
The Resolved Mutual Information Function as a Structural Fingerprint of Biomolecular Sequences for Interpretable Machine Learning Classifiers

 , , , and
, , , and
Abstract
:1. Introduction
2. Variants of Mutual Information Functions as Biomolecular Sequence Signatures
2.1. The Resolved Mutual Information Function Based on the Shannon Entropy
2.2. Rényi -Entropy and Related Mutual Information Functions
2.3. Tsallis -Entropy and Related Mutual Information Functions
3. Interpretable Classification Learning by Learning Vector Quantization
4. Applications of Mutual Information Functions for Sequence Classification
4.1. Data Sets
4.1.1. Quadruplex Detection
4.1.2. lncRNA vs. mRNA
4.1.3. COVID Types
4.2. Feature Generation
4.2.1. Natural Vectors
4.2.2. Mutual Information Functions
4.2.3. Handling of Ambiguous Characters
4.3. Classification
5. Results and Discussion
5.1. Classification Performance
5.2. Visualization of MIF Variants
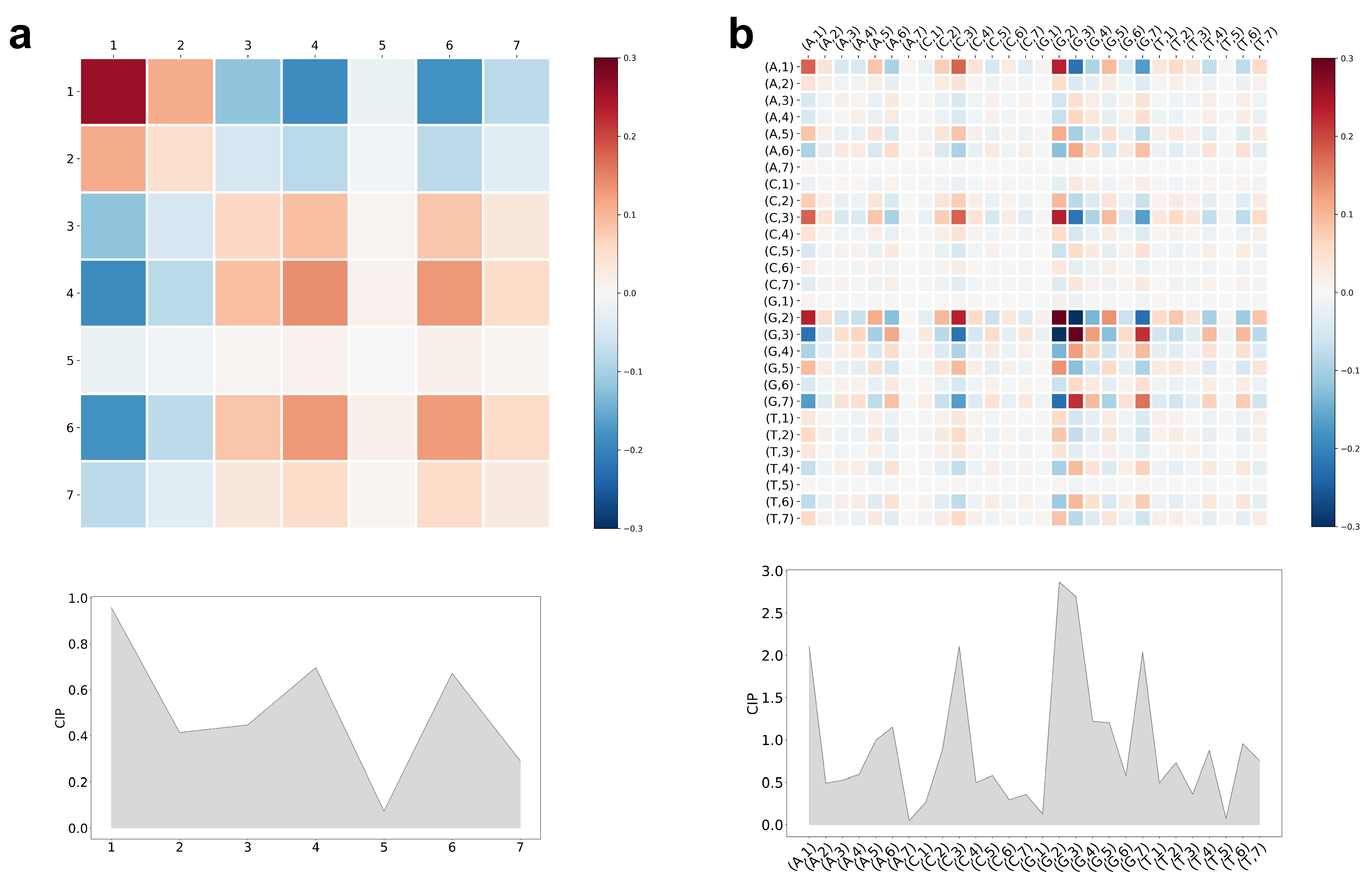
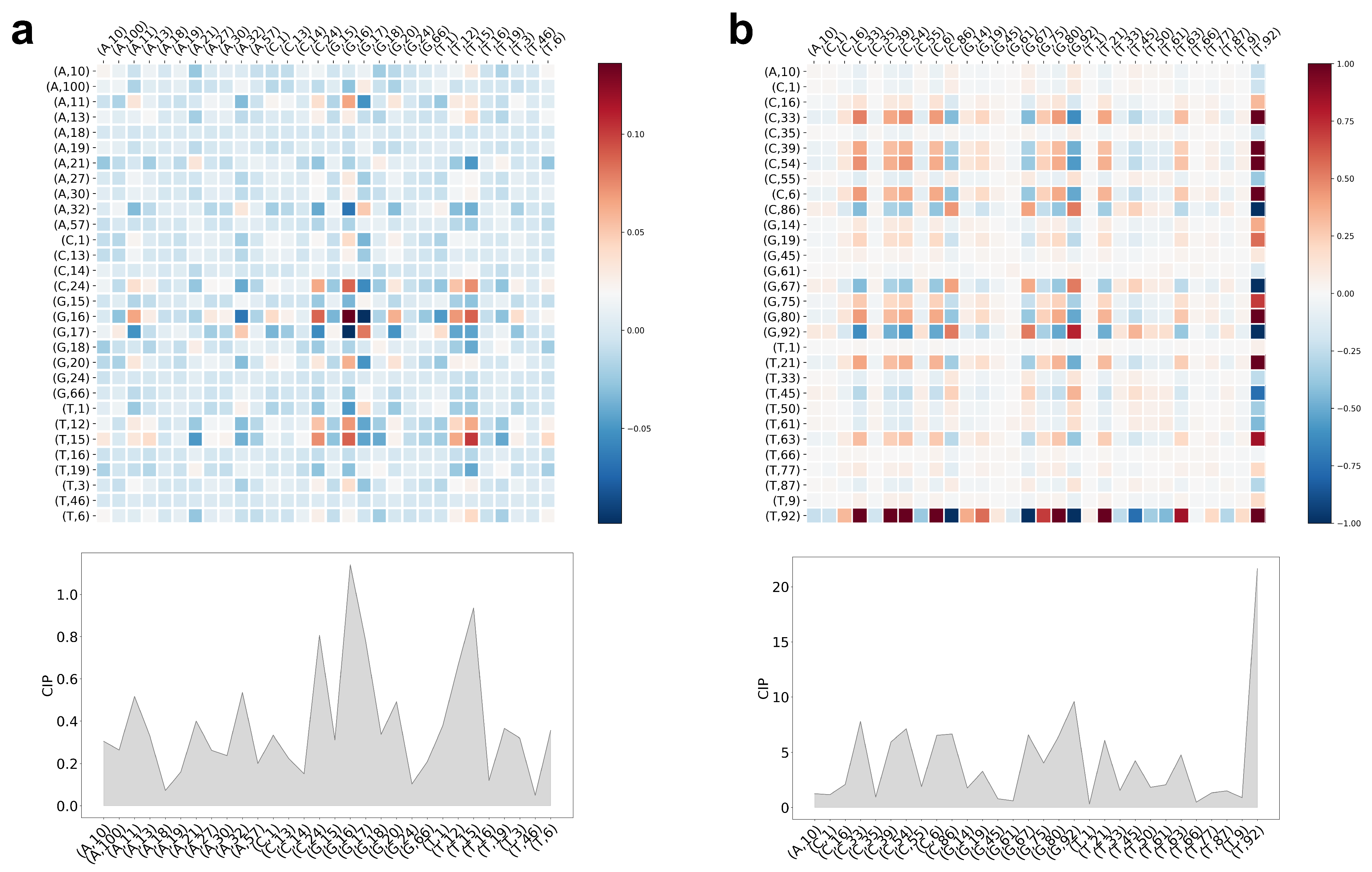
5.3. Interpretation of CCM and CIP of the Trained LiRaM-LVQ Model
6. Conclusions, Remarks, and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMI | Average Mutual Information |
| CCM | Classification Correlation Matrix |
| CIP | Classification Influence Profile |
| GISAID | Global Initiative on Sharing Avian Influenza Data |
| GLVQ | Generalized Matrix Learning Vector Quantization |
| IUPAC | International Union of Pure and Applied Chemistry |
| lncRNA | Long Non-Coding RNA |
| LiRaM-LVQ | Limited Rank Matrix Learning Vector Quantization |
| LVQ | Learning Vector Quantization |
| MIF | Mutual Information Function |
| mRNA | messenger RNA |
| NV | Natural Vectors |
| rMIF | resolved Mutual Information Function |
| RMIF | Rényi Mutual Information Function |
| rRMIF | resolved Rényi Mutual Information Function |
| rTMIF | resolved Tsallis Mutual Information Function |
| SGDL | Stochastic Gradient Descent Learning |
| TMIF | Tsallis Mutual Information Function |
References
- Schrödinger, E. What Is Life? Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Eigen, M.; Schuster, P. Stages of emerging life —Five principles of early organization. J. Mol. Evol. 1982, 19, 47–61. [Google Scholar] [CrossRef]
- Haken, H. Synergetics—An Introduction Nonequilibrium Phase Transitions and Self-Organization in Physics, Chemistry and Biology; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]
- Haken, H. Information and Self-Organization; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Baldi, P.; Brunak, S. Bioinformatics, 2nd ed.; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Gatlin, L. The information content of DNA. J. Theor. Biol. 1966, 10, 281–300. [Google Scholar] [CrossRef]
- Gatlin, L. The information content of DNA. II. J. Theor. Biol. 1968, 18, 181–194. [Google Scholar] [CrossRef]
- Chanda, P.; Costa, E.; Hu, J.; Sukumar, S.; Hemert, J.V.; Walia, R. Information Theory in Computational Biology: Where We Stand Today. Entropy 2020, 22, 627. [Google Scholar] [CrossRef] [PubMed]
