1. Introduction
The demand for electrical energy has been steadily increasing in recent years, and, along with it, the importance of environmentally friendly energy converters. Electrochemical systems, such as fuel cells, are such alternative technologies for directly converting the chemical internal energy of a fuel into electrical energy. Hydrogen as an energy carrier becomes inexhaustible due to its simple production using solar-based carbon free energy sources and enables its use without direct CO2 emissions, making it environmentally friendly, also with regard to the Paris Climate Agreement.
The solid oxide fuel cell (SOFC) is a promising technology due to its superior performance indicators. These include the integration into the current energy supply system through the use of carbon-containing fuels, such as natural gas, the variability of the purity of the hydrogen required, and high efficiency due to the high operating temperatures. According to the current state of development, the electrical efficiency of SOFCs reaches values of up to 60–65% [
1]. For the development of improved SOFCs, it is of great importance to be able to simulate the processes and the operating behavior taking place inside of the cells with the help of models. This is especially true as an experimental assessment of the ongoing processes inside the cell is prohibitive because of the temperature level around 1000 K. Many models already exist in the literature. These range from modeling individual layers within one cell to modeling the SOFC as a stack system. Three-dimensional models were set up in the work of Anderson et al. [
2], Mauro et al. [
3], Yakabe et al. [
4], and Peksen et al. [
5], in which the SOFCs are modeled based on the 3D finite element method. The momentum transport is described in these works with the help of the Navier–Stokes equations. In the work of Peksen et al., the SOFC is modeled, including peripheral components, such as the heat exchangers and the recirculation loops. Individual layers were investigated, for example, by Xu and Dang [
6], Niu et al. [
7], and Joos [
8]. They model the porous electrodes of the SOFC in terms of their exact microstructure and investigate the relationship between the microstructure and the electrochemical reactions at the three-phase boundaries. In all these models, the transport processes are described using the classical empirical equations for the transport of heat, charges, molecules, and momentum. Why these empirical equations may not be sufficient for an exact description of the transport mechanisms in an SOFC is addressed in this work.
The performance and lifetime in an SOFC are determined by large temporal and spatial temperature gradients. Zeng et al. [
9] shows an extensive literature review on heat transfer in SOFC stacks and derived thermal management methods. The study shows that excessive temperature gradients in SOFCs can lead to delamination and cracks in the electrolyte and electrodes. Depending on the operating point, temperature gradients in a planar co-flow SOFC along the electrolyte in the main flow stream of up to 30 K/cm can be reached. To avoid damage in the SOFC, a temperature gradient of 10 K/cm should not be exceeded [
10]. Such temperature gradients can be reduced in the system by means of an effective heat management.
Temperature gradients are also caused by local heat generation and by flow field arrangement. Two types of heat generation are relevant in an SOFC. Reversible heat describes the reaction entropy of the system as a function of temperature, while irreversible heat is caused by dissipated energy. Dissipated energy arises due to ohmic resistance and overvoltages at the electrodes. The heat generation due to an electric current is described by means of the first Joule’s law, which is why the irreversible heat is also called Joule’s heat. In contrast, the electrochemical reaction at the three-phase interface leads to a thermoelectric effect in which reversible heat is released or absorbed at each electrode, also called Peltier heat.
In addition to a temperature gradient or a gradient in the electric potential, a gradient in the chemical potential of a species can also lead to a heat flow, called the Dufour heat. The influence of this coupling of multiple transport mechanisms caused by gradients of different intensive variables on the heat flux in a PEMFC is investigated in Reference [
11], where individual effects on heat generation (Fourier, Peltier, and Dufour) are examined. As expected, at typical current densities, there is a net heat flux pointing out of the anode and cathode. An increasing current density leads to a higher potential difference and a higher electro-osmotic effect, which increases the Peltier and the Dufour effect. However, above a current density of
, Fourier heat dominates due to the temperature rise of the cathode surface caused by the irreversible activation overvoltage. Valadez Huerta [
12] investigates, among other things, the contribution of the Peltier effect to the heat fluxes in an SOFC. A significant contribution is shown to come from the electrolyte, where the heat flux can flow from a lower to a higher temperature within the electrolyte due to the coupling effects mentioned above.
At the same time, the temperature gradients which develop affect the charge flow (Seebeck effect) and the material flow (Soret effect). The Seebeck effect describes a charge transport caused by a temperature gradient. Thus, due to this thermoelectric effect, a voltage can be measured between two different conductors when a temperature gradient is imposed on them. The magnitude of this voltage is described by the Seebeck coefficient, which depends on the material and the temperature. In the works of Kjelstrup et al. [
13,
14], a temperature gradient is applied to an electrochemical cell in order to measure the Seebeck coefficient. From this, the entropy of the oxygen ions transported in the electrolyte can be determined, which can also be used to calculate the Peltier coefficient.
Fick’s law, the Stefan–Maxwell diffusion, or the Dusty-Gas model (DGM) are widely used to describe mass transport within a cell. In Reference [
12], the coupling of the mass flow with other flows is neglected. Using Fick’s law, the concentrations of the individual components are calculated using multi-component diffusion coefficients from the work of Costamagna et al. [
15]. In the work of Suwanwarangkul et al., the approaches for describing the mass transport by means of Fick’s law, Stefan–Maxwell diffusion, and the DGM for determining the concentration overvoltage at the anode are examined. The modeling with the DGM achieves the best results, whereas the approaches using Fick’s law and Stefan–Maxwell diffusion are good approximations, especially for small electrical current densities and large pore diameters [
16].
In Reference [
11], the modeling of a PEMFC using non-equilibrium thermodynamics (NET) takes into account the Soret effect, whereby a material flow is driven by a temperature gradient. In operating ranges of high electric current densities, a significant influence of the Soret effect is observed in the membrane of the PEMFC. This is due to the strongly increasing temperature gradients in the membrane with increasing electric current density.
