version]
- Caplan, M.E.; Horowitz, C.J. Colloquium: Astromaterial science and nuclear pasta. Rev. Mod. Phys. 2017, 89, 041002. [Google Scholar] [CrossRef] [Green Version]
- Caplan, M.E.; Schneider, A.S.; Horowitz, C.J. Elasticity of Nuclear Pasta. Phys. Rev. Lett. 2018, 121, 132701. [Google Scholar] [CrossRef] [Green Version]
- Coslovich, D.; Strauss, L.; Kahl, G. Hopping and microscopic dynamics of ultrasoft particles in cluster crystals. Soft Matter 2011, 7, 2127. [Google Scholar] [CrossRef] [Green Version]
- Mladek, B.; Kahl, G.; Likos, C. Computer assembly of cluster-forming amphiphilic dendrimers. Phys. Rev. Lett. 2008, 100, 028301. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Méndez, R.; Pupillo, G.; Mezzacapo, F.; Wallin, M.; Lidmar, J.; Babaev, E. Phase-change switching in 2D via soft interactions. Soft Matter 2019, 15, 355–358. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Charbonneau, P. Communication: Microphase equilibrium and assembly dynamics. J. Chem. Phys. 2017, 147, 091102. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, B.A.; Dutta, S.; Jadrich, R.B.; Milliron, D.J.; Truskett, T.M. Interactions and design rules for assembly of porous colloidal mesophases. Soft Matter 2017, 13, 1335–1343. [Google Scholar] [CrossRef]
- Likos, C.N.; Sciortino, F.; Zaccarelli, E.; Ziherl, P. Soft Matter Self-Assembly; IOS: Amsterdam, The Netherlands; SIF: Bologna, Italy, 2016. [Google Scholar]
- Colla, T.; Mohanty, P.S.; Nöjd, S.; Bialik, E.; Riede, A.; Schurtenberger, P.; Likos, C.N. Self-Assembly of Ionic Microgels Driven by an Alternating Electric Field: Theory, Simulations, and Experiments. ACS Nano 2018, 12, 4321–4337. [Google Scholar] [CrossRef] [PubMed]
- Jochum, C.; Adžić, N.; Stiakakis, E.; Derrien, T.L.; Luo, D.; Kahl, G.; Likos, C.N. Structure and stimuli-responsiveness of all-DNA dendrimers: Theory and experiment. Nanoscale 2019, 11, 1604–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiakakis, E.; Jung, N.; Adžić, N.; Balandin, T.; Kentzinger, E.; Rücker, U.; Biehl, R.; Dhont, J.K.; Jonas, U.; Likos, C.N. Self assembling cluster crystals from DNA based dendritic nanostructures. Nat. Commun. 2021, 12, 7167. [Google Scholar] [CrossRef]
- Likos, C.N.; Lang, A.; Watzlawek, M.; Löwen, H. Criterion for determining clustering versus reentrant melting behavior for bounded interaction potentials. Phys. Rev. E 2001, 63, 031206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhaus, T.; Likos, C.N. Phonon dispersions of cluster crystals. J. Phys. Condens. Matter 2011, 23, 234112. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Charbonneau, P.; Mladek, B.M. Reentrant and Isostructural Transitions in a Cluster-Crystal Former. Phys. Rev. Lett. 2010, 105, 245701. [Google Scholar] [CrossRef] [Green Version]
- Wilding, N.B.; Sollich, P. A Monte Carlo method for chemical potential determination in single and multiple occupancy crystals. EPL (Europhys. Lett.) 2013, 101, 10004. [Google Scholar] [CrossRef] [Green Version]
- Wilding, N.B.; Sollich, P. Demixing cascades in cluster crystals. J. Chem. Phys. 2014, 141, 094903. [Google Scholar] [CrossRef] [Green Version]
- Prestipino, S.; Gazzillo, D.; Tasinato, N. Probing the existence of phase transitions in one-dimensional fluids of penetrable particles. Phys. Rev. E 2015, 92, 000138. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Mello, M.; Díaz-Méndez, R.; Mendoza-Coto, A. Ultrasoft Classical Systems at Zero Temperature. Entropy 2023, 25, 356. https://doi.org/10.3390/e25020356
de Mello M, Díaz-Méndez R, Mendoza-Coto A. Ultrasoft Classical Systems at Zero Temperature. Entropy. 2023; 25(2):356. https://doi.org/10.3390/e25020356
Chicago/Turabian Stylede Mello, Matheus, Rogelio Díaz-Méndez, and Alejandro Mendoza-Coto. 2023. "Ultrasoft Classical Systems at Zero Temperature" Entropy 25, no. 2: 356. https://doi.org/10.3390/e25020356