- Adami, C. Information Theory in Molecular Biology. Phys. Life Rev. 2004, 1, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Vinga, S. Information Theory Applications for Biological Sequence Analysis. Briefings Bioinform. 2014, 15, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Uda, S. Application of Information Theory in Systems Biology. Biophys. Rev. 2020, 12, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Smith, M. DNA Sequence Analysis in Clinical Medicine, Proceeding Cautiously. Front. Mol. Biosci. 2017, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Bioinformatics and Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 1709–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G. ImageNet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems 25 (NIPS); Curran Associates, Inc.: San Diego, CA, USA, 2012; pp. 1097–1105. [Google Scholar]
- Schölkopf, B.; Smola, A. Learning with Kernels; MIT Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep Learning for Computational Biology. Mol. Sys. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2016, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.; Tran, V.; Ngo, D.; Phan, D.; Lumbanraja, F.; Faisal, M.; Abapihi, B.; Kubo, M.; Satou, K. DNA Sequence Classification by Convolutional Neural Network. J. Biomed. Sci. Eng. 2016, 9, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Jaakkola, T.; Diekhans, M.; Haussler, D. A discrimitive framework for detecting remote protein homologies. J. Comput. Biol. 2000, 7, 95–114. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nat. 2021, 596, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Samek, W.; Monatvon, G.; Vedaldi, A.; Hansen, L. Explainable AI: Interpreting, Explaining and Visualizing Deep Learning; Number 11700 in LNAI; Müller, K.R., Ed.; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Ustun, B.; Rudin, C. Interpretable classification models for recidivism prediction. J. R. Stat. Soc. Ser. A 2017, 180, 1–34. [Google Scholar] [CrossRef]
- Lisboa, P.; Saralajew, S.; Vellido, A.; Villmann, T. The coming of age of interpretable and explainable machine learning models. In Proceedings of the 29th European Symposium on Artificial Neural Networks, Computational Intelligence and Machine Learning (ESANN’2021), Bruges, Belgium, 6–8 October 2021; Verleysen, M., Ed.; i6doc.com: Louvain-La-Neuve, Belgium, 2021; pp. 547–556. [Google Scholar]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-Free Sequence Comparison: Benefits, Applications, and Tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [Green Version]
- Just, W. Computational Complexity of Multiple Sequence Alignment with SP-Score. J. Comput. Biol. 2001, 8, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Kucherov, G. Evolution of Biosequence Search Algorithms: A Brief Survey. Bioinformatics 2019, 35, 3547–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubold, B. Alignment-Free Phylogenetics and Population Genetics. Briefings Bioinform. 2014, 15, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.X.; Bernard, G.; Poirion, O.; Hogan, J.M.; Ragan, M.A. Inferring Phylogenies of Evolving Sequences without Multiple Sequence Alignment. Sci. Rep. 2014, 4, 6504. [Google Scholar] [CrossRef] [Green Version]
- Hatje, K.; Kollmar, M. A Phylogenetic Analysis of the Brassicales Clade Based on an Alignment-Free Sequence Comparison Method. Front. Plant Sci. 2012, 3, 192. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Ye, Y. A Novel Abundance-Based Algorithm for Binning Metagenomic Sequences Using l-Tuples. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2011, 18, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Leung, G.; Eisen, M.B. Identifying Cis-Regulatory Sequences by Word Profile Similarity. PLoS ONE 2009, 4, e6901. [Google Scholar] [CrossRef] [Green Version]