Considering these results, the coupling effects for a reliable description of the transport mechanisms in fuel cells cannot be neglected. It is not sufficient to address these coupling effects individually and simply add them up. It is very important to take an integrated approach as will be shown below. An integrated coupling of several transport processes can be based on theory of non-equilibrium thermodynamics (NET), which has only been considered in a few models so far. The first known model based on NET is the 1D model by Kjelstrup and Bedeaux [
17], which can be used to calculate the potential field and the temperature curve of the cell. Within the NET theory, a generalized flux
results from the linear combination of all occurring forces
[
18]:
where the phenomenological coefficients
, also called conductances, are the connection between forces and fluxes. Forces are gradients of intensive variables. From the phenomenological equations, a matrix with the
conductances
necessary for modeling is obtained. By Onsager’s reciprocity relation
, the problem reduces to the upper and lower triangular matrices of this
matrix, including the coefficients
of the main diagonal [
19].
For the optimization of a technical system, it is of great importance to understand these local transport mechanisms and their driving forces, as they all dissipate energy, i.e., create entropy. With the help of the local entropy production rate, loss mechanisms can be identified and quantified as exergy losses. In terms of NET, the local volume specific entropy production rate
due to the transport process is generally calculated by multiplying a flux
by the respective corresponding force
[
20]:
thus allowing the second law of thermodynamics to be integrated into the model approach. However, if several fluxes are present simultaneously in a process, the total local entropy production is calculated by summing all products of occurring fluxes
with the respective corresponding force
[
20]:
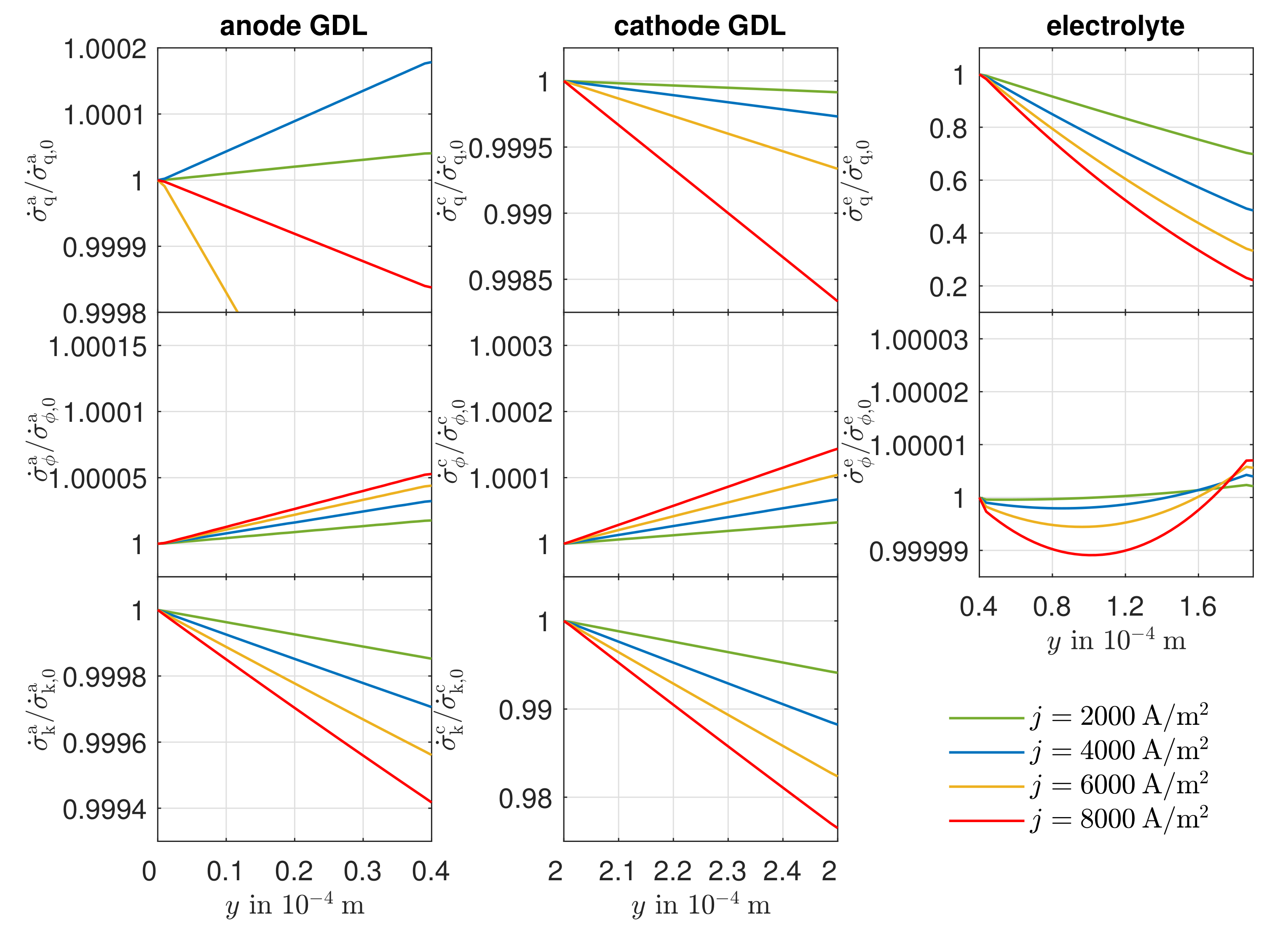
Sauermoser et al. concluded that, in a PEMFC, the total local entropy production rate is highest at the cathode, which is explained by the potential profile. In contrast, Valadez et al. break down the total local entropy production rate of an SOFC into its individual contributions. In the gas diffusion layers (GDL), the diffusion process provides the largest contribution to the local entropy production rate, with losses at the cathode GDL being higher than losses at the anode GDL. In the electrolyte, the local entropy production rate is dominated by ionic conduction. If the Peltier effect is neglected, the GDLs show reduced values for the heat flux and potential contribution. As a result, a change of direction of the heat flux in the electrolyte happens. In both cases, the highest local entropy production rate results in the electrolyte, with up to 73%.