- de Lima Nichio, B.T.; de Oliveira, A.M.R.; de Pierri, C.R.; Santos, L.G.C.; Lejambre, A.Q.; Vialle, R.A.; da Rocha Coimbra, N.A.; Guizelini, D.; Marchaukoski, J.N.; de Oliveira Pedrosa, F.; et al. RAFTS3G: An Efficient and Versatile Clustering Software to Analyses in Large Protein Datasets. BMC Bioinform. 2019, 20, 392. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de Novo Short Read Assembly Using de Bruijn Graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajuste, F.D.; Kaplinski, L.; Möls, M.; Puurand, T.; Lepamets, M.; Remm, M. FastGT: An Alignment-Free Method for Calling Common SNVs Directly from Raw Sequencing Reads. Sci. Rep. 2017, 7, 2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Lee, W.; Jia, L.; Ji, F.; Tsai, L. Statistical correlatation of nucleotides in a DNA sequence. Phys. Rev. E 1998, 58, 861–871. [Google Scholar] [CrossRef]
- Luo, L.; Li, H. The statistical correlation of nucleotides in protein-coding DNA sequences. Bull. Math. Biol. 1991, 53, 345–353. [Google Scholar] [CrossRef]
- Jeffrey, H. Chaos Game Representation of Gene Structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Adjeroh, D.; Jiang, B.H.; Jiang, Y. K2 and K2*: Efficient alignment-free sequence similarity measurement based on Kendall statistics. Bioinformatics 2018, 34, 1682–1689. [Google Scholar] [CrossRef]
- Li, W. The study of correlation structures of DNA sequences: A critical review. Comput. Chem. 1997, 21, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.K.; Buldyrev, S.V.; Goldberger, A.L.; Havlin, S.; Sciortino, F.; Simons, M.; Stanley, H.E. Long-Range Correlations in Nucleotide Sequences. Nature 1992, 356, 168–170. [Google Scholar] [CrossRef]
- Voss, R. Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys. Rev. A 1992, 68, 3805–3808. [Google Scholar] [CrossRef]
- Deng, M.; Yu, C.; Liang, Q.; He, R.L.; Yau, S.S.T. A Novel Method of Characterizing Genetic Sequences: Genome Space with Biological Distance and Applications. PLoS ONE 2011, 6, e17293. [Google Scholar] [CrossRef]
- Li, Y.; Tian, K.; Yin, C.; He, R.; Yau, S.T. Virus classification in 60-dimensional protein space. Mol. Phylogenet. Evol. 2016, 99, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tian, K.; Yau, S. Proteine Sequence Classification using natural vectors and the convex hull method. J. Comput. Biol. 2019, 26, 315–321. [Google Scholar] [CrossRef]
- Li, W. Mutual information functions versus correlation function. J. Stat. Phys. 1990, 60, 823–837. [Google Scholar] [CrossRef]
- Herzel, H.; Grosse, I. Maesuring correlations in symbol sequences. Phys. A 1995, 216, 518–542. [Google Scholar] [CrossRef]
- Berryman, M.; Allison, A.; Abbott, D. Mutual information for examining correlataions in DNA. Fluct. Noise Lett. 2004, 4, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Swati, D. Use of Mutual Information Function and Power Spectra for Analyzing the Structure of Some Prokaryotic Genomes. Am. J. Math. Manag. Sci. 2007, 27, 179–198. [Google Scholar] [CrossRef]
- Bauer, M.; Schuster, S.; Sayood, K. The average mutual information profile as a genomic signature. BMC Bioinform. 2008, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gregori-Puigjané, E.; Mestres, J. SHED: Shannon Entropy Descriptors from Topological Feature Distributions. J. Chem. Inf. Model. 2006, 46, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Dehnert, M.; Helm, W.E.; Hütt, M.T. Information Theory Reveals Large-Scale Synchronisation of Statistical Correlations in Eukaryote Genomes. Gene 2005, 345, 81–90. [Google Scholar] [CrossRef]
- Grosse, I.; Herzel, H.; Buldyrev, S.V.; Stanley, H.E. Species Independence of Mutual Information in Coding and Noncoding DNA. Phys. Rev. E 2000, 61, 5624–5629. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.T.; Farber, R.M.; Wolpert, D.H.; Lapedes, A.S. Covariation of Mutations in the V3 Loop of Human Immunodeficiency Virus Type 1 Envelope Protein: An Information Theoretic Analysis. Proc. Natl. Acad. Sci. USA 1993, 90, 7176–7180. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, F.; Antoneli, F.; Briones, M.R.S. MIA: Mutual Information Analyzer, a Graphic User Interface Program That Calculates Entropy, Vertical and Horizontal Mutual Information of Molecular Sequence Sets. BMC Bioinform. 2015, 16, 409. [Google Scholar] [CrossRef] [Green Version]
- Nalbantoglu, Ö.U.; Russell, D.J.; Sayood, K. Data Compression Concepts and Algorithms and Their Applications to Bioinform. Entropy 2010, 12, 34–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, C. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–432. [Google Scholar] [CrossRef] [Green Version]
- Rényi, A. On measures of entropy and information. In Proceedings of the Fourth Berkeley Symposium on Mathematical Statistics and Probability, Berkeley, CA, USA, 20–30 July 1960; Neyman, J., Ed.; University of California Press: Berkeley, CA, USA, 1961. [Google Scholar]
- Rényi, A. Probability Theory; North-Holland Publishing Company: Amsterdam, The Netherlands, 1970. [Google Scholar]
- Tsallis, C. Possible generalization of Bolzmann-Gibbs statistics. J. Math. Phys. 1988, 52, 479–487. [Google Scholar]
- Sparavigna, A. Mutual Information and Nonadditive Entropies: The Case of Tsallis Entropy. Int. J. Sci. 2015, 4. [Google Scholar] [CrossRef]
- Villmann, T.; Geweniger, T. Multi-class and Cluster Evaluation Measures Based on Rényi and Tsallis Entropies and Mutual Information. In Proceedings of the 17th International Conference on Artificial Intelligence and Soft Computing-ICAISC, Zakopane; Rutkowski, L., Scherer, R., Korytkowski, M., Pedrycz, W., Tadeusiewicz, R., Zurada, J., Eds.; Springer International Publishing: Cham, Switzerland, 2018; LNCS 10841; pp. 724–735. [Google Scholar] [CrossRef]
- Vinga, S.; Almeida, J. Local Rényi entropic profiles of DNA sequences. BMC Bioinform. 2007, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinga, S.; Almeida, J. Rényi continuous entropy of DNA sequences. J. Theor. Biol. 2004, 231, 377–388. [Google Scholar] [CrossRef]
- Delgado-Soler, L.; Toral, R.; Tomás, M.; Robio-Martinez, J. RED: A Set of Molecular Descriptors Based on Rényi Entropy. J. Chem. Inf. Model. 2009, 49, 2457–2468. [Google Scholar] [CrossRef]
- Papapetrou, M.; Kugiumtzis, D. Tsallis conditional mutual information in investigating long range correlation in symbol sequences. Phys. A 2020, 540, 1–13. [Google Scholar] [CrossRef]
- Gao, Y.; Luo, L. Genome-based phylogeny of dsDNA viruses by a novel alignment-free method. Gene 2012, 492, 309–314. [Google Scholar] [CrossRef]
- Schneider, P.; Biehl, M.; Hammer, B. Adaptive Relevance Matrices in Learning Vector Quantization. Neural Comput. 2009, 21, 3532–3561. [Google Scholar] [CrossRef] [Green Version]