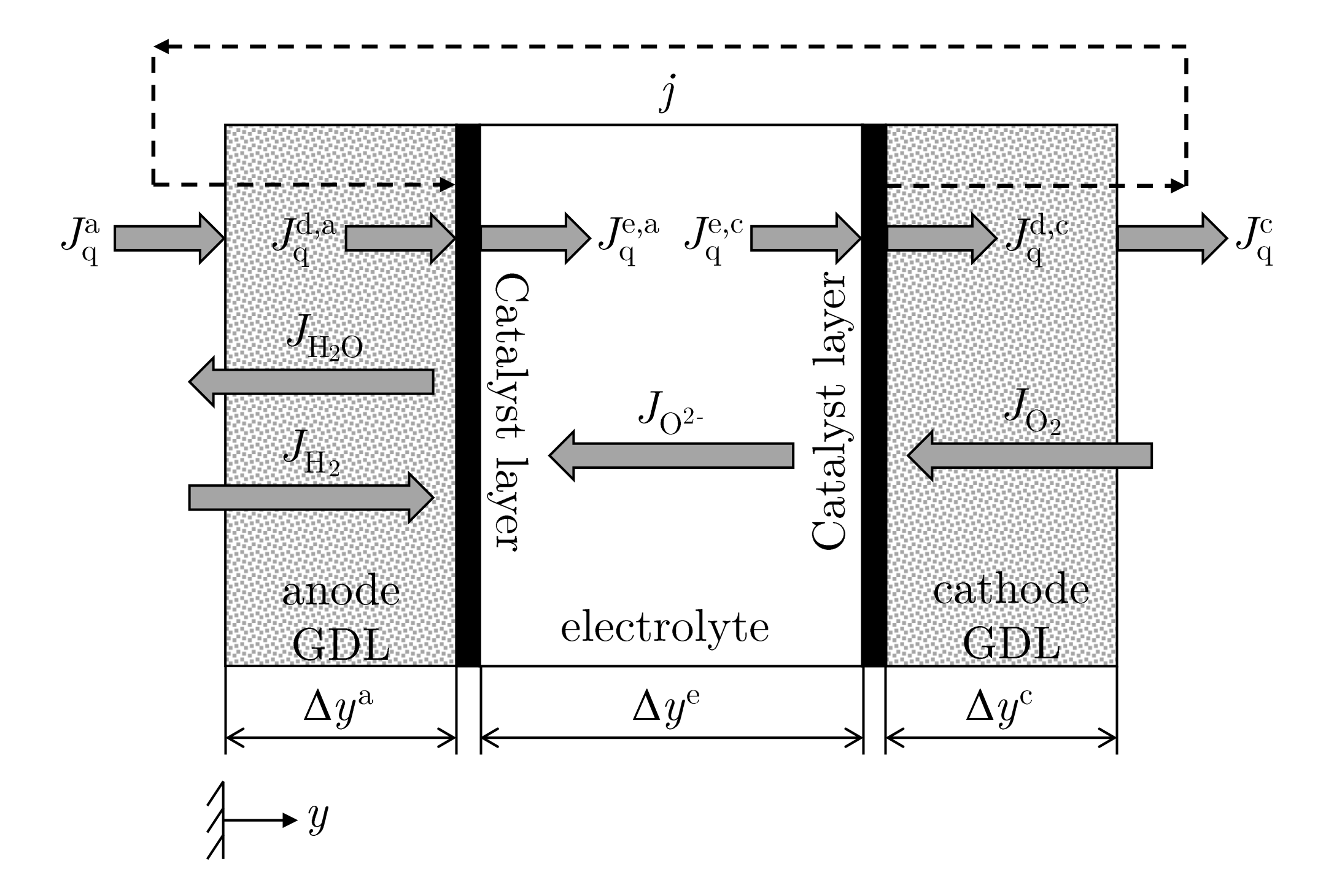
Our work aims at providing a consistent model approach for a single solid oxide fuel cell based on the NET theory. The 1D NET model by Valadez Huerta [
12] provides the starting model for our approach. However, the mass transport described by Fick’s law is extended by transport equations that consider the coupling of several transport mechanisms. In addition, the phenomenological equations of the electrolyte are transformed to model the electrolyte with measurable coefficients. With the localization of the individual loss mechanisms, the system is then evaluated exegetically.
4. Conclusions
The aim of this study is to theoretically determine the local entropy production rate of an SOFC single cell at an operating temperature of
K and an operating pressure of