- Saralajew, S.; Holdijk, L.; Villmann, T. Fast Adversarial Robustness Certification of Nearest Prototype Classifiers for Arbitrary Seminorms. In Proceedings of the 34th Conference on Neural Information Processing Systems (NeurIPS 2020), Virtual-only Conference, 6–12 December 2020; Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M., Lin, H., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2020; Volume 33, pp. 13635–13650. [Google Scholar]
- Cichocki, A.; Amari, S. Families of Alpha- Beta- and Gamma-Divergences: Flexible and Robust Measures of Similarities. Entropy 2010, 12, 1532–1568. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D. Inf. Theory, Inference Learn. Algorithms; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Kullback, S.; Leibler, R. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Kantz, H.; Schreiber, T. Nonlinear Time Series Analysis; Cambridge Nonlinear Science Series; Cambridge University Press: Cambridge, UK, 1997; Volume 7. [Google Scholar]
- Fraser, A.; Swinney, H. Independent coordinates for strange attractors from mutual information. Phys. Rev. A 1986, 33, 1134–1140. [Google Scholar] [CrossRef]
- Li, W. Mutual Information Functions of Natural Language Texts; Technical Report SFI-89-10-008; Santa Fe Institute: Santa Fe, NM, USA, 1989. [Google Scholar]
- Golub, G.; Loan, C.V. Matrix Computations, 4th ed.; Johns Hopkins Studies in the Mathematical Sciences; John Hopkins University Press: Baltimore, MD, USA, 2013. [Google Scholar]
- Horn, R.; Johnson, C. Matrix Analysis, 2nd ed.; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Erdogmus, D.; Principe, J.; II, K.H. Beyond second-order statistics for learning: A pairwise interaction model for entropy estimation. Nat. Comput. 2002, 1, 85–108. [Google Scholar] [CrossRef]
- Hild, K.; Erdogmus, D.; Principe, J. Blind Source Separation Using Rényi’s Mutual Information. IEEE Signal Process. Lett. 2001, 8, 174–176. [Google Scholar] [CrossRef]
- Jenssen, R.; Principe, J.; Erdogmus, D.; Eltoft, T. The Cauchy-Schwarz divergence and Parzen windowing: Connections to graph theory and Mercer kernels. J. Frankl. Inst. 2006, 343, 614–629. [Google Scholar] [CrossRef]
- Lehn-Schiøler, T.; Hegde, A.; Erdogmus, D.; Principe, J. Vector quantization using information theoretic concepts. Nat. Comput. 2005, 4, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Principe, J. Information Theoretic Learning; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Singh, A.; Principe, J. Information theoretic learning with adaptive kernels. Signal Process. 2011, 91, 203–213. [Google Scholar] [CrossRef]
- Villmann, T.; Haase, S. Divergence based vector quantization. Neural Comput. 2011, 23, 1343–1392. [Google Scholar] [CrossRef] [PubMed]
- Mwebaze, E.; Schneider, P.; Schleif, F.M.; Aduwo, J.; Quinn, J.; Haase, S.; Villmann, T.; Biehl, M. Divergence based classification in Learning Vector Quantization. Neurocomputing 2011, 74, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Bunte, K.; Haase, S.; Biehl, M.; Villmann, T. Stochastic Neighbor Embedding (SNE) for Dimension Reduction and Visualization Using Arbitrary Divergences. Neurocomputing 2012, 90, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Csiszár, I. Axiomatic Characterization of Information Measures. Entropy 2008, 10, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Fehr, S.; Berens, S. On the Conditional Rényi Entropy. IEEE Trans. Inf. Theory 2014, 60, 6801–6810. [Google Scholar] [CrossRef]