bar. Local entropy production rates can be identified as loss mechanisms, so that this knowledge is of great importance for cell development and optimization of operating strategies. For this purpose, the mass transport in the GDLs of a 1D SOFC model from Reference [
12] is described by improved transport equations. The modeling of the GDLs and the electrolyte are based on the NET approach, whereas the reactivity layers are described with the Butler–Volmer approach. The mass transport starting from pure Stefan–Maxwell diffusion is gradually superimposed with further transport mechanisms (Knudsen diffusion, convection, and thermal diffusion) in order to investigate the influences of the individual transport mechanisms. For a comparison of all approaches, the mass transport is additionally modeled with Fick’s law. Furthermore, the transport equations are described as a function of the simulated phenomenological coefficients from Reference [
12], with the definitions from Reference [
20] as a function of the empirical coefficients. The data for thermal conductivities and ionic conductivity are taken from the literature. The Peltier coefficient is calculated with the approach of Ratkje et al. [
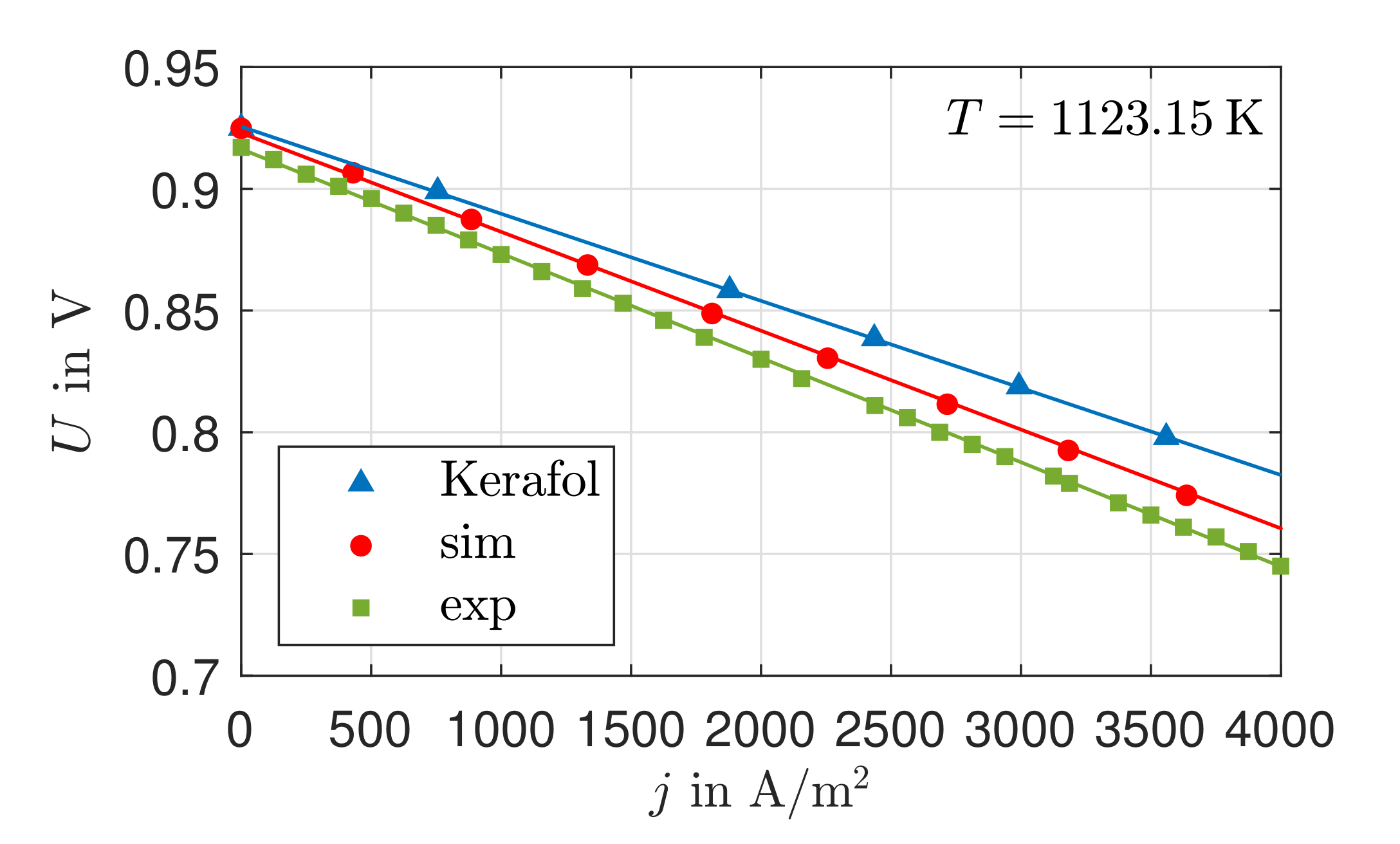
14]. For the validation, data for a
-characteristic curve of the single cell KeraCell II from the manufacturer Kerafol GmbH and, additionally, self-measured data for the same single cell are used. The calculated curves of temperature, heat flow, electrical potential, and local entropy production rates are discussed. An exergetic analysis is used to assess the efficiency of the cell.
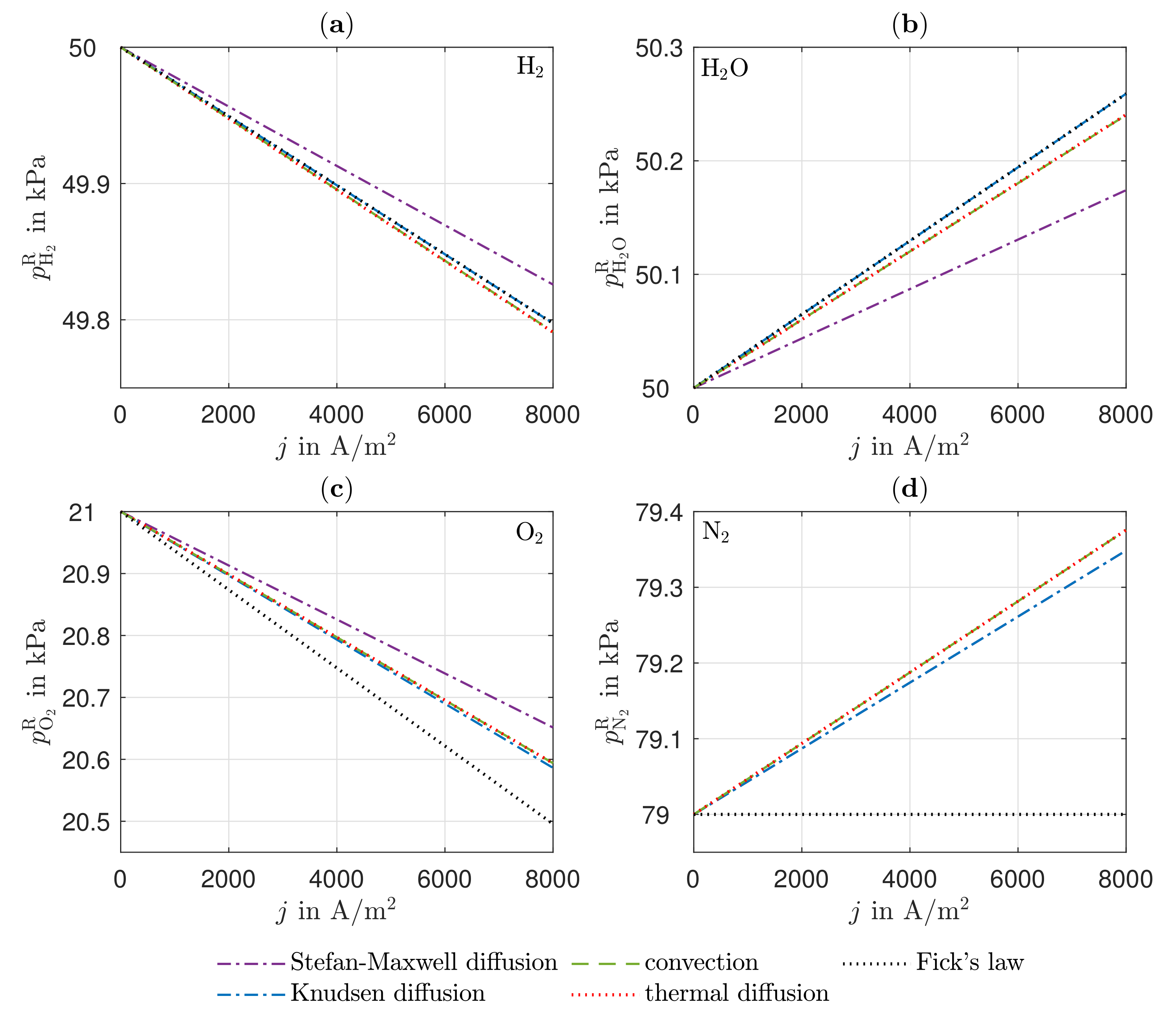
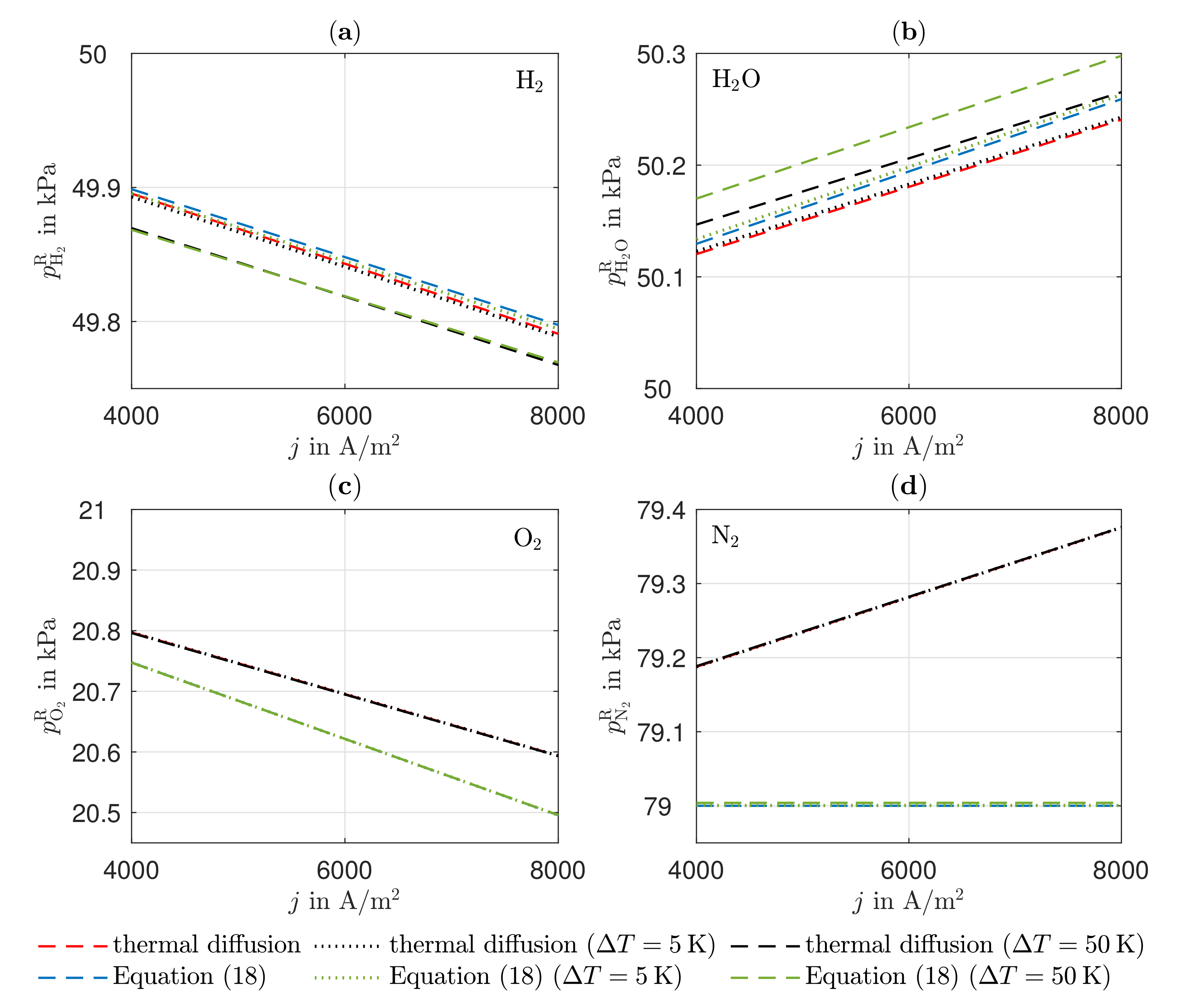
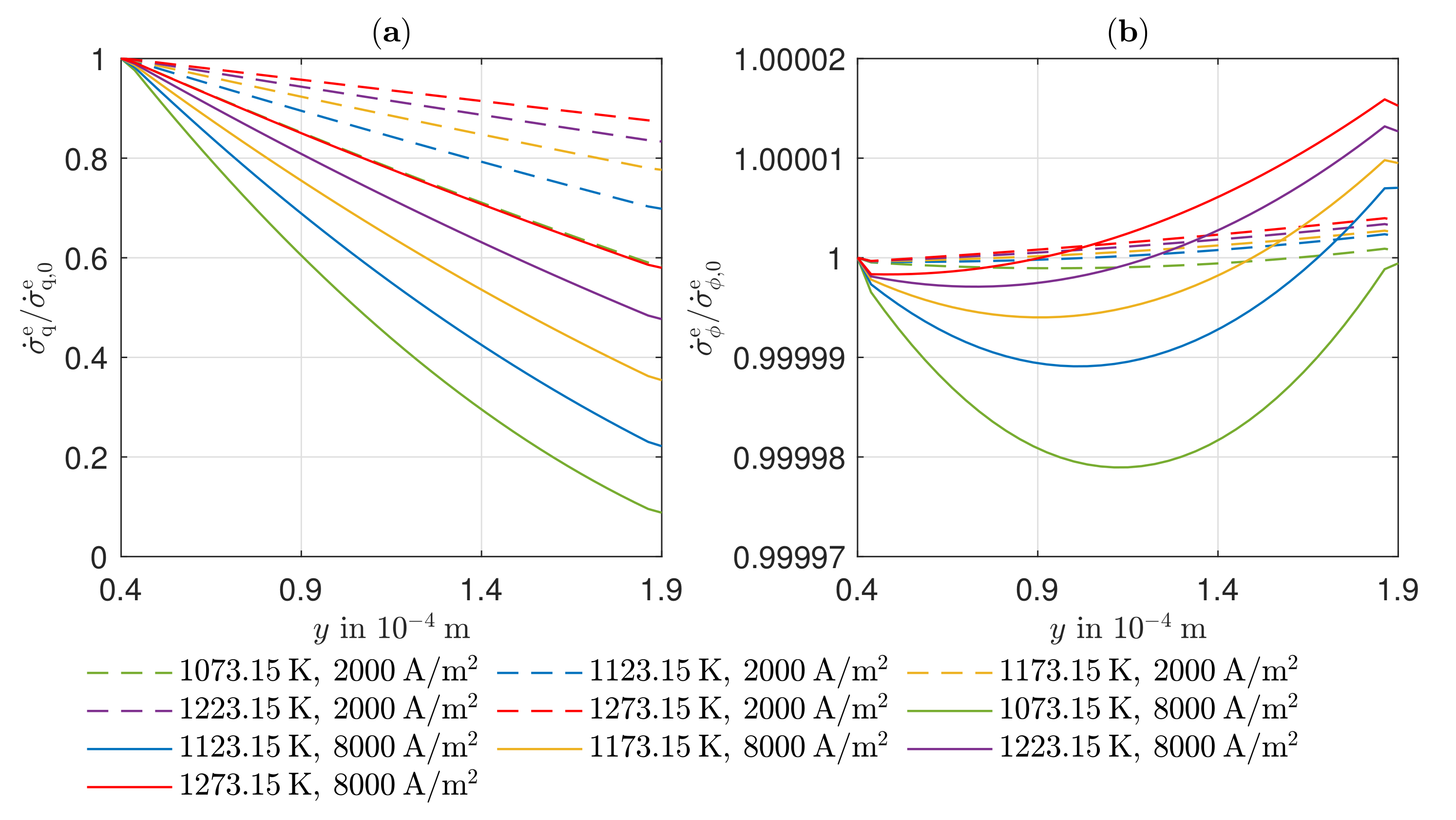
Due to the equimolar composition of the hydrogen and water in the anode, the DGM shows no advantages over the simplified Fick’s law in this particular case. The effect of convection is small compared to the other transport mechanisms because there are low gradients in the total pressure in the anode. There is a higher total pressure in the anode reaction layer than in the gas channel, which is why the gas mixture moves in the direction of the gas channel as a result of convection, thus hindering the mass transport of the hydrogen and improving the mass transport of the water. The influence of Stefan–Maxwell diffusion on the cathode gases is larger, as a significantly smaller effective binary diffusion coefficient results for the cathode-side gas mixture. The improved mass transfer of oxygen by convection is very small in its effect since the gas mixture on the cathode side consists predominantly of nitrogen. On the anode and cathode sides, thermal diffusion has almost no influence on mass transport under the operating conditions considered. The differences between the DGM and the NET approach are negligible in this case.
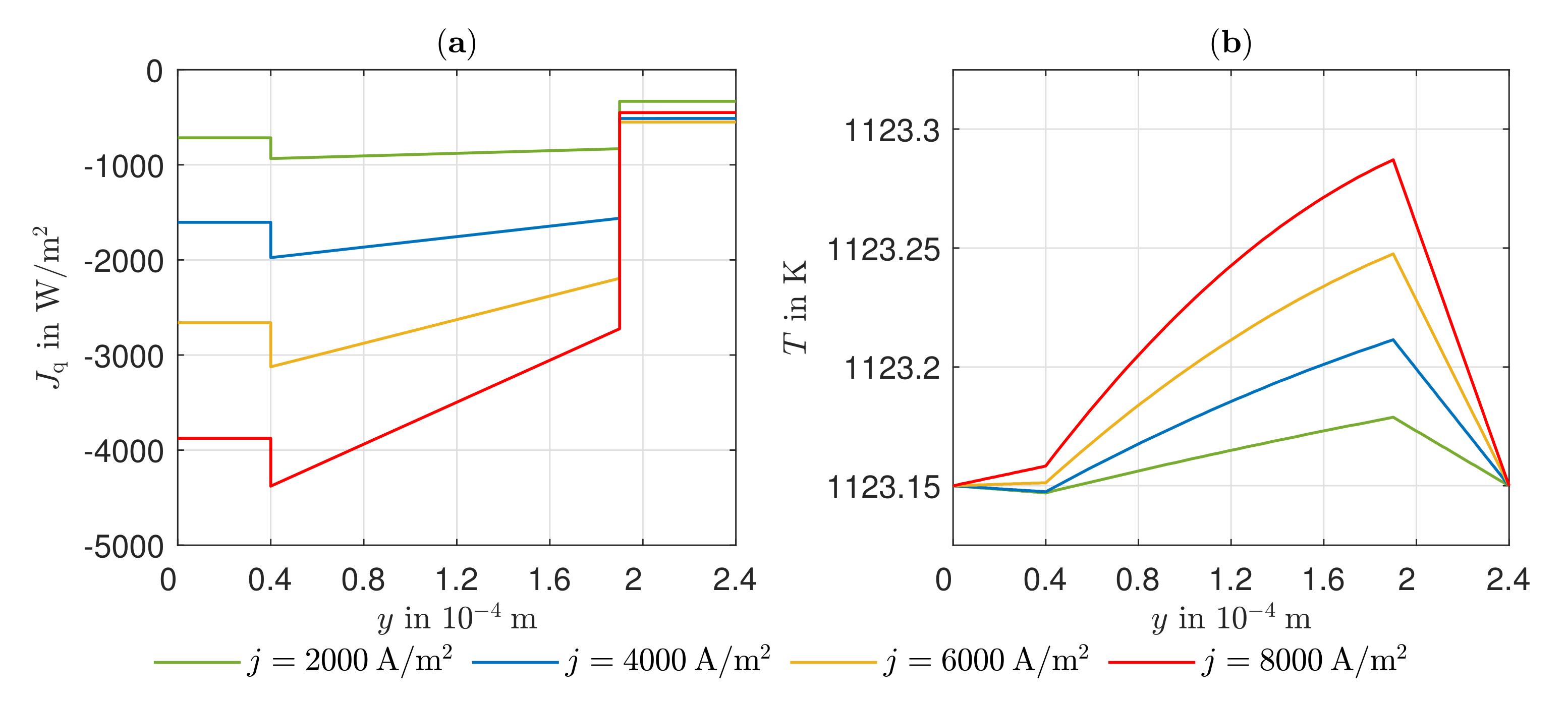
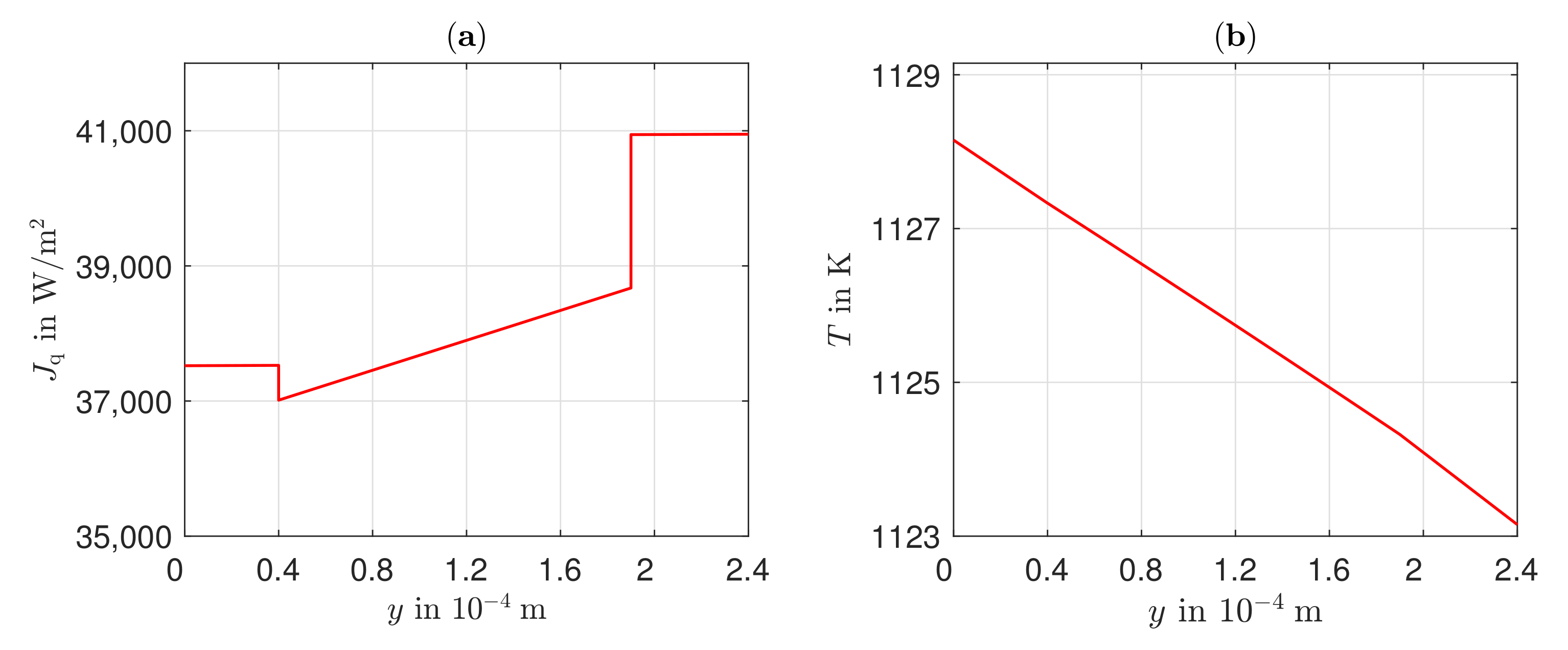
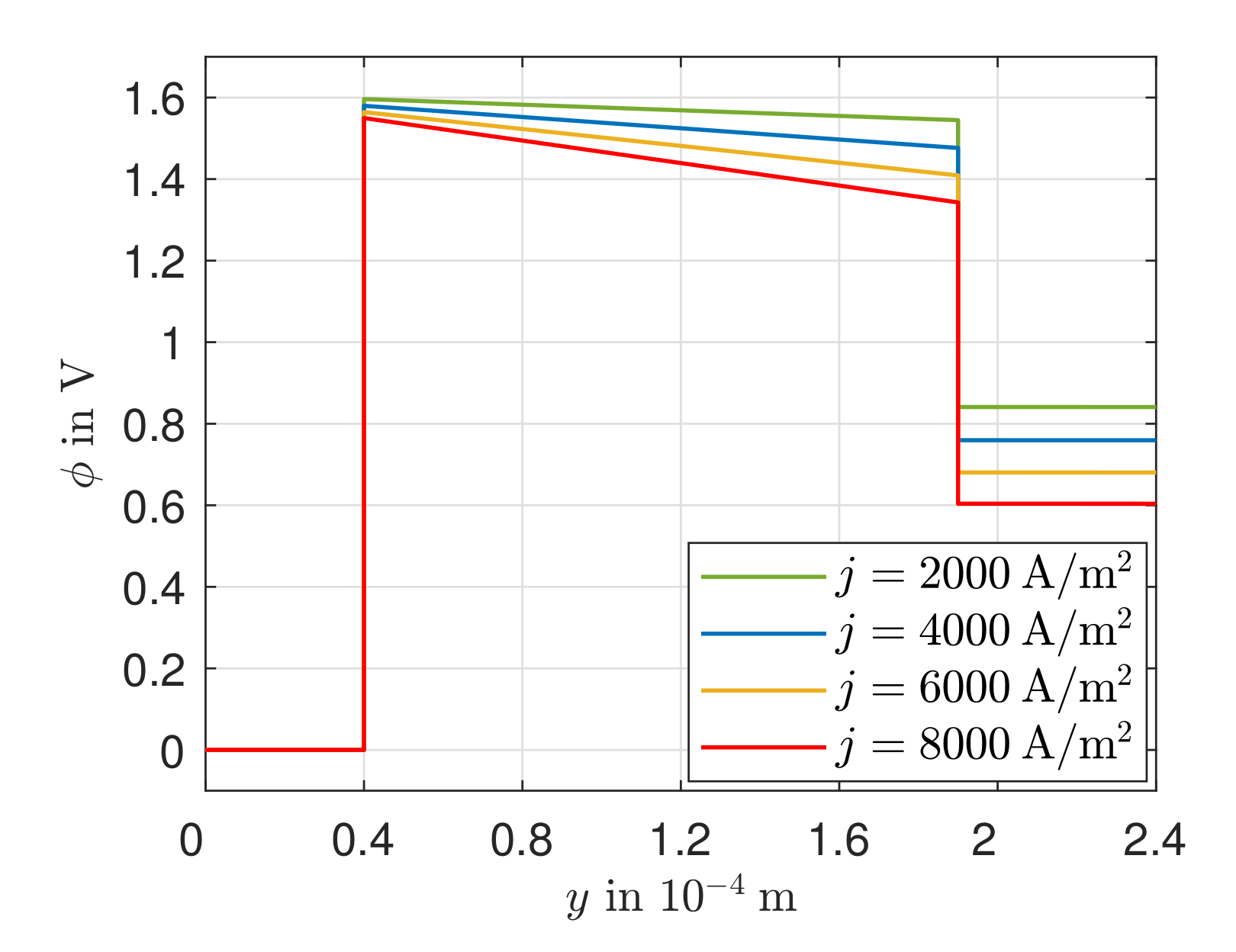
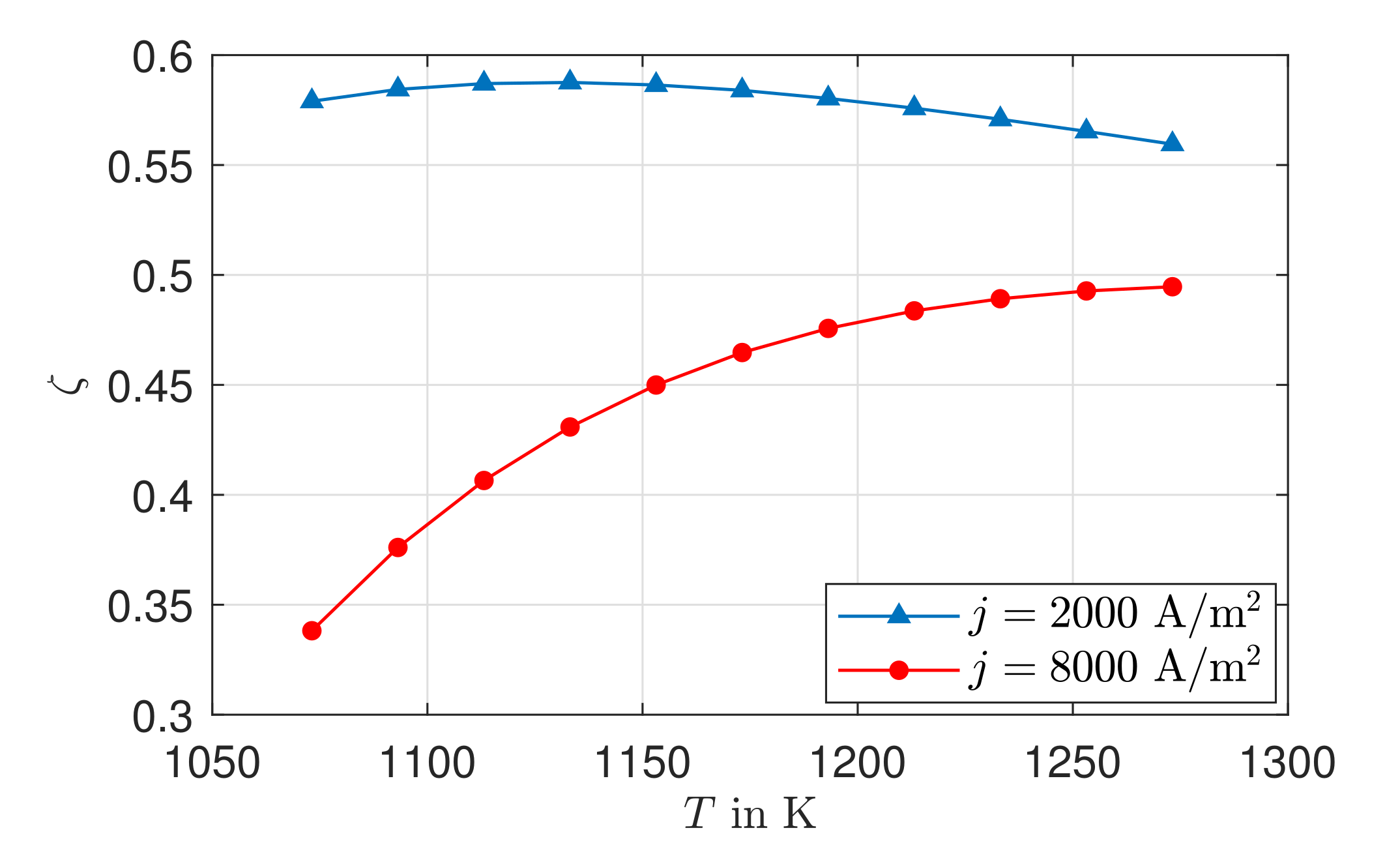
The existing model is able to represent the -characteristics with a minimum error of 0.08% and a maximum error of 0.93% in a range of the electric current density of –4000 A/. Due to the negative reaction entropy of the cathode reaction and the irreversibilities, a temperature maximum occurs at the cathode reaction layer. In the cathode GDL, the heat flows toward higher temperatures, which illustrates the significance of the Peltier effects. At the anode reaction layer, net heat is absorbed by the positive reaction entropy. Under an electric current density of A/, Peltier heat absorption outweighs Fourier heat absorption. The electric potential field is significantly influenced by the ohmic losses in the electrolyte. These are always greater than the overvoltages at the electrodes and cause more than half of the voltage losses. This is also shown in the curves of the local entropy production rates. The entropy production rates as a result of the charge transport in the electrolyte are significantly larger in amount than all other losses. The irreversibilities cause 64.44% of the total losses in the cell in the electrolyte at an electric current density of A/ and an operating temperature of . The irreversibilities at the electrodes, together, cause one third of the losses. As the entropy productions in the GDLs are very low, the primary need is to investigate alternative electrolyte materials.
The exergetic efficiency is fundamentally influenced by the operating temperature and the electrical current density. An increase in exergetic efficiency can be achieved through low current densities and higher operating temperatures.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}