- Teixeira, A.; Matos, A.; Antunes, L. Conditional Rényi Entropies. IEEE Trans. Inf. Theory 2012, 58, 4273–4277. [Google Scholar] [CrossRef]
- Iwamoto, M.; Shikata, J. Revisiting Conditional Rényi Entropies and Generalizing Shannons Bounds in Information Theoretically Secure Encryption; Technical Report; Cryptology ePrint Archive 440/2013; International Association for Cryptologic Research (IACR): Lyon, France, 2013. [Google Scholar]
- Ilić, V.; Djordjević, I.; Stanković, M. On a General Definition of Conditional Rényi Entropies. In Proceedings of the 4th International Electronic Conference on Entropy and Its Application (ECEA 2017), Online, 21 November–1 December 2017; Volume 2, pp. 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jizba, P.; Arimitsu, T. The world according to Rényi: Thermodynamics of multifractal systems. In AIP Conference Proceedings; American Institute of Physics (AIP): Ellipse College Park, MD, USA, 2001; Volume 597, pp. 341–348. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Verdú, S. Conditional Rényi divergence saddlepoint and the maximization of α-mutual information. Entropy 2020, 21, 316. [Google Scholar] [CrossRef] [Green Version]
- Havrda, J.; Charvát, F. Quantification method of classification processes: Concept of structrual α-entropy. Kybernetika 1967, 3, 30–35. [Google Scholar]
- Vila, M.; Bardera, A.; Sbert, M.F.M. Tsallis Mutual Information for Document Classification. Entropy 2011, 13, 1694–1707. [Google Scholar] [CrossRef]
- Kohonen, T. Learning Vector Quantization. Neural Networks 1988, 1, 303. [Google Scholar]
- Kohonen, T. Self-Organizing Maps; Springer Series in Information Sciences; Springer: Berlin/Heidelberg, Germany, 1995; Volume 30. [Google Scholar]
- Biehl, M.; Hammer, B.; Villmann, T. Prototype-based Models for the Supervised Learning of Classification Schemes. Proc. Int. Astron. Union 2017, 12, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Yamada, K. Generalized learning vector quantization. In Advances in Neural Information Processing Systems 8, Proceedings of the 1995 Conference; Touretzky, D.S., Mozer, M.C., Hasselmo, M.E., Eds.; MIT Press: Cambridge, MA, USA, 1996; pp. 423–429. [Google Scholar]
- Bunte, K.; Schneider, P.; Hammer, B.; Schleif, F.M.; Villmann, T.; Biehl, M. Limited Rank Matrix Learning, discriminative dimension reduction and visualization. Neural Netw. 2012, 26, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Villmann, T.; Bohnsack, A.; Kaden, M. Can Learning Vector Quantization be an Alternative to SVM and Deep Learning? J. Artif. Intell. Soft Comput. Res. 2017, 7, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Villmann, T. Generalized Relevance Learning Vector Quantization. Neural Netw. 2002, 15, 1059–1068. [Google Scholar] [CrossRef]
- Biehl, M.; Hammer, B.; Villmann, T. Prototype-based models in machine learning. Wiley Interdiscip. Rev. Cogn. Sci. 2016, 7, 92–111. [Google Scholar] [CrossRef]
- Crammer, K.; Gilad-Bachrach, R.; Navot, A.; Tishby, A. Margin analysis of the LVQ algorithm. In Advances in Neural Information Processing (Proc. NIPS 2002); Becker, S., Thrun, S., Obermayer, K., Eds.; MIT Press: Cambridge, MA, USA, 2003; Volume 15, pp. 462–469. [Google Scholar]
- Garant, J.M.; Perreault, J.P.; Scott, M.S. Motif Independent Identification of Potential RNA G-Quadruplexes by G4RNA Screener. Bioinformatics 2017, 33, 3532–3537. [Google Scholar] [CrossRef] [Green Version]
- Garant, J.M.; Luce, M.J.; Scott, M.S.; Perreault, J.P. G4RNA: An RNA G-Quadruplex Database. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Liu, Y.; Shi, Y.; Huang, H.; Deng, B.; Xiao, X. A Classification Model for lncRNA and mRNA Based on K-Mers and a Convolutional Neural Network. BMC Bioinform. 2019, 20, 469. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic Network Analysis of SARS-CoV-2 Genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 9241–9243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Ho, Y.k.; Yau, S. Clustering DNA Sequences by Feature Vectors. Mol. Phylogenet. Evol. 2006, 41, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. Nomenclature for Incompletely Specified Bases in Nucleic Acid Sequences: Recommendations 1984. Nucleic Acids Res. 1985, 13, 3021–3030. [Google Scholar] [CrossRef]
- Yu, C.; Hernandez, T.; Zheng, H.; Yau, S.C.; Huang, H.H.; He, R.L.; Yang, J.; Yau, S.S.T. Real Time Classification of Viruses in 12 Dimensions. PLoS ONE 2013, 8, e64328. [Google Scholar] [CrossRef]
- Blaisdell, B.E. Average Values of a Dissimilarity Measure Not Requiring Sequence Alignment Are Twice the Averages of Conventional Mismatch Counts Requiring Sequence Alignment for a Variety of Computer-Generated Model Systems. J. Mol. Evol. 1989, 29, 538–547. [Google Scholar] [CrossRef]
- Goldberg, Y. Neural Network Methods for Natural Language Processing. Synth. Lect. Hum. Lang. Technol. 2017, 10, 1–309. [Google Scholar] [CrossRef]
- Kaden, M.; Bohnsack, K.S.; Weber, M.; Kudła, M.; Gutowska, K.; Blazewicz, J.; Villmann, T. Learning Vector Quantization as an Interpretable Classifier for the Detection of SARS-CoV-2 Types Based on Their RNA Sequences. Neural Comput. Appl. 2021, 1–12. [Google Scholar] [CrossRef]
- Riley, P. Three pitfalls to avoid in machine learning. Nature 2019, 572, 27–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csiszár, I. Information-type measures of differences of probability distributions and indirect observations. Studia Sci. Math. Hungaria 1967, 2, 299–318. [Google Scholar]
- Hnizdo, V.; Tan, J.; Killian, B.J.; Gilson, M.K. Efficient Calculation of Configurational Entropy from Molecular Simulations by Combining the Mutual-Information Expansion and Nearest-Neighbor Methods. J. Comput. Chem. 2008, 29, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Kolekar, P.; Kale, M.; Kulkarni-Kale, U. Alignment-Free Distance Measure Based on Return Time Distribution for Sequence Analysis: Applications to Clustering, Molecular Phylogeny and Subtyping. Mol. Phylogenet. Evol. 2012, 65, 510–522. [Google Scholar] [CrossRef]
- Wei, D.; Jiang, Q.; Wei, Y.; Wang, S. A Novel Hierarchical Clustering Algorithm for Gene Sequences. BMC Bioinform. 2012, 13, 174. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, X.; Li, X.; Ma, B.; Vitanyi, P. The Similarity Metric. IEEE Trans. Inf. Theory 2004, 50, 3250–3264. [Google Scholar] [CrossRef]
- Yin, C.; Chen, Y.; Yau, S.S.T. A Measure of DNA Sequence Similarity by Fourier Transform with Applications on Hierarchical Clustering. J. Theor. Biol. 2014, 359, 18–28. [Google Scholar] [CrossRef]
- Bao, J.; Yuan, R. A Wavelet-Based Feature Vector Model for DNA Clustering. Genet. Mol. Res. 2015, 14, 19163–19172. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.A.; Mitra, S.K.; Carli, M.; Neri, A. New Approaches to Genome Sequence Analysis Base Don Digital Signal Processing. Proceedings of IEEE Workshop on Genomic Signal Processing and Statistics (GENSIPS), Raleigh, NC, USA, 12–13 October 2002; p. 4. [Google Scholar]
- Almeida, J.S.; Vinga, S. Universal Sequence Map (USM) of Arbitrary Discrete Sequences. BMC Bioinform. 2002, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vellido, A. The importance of interpretability and visualization in machine learning for applications in medicine and health care. Neural Netw. Appl. 2020, 32, 18069–18083. [Google Scholar] [CrossRef] [Green Version]
- Bittrich, S.; Kaden, M.; Leberecht, C.; Kaiser, F.; Villmann, T.; Labudde, D. Application of an Interpretable Classification Model on Early Folding Residues during Protein Folding. Biodata Min. 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, L.; Hammer, B.; Wersing, H. Efficient rejection strategies for prototype-based classification. Neurocomputing 2015, 169, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Villmann, A.; Kaden, M.; Saralajew, S.; Villmann, T. Probabilistic Learning Vector Quantization with Cross-Entropy for Probabilistic Class Assignments in Classification Learning. In Proceedings of the 17th International Conference on Artificial Intelligence and Soft Computing-ICAISC, Zakopane, Zakopane, Poland, 3–7 June 2018; Rutkowski, L., Scherer, R., Korytkowski, M., Pedrycz, W., Tadeusiewicz, R., Zurada, J., Eds.; Springer International Publishing: Cham, Switzerland, 2018; LNCS 10841; pp. 736–749. [Google Scholar] [CrossRef]
- Saralajew, S.; Holdijk, L.; Rees, M.; Asan, E.; Villmann, T. Classification-by-Components: Probabilistic Modeling of Reasoning over a Set of Components. In Proceedings of the 33rd Conference on Neural Information Processing Systems (NeurIPS 2019), Vancouver, BC, Canada, 8–14 December 2019; MIT Press: Cambridge, MA, USA, 2019; pp. 2788–2799. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Data Set | Classes | Sequences | Per Class * | Mean Length | Std. Length |
|---|---|---|---|---|---|
| Quadruplex detection | 2 | 368 | 175/193 | 62.1 | 43.7 |
| lncRNA vs. mRNA | 2 | 20,000 | 10,000 each | 1197.3 | 710.8 |
| COVID types | 3 | 156 | 44/90/22 | 29,862.9 | 34.1 |
| MIF | rMIF | |
|---|---|---|
| Shannon | ||
| Rényi |
| Data Set | NV | MIF | rMIF | ||
|---|---|---|---|---|---|
| Shannon | Rényi | Shannon | Rényi | ||
| Quadruplex detection | |||||
| lncRNA vs. mRNA | |||||
| COVID types | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohnsack, K.S.; Kaden, M.; Abel, J.; Saralajew, S.; Villmann, T. The Resolved Mutual Information Function as a Structural Fingerprint of Biomolecular Sequences for Interpretable Machine Learning Classifiers. Entropy 2021, 23, 1357. https://doi.org/10.3390/e23101357
Bohnsack KS, Kaden M, Abel J, Saralajew S, Villmann T. The Resolved Mutual Information Function as a Structural Fingerprint of Biomolecular Sequences for Interpretable Machine Learning Classifiers. Entropy. 2021; 23(10):1357. https://doi.org/10.3390/e23101357
Chicago/Turabian StyleBohnsack, Katrin Sophie, Marika Kaden, Julia Abel, Sascha Saralajew, and Thomas Villmann. 2021. "The Resolved Mutual Information Function as a Structural Fingerprint of Biomolecular Sequences for Interpretable Machine Learning Classifiers" Entropy 23, no. 10: 1357. https://doi.org/10.3390/e23